

Abstract
Background
Presynaptic effects of general anaesthetics are not well characterized. We tested the hypothesis that isoflurane exhibits transmitter-specific effects on neurotransmitter release from neurochemically and functionally distinct isolated mammalian nerve terminals.
Methods
Nerve terminals from adult male rat brain were prelabelled with [3H]glutamate and [14C]GABA (cerebral cortex), [3H]norepinephrine (hippocampus), [14C]dopamine (striatum), or [3H]choline (precursor of [3H]acetylcholine; striatum). Release evoked by depolarizing pulses of 4-aminopyridine (4AP) or elevated KCl was quantified using a closed superfusion system.
Results
Isoflurane at clinical concentrations (<0.7 mM; ∼2 times median anaesthetic concentration) inhibited Na+ channel-dependent 4AP-evoked release of the five neurotransmitters tested in a concentration-dependent manner. Isoflurane was a more potent inhibitor [expressed as IC50 (sem)] of glutamate release [0.37 (0.03) mM; P<0.05] compared with the release of GABA [0.52 (0.03) mM], norepinephrine [0.48 (0.03) mM], dopamine [0.48 (0.03) mM], or acetylcholine [0.49 (0.02) mM]. Inhibition of Na+ channel-independent release evoked by elevated K+ was not significant at clinical concentrations of isoflurane, with the exception of dopamine release [IC50=0.59 (0.03) mM].
Conclusions
Isoflurane inhibited the release of the major central nervous system neurotransmitters with selectivity for glutamate release, consistent with both widespread inhibition and nerve terminal-specific presynaptic effects. Glutamate release was most sensitive to inhibition compared with GABA, acetylcholine, dopamine, and norepinephrine release due to presynaptic specializations in ion channel expression, regulation, and/or coupling to exocytosis. Reductions in neurotransmitter release by volatile anaesthetics could contribute to altered synaptic transmission, leading to therapeutic and toxic effects involving all major neurotransmitter systems.
Keywords: acetylcholine, γ-aminobutyric acid, anaesthetics, dopamine, exocytosis, glutamate, Na+ channels, nerve terminal, neurotransmitter release, norepinephrine
Editor's key points.
The effects of isoflurane on neurotransmitter release were studied in isolated rat nerve terminals in vitro.
Isoflurane inhibited presynaptic release of five major neurotransmitters.
Such effects have implications for therapeutic and toxic effects of volatile anaesthetic agents involving altered synaptic transmission.
General anaesthetics exert profound effects on the central nervous system (CNS) primarily by altering synaptic transmission.1 These effects involve both presynaptic, postsynaptic, and extrasynaptic actions that vary between anaesthetic agents and brain regions. These pharmacological actions lead to general depression of fast excitatory and/or enhancement of fast inhibitory synaptic transmission mediated by the principal excitatory and inhibitory neurotransmitters glutamate and γ-aminobutyric acid (GABA), respectively.2 The relative importance and mechanisms of anaesthetic effects on the balance between excitatory and inhibitory synaptic transmission, mediated primarily by glutamate and GABA and also by other neurotransmitters, are unknown.
Volatile inhaled anaesthetics at clinical concentrations inhibit depolarization-evoked release of the major excitatory (glutamate) and inhibitory (GABA) neurotransmitters from isolated brain and spinal cord nerve terminals.3–5 Inhibition of the release of the excitatory neurotransmitter glutamate occurs with greater potency than the release of the inhibitory neurotransmitter GABA, consistent with greater depression of excitatory relative to inhibitory signalling.5,6 This presynaptic selectivity, in conjunction with potentiation of postsynaptic and extrasynaptic GABAA receptors, provides potentially synergistic pathways to CNS depression by volatile anaesthetics based on electrophysiological data obtained in isolated cellular and brain slice preparations.1,7,8 The relative potencies of anaesthetic effects on the release of other major CNS neurotransmitters have not been reported.
The release of neurotransmitters of different classes involves distinct cellular and molecular mechanisms between fast transmitters, catecholamines, and neuropeptides.9–11 In order to gain further insight into the specificity of the presynaptic actions of anaesthetics, we compared the effects of the model volatile anaesthetic isoflurane on the release of three classical fast neurotransmitters packaged in small synaptic vesicles (glutamate, GABA, acetylcholine), and of two catecholamine neurotransmitters packaged in small dense-core vesicles (dopamine and norepinephrine). Release was evoked by 4-aminopyridine (4AP), a stimulus that mimics action-potential-evoked neurotransmitter release in requiring sequential activation of nerve terminal voltage-gated Na+ and Ca2+ channels, or by elevated K+ that only requires activation of Ca2+ channels, independent of Na+ channel activation.12 We tested the hypothesis that isoflurane differentially inhibits the release of specific neurotransmitters from neurochemically and functionally distinct nerve terminals prepared from three different regions of adult rat brain. This approach provides the most accessible approach to studying stimulus–secretion coupling of multiple transmitter types from different brain regions in isolation of interfering postsynaptic effects.13
Methods
Materials
Isoflurane was from Abbott Laboratories (North Chicago, IL, USA). 4AP, choline oxidase, buffer constituents, and other drugs were from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). l-[3H]Glutamate (60 Ci mmol−1), [14C]GABA (55 mCi mmol−1), and [14C]dopamine (55 mCi mmol−1) were from American Radiolabel Chemicals Inc. (St Louis, MO, USA), and [3H]norepinephrine (15 Ci mmol−1) and [3H]choline (86 Ci mmol−1) were from PerkinElmer Inc. (Boston, MA, USA).
Nerve terminal preparation and neurotransmitter loading
Experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals as approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee and conformed to ARRIVE guidelines. Efforts were made to minimize the numbers and suffering of animals used; animals were allowed food and water ad libidum, and were housed in an approved facility with environmental controls and 12 h light/dark cycling. Synaptosomes were prepared as previously described14,15 from the cerebral cortex, hippocampus, or stratum from adult (200–300 g) male Sprague–Dawley rats (Charles River Laboratories, Troy, NY, USA) killed by inhalation of 80% CO2 in oxygen followed by decapitation. Demyelinated synaptosomes from regions rich in nerve terminals containing specific neurotransmitters were suspended in Krebs–HEPES buffer (KHB, composition in mM: NaCl 140, KCl 5, HEPES 20, MgCl2 1, Na2HPO4 1.2, NaHCO3 5, EGTA 0.1, and d-glucose 10, pH 7.4 with NaOH) and loaded with radiolabelled neurotransmitters as follows: cerebrocortical synaptosomes with both 10 nM l-[3H]glutamate and 500 nM [14C]GABA for 15 min at 30°C; hippocampal synaptosomes with 200 nM [3H]norepinephrine for 45 min at 35°C; and striatal synaptosomes with either 5 µM [14C]dopamine for 45 min at 35°C, or with 20 nM [3H]choline for 30 min at 35°C. Internalized [3H]choline is rapidly converted to [3H]acetylcholine by choline acetyltransferase.16 Experiments involving dopamine and norepinephrine included 10 µM pargyline to minimize metabolism, and either 100 nM desipramine or 10 nM GBR-12935, respectively, to prevent non-specific uptake, as demonstrated in control experiments (data not shown). After loading, synaptosomes were collected by centrifugation for 10 min at 20 000g at 4°C, resuspended in ice-cold 0.32 M sucrose, and loaded into release chambers.
Transmitter release assays
Release of all neurotransmitter was assayed as we have previously described14 by continuous superfusion at 0.5 ml min−1 with KHB plus 0.1 mM EGTA at 37°C using a customized Brandel SF12 superfusion apparatus (Gaithersburg, MD, USA) set to collect 1 min fractions. Stock solutions of isoflurane (∼12 mM) were prepared in KHB, diluted to aqueous concentrations equivalent to 0.1–8 times median alveolar concentration (MAC=0.35 mM for isoflurane in rat at 37°C),17 and stored in gas-tight glass syringes connected to the superfusion apparatus. Isoflurane solutions, quantified by gas chromatography, were perfused for 12 min before, during, and after, 2 min depolarizing pulses of either 100 µM 4AP, 1 mM 4AP, or 15 mM KCl (with additional KCl replacing equimolar NaCl in KHB) in the absence or presence of 1.9 mM free extracellular Ca2+. Experiments were terminated by synaptosomal lysis after superfusion with 0.2 M perchloric acid. Radioactivity in each 1 min fraction was quantified by liquid scintillation spectrometry with single or dual isotope quench correction. Released acetylcholine was separated from the precursor [3H]choline in each fraction by enzymatic conversion of choline to betaine lysed synaptosome supernatant adjusted to pH 7.4–8.0 with NaOH using choline oxidase (150 mU) at 37°C for 30 min. [3H]Acetylcholine was extracted into 500 µl of tetraphenylborate/butyronitrile (10:1 w/v) and radioactivity quantified in 200 µl samples by liquid scintillation spectrometry. Extracted tritium radioactivity was confirmed as >95% acetylcholine by cellulose chromatography (data not shown).
Secretogogues
Depolarization-evoked neurotransmitter release was stimulated with either 100 µM 4AP, 1 mM 4AP, or 15 mM KCl. 4AP-evoked release requires activation of both voltage-gated Na+ (Nav) and Ca2+ (Cav) channels by inducing repetitive spontaneous nerve terminal depolarizations.12 The use of 1 mM 4AP is often employed to maximize release, but is less dependent on Nav function than release evoked by 100 µM 4AP, but is included to facilitate comparisons between secretogogues at equi-effective concentrations.18 Depolarization of isolated nerve terminals by elevated extracellular K+ concentration evokes Ca2+-dependent neurotransmitter release that requires Cav activation but is independent of Nav activation.12 Depolarization by 4AP or elevated K+ was used to distinguish between Na+ channel- and/or Ca2+ channel-mediated pharmacological effects on neurotransmitter release from isolated nerve terminals.12
Data analysis
Labelled transmitter released in each 1 min sample of superfusate was expressed as fractional release (FR), or the radioactivity released as a fraction of the total synaptosomal radioactivity present before that sample. Stimulus-evoked release was determined by subtracting baseline FR from cumulative FR values over the duration of the release pulse (sum ΔFR).5 Sum ΔFR data (area under the FR time curve) from each experiment were normalized by the ratio of each assay control to mean control release in the presence of Ca2+ [mean (sd)] from all experiments before curve fitting or statistical analysis (Prism 5.0; GraphPad Software, San Diego, CA, USA).
Data for inhibition of evoked release were fitted to concentration–effect curves by least-squares analysis to estimate IC50 (se) and analysed as described.14 Significant differences between curve fits were determined by F-test comparisons between best-fit values derived from separate curve fits to global fits with the parameter shared. Ca2+ dependence was determined as the percentage difference of the mean normalized sum ΔFR in the absence of Ca2+ and that in the presence of Ca2+ expressed as mean (sd). Control-evoked release and Ca2+ dependence data were analysed by Student's t-test or one-way analysis of variance (anova) with the Tukey post hoc test for multiple comparisons, respectively.
Results
Each of the five transmitters analysed was released in response to depolarization with both secretogogues, but with different degrees of Ca2+ dependence both between secretogogues and transmitters (Table 1). Ca2+ dependence is a property of evoked transmitter release in vivo, and was greatest for release evoked by 100 µM 4AP and 15 mM KCl. These conditions were used for comparisons of the effects of isoflurane on Na+ channel-dependent (4AP-evoked) with Na+ channel-independent (KCl-evoked) release of each transmitter in the presence of extracellular Ca2+.
Table 1.
Ca2+ dependence of neurotransmitter release evoked by different secretogogues. Data shown as mean (sd) (n). Ca2+ dependence was determined as the percentage of the mean normalized sum ΔFR in the absence of Ca2+ compared to that in the presence of Ca2+. ND, not determined. *P<0.05, ***P<0.001 vs glutamate; †††P<0.001 vs GABA; ‡‡‡ P<0.001 vs norepinephrine, by one-way anova with the Tukey post hoc test
| Neurotransmitter | 100 μM 4AP (%) | 1.0 mM 4AP (%) | 15 mM KCl (%) |
|---|---|---|---|
| Glutamate | 68 (6) (9) | 59 (11) (33) | 42 (12) (9) |
| GABA | 78 (3) (9)*** | 72 (6) (33)*** | 56 (12) (9)* |
| Acetylcholine | 97 (5) (6)***,††† | ND | 90 (5) (6)***,††† |
| Norepinephrine | 90 (2) (6)***,††† | 81 (6) (8)***,††† | 90 (5) (6)***,††† |
| Dopamine | 91 (5) (6)***,††† | 37 (9) (10)***,†††,‡‡‡ | 86 (10) (6)***,††† |
Glutamate release
A 2 min stimulus pulse of 100 µM 4AP evoked release of glutamate from cortical nerve terminals [0.029 (0.008) sum ΔFR, n=15] that was completely inhibited by isoflurane in a concentration-dependent manner [Fig. 1; IC50=0.37 (0.03) mM]. Stimulation with a 2 min pulse of 1 mM 4AP evoked significantly more release of glutamate [0.058 (0.008) sum ΔFR, n=18; P<0.0001] compared with that evoked by 100 µM 4AP, which isoflurane completely inhibited with comparable potency [IC50=0.42 (0.03 mM)]. A 2 min pulse of 15 mM KCl also stimulated greater release of glutamate [0.064 (0.010) sum ΔFR, n=5; P<0.0001] than that evoked by 100 µM 4AP, but comparable with 1 mM 4AP-evoked release. Isoflurane inhibited KCl-evoked release with reduced potency compared with 4AP-evoked release [IC50=3.0 (0.97) mM]. The Ca2+ dependence of glutamate release, which was greatest for 100 μM 4AP-evoked release, was significantly lower than that observed for the other four transmitters (Table 1).
Fig 1.
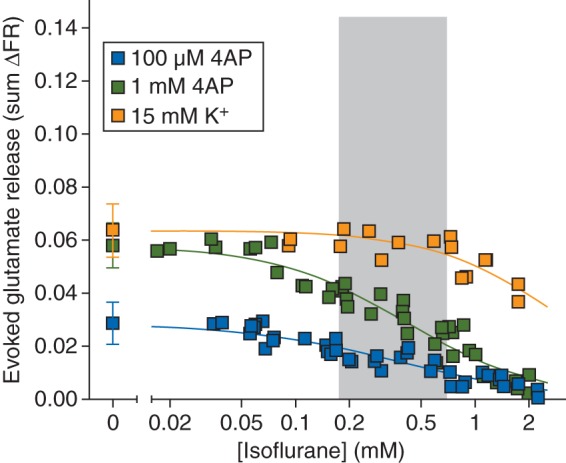
Inhibition by isoflurane of glutamate release evoked by 100 µM or 1 mM 4AP, or by 15 mM extracellular K+. Control release values are shown on the left [mean (sd)]. Shaded area indicates the clinical concentration range of isoflurane for general anaesthesia in rat (0.5–2 times MAC).
GABA release
A 2 min stimulus pulse of 100 µM 4AP evoked release of GABA from cortical nerve terminals (0.064 [0.016] sum ΔFR, n=16) that was completely inhibited by isoflurane in a concentration-dependent manner [Fig. 2; IC50=0.52 (0.03) mM]. Stimulation with a 2 min pulse of 1 mM 4AP evoked significantly more release of GABA [0.11 (0.01) sum ΔFR, n=17; P<0.0001] compared with that evoked by 100 µM 4AP, and isoflurane completely inhibited release with comparable potency [IC50=0.56 (0.02) mM]. A 2 min pulse of 15 mM KCl stimulated release of GABA [0.072 (0.011) sum ΔFR; n=5] comparable with that evoked by 100 µM 4AP. Isoflurane inhibited KCl-evoked release with reduced potency compared with 4AP-evoked release [IC50=1.9 (0.20) mM]. The Ca2+ dependence of GABA release, which was greatest for 100 μM 4AP-evoked GABA release, was significantly lower than that for acetylcholine, norepinephrine, and dopamine but greater than that of glutamate (Table 1).
Fig 2.
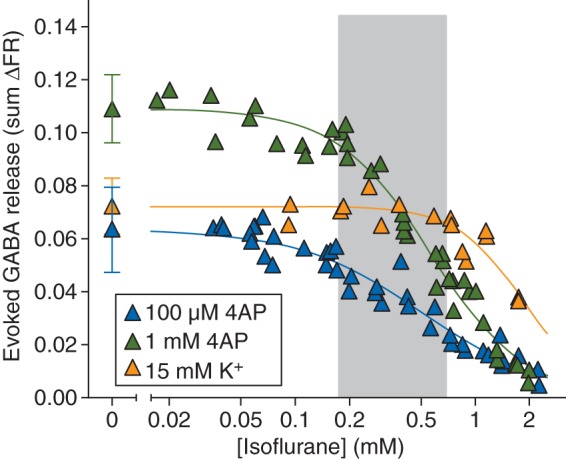
Inhibition by isoflurane of GABA release evoked by 100 µM or 1 mM 4AP, or by 15 mM extracellular K+. Control release values are shown on the left [mean (sd)]. Shaded area indicates the clinical concentration range of isoflurane for general anaesthesia in rat (0.5–2 times MAC).
Acetylcholine release
A 2 min stimulus pulse of 100 µM 4AP evoked release of acetylcholine from striatal nerve terminals [0.097 (0.014) sum ΔFR, n=12] that was completely inhibited by isoflurane in a concentration-dependent manner [Fig. 3; IC50=0.49 (0.02) mM]. A 2 min pulse of 15 mM KCl evoked release of acetylcholine [0.083 (0.020); n=8] comparable with that evoked by 100 µM 4AP. Isoflurane inhibited KCl-evoked release with reduced potency compared with 4AP-evoked release [IC50=0.97 (0.04) mM), but with greater potency relative to KCl-evoked glutamate release. Acetylcholine release evoked by 4AP or KCl was highly Ca2+ dependent (Table 1).
Fig 3.
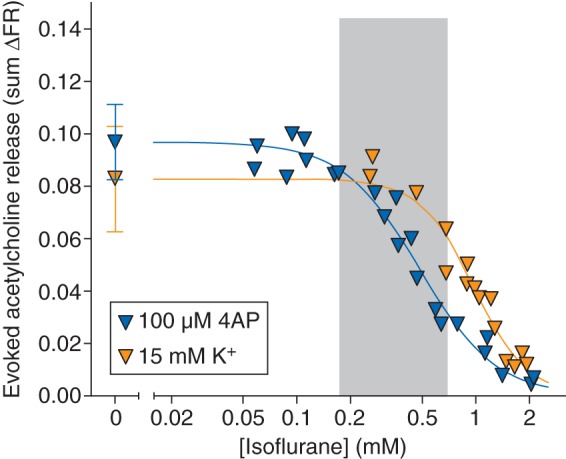
Inhibition by isoflurane of acetylcholine release evoked by 100 µM 4AP, or by 15 mM extracellular K+. Control release values are shown on the left [mean (sd)]. Shaded area indicates the clinical concentration range of isoflurane for general anaesthesia in rat (0.5–2 times MAC).
Norepinephrine release
A 2 min stimulus pulse of 100 µM 4AP evoked release of norepinephrine from hippocampal nerve terminals [0.080 (0.022) sum ΔFR, n=8] that was completely inhibited by isoflurane in a concentration-dependent manner [Fig. 4; IC50=0.48 (0.03) mM]. Stimulation with a 2 min pulse of 1 mM 4AP evoked significantly more release of norepinephrine [0.11 (0.05) sum ΔFR, n=29; P<0.0001] compared with that evoked by 100 µM 4AP; isoflurane completely inhibited release with reduced potency [IC50=0.95 (0.08) mM]. A 2 min pulse of 15 mM KCl stimulated release of norepinephrine [0.10 (0.01) sum ΔFR; n=6] comparable with that evoked by 100 µM 4AP. Isoflurane inhibited KCl-evoked release with greater potency relative to glutamate release and with similar potency compared with 1 mM 4AP-evoked release [IC50=1.1 (0.04) mM]. Norepinephrine release evoked by 4AP or KCl was highly Ca2+ dependent (Table 1).
Fig 4.
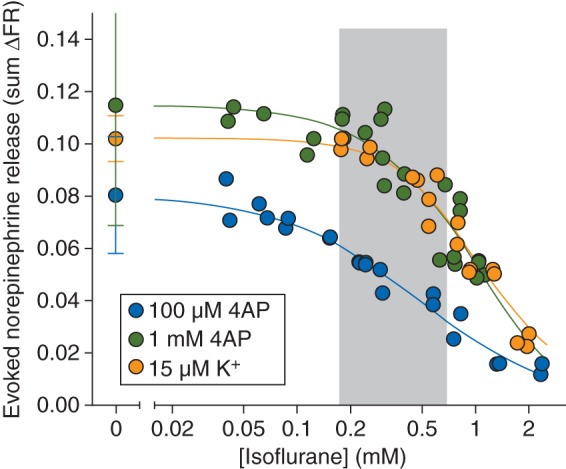
Inhibition by isoflurane of norepinephrine release evoked by 100 µM or 1 mM 4AP, or by 15 mM extracellular K+. Control release values are shown on the left [mean (sd)]. Shaded area indicates the clinical concentration range of isoflurane for general anaesthesia in rat (0.5–2 times MAC).
Dopamine release
A 2 min stimulus pulse of 100 µM 4AP evoked release of dopamine from striatal nerve terminals [0.052 (0.020) sum ΔFR, n=8] that was completely inhibited by isoflurane in a concentration-dependent manner [Fig. 5; IC50=0.48 (0.03) mM]. Stimulation with a 2 min pulse of 1 mM 4AP evoked significantly more release of dopamine [0.14 (0.05) sum ΔFR, n=9; P<0.0001] compared with that evoked by 100 µM 4AP; isoflurane completely inhibited release with reduced potency [IC50=1.2 (0.12) mM]. A 2 min pulse of 15 mM KCl stimulated release of dopamine [0.064 (0.012) sum ΔFR; n=6] comparable with that evoked by 100 µM 4AP. Isoflurane inhibited KCl-evoked release with greater potency relative to glutamate release and with greater potency compared with 1 mM 4AP-evoked release [IC50=0.59 (0.03) mM]. Dopamine release evoked by 100 µM 4AP or KCl was highly Ca2+-dependent (Table 1).
Fig 5.
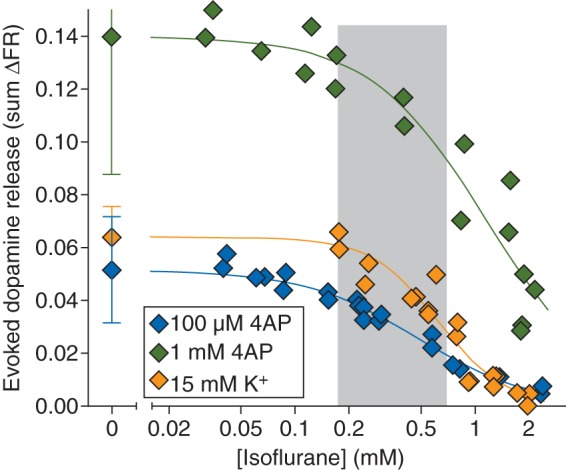
Inhibition by isoflurane of dopamine release evoked by 100 µM or 1 mM 4AP, or by 15 mM extracellular K+. Control release values are shown on the left [mean (sd)]. Shaded area indicates the clinical concentration range of isoflurane for general anaesthesia in rat (0.5–2 times MAC).
Comparisons between isoflurane effects
Glutamate release evoked by 100 µM 4AP was inhibited by isoflurane more potently than the release of GABA from cortical terminals (P=0.016), norepinephrine from hippocampal terminals (P=0.031), dopamine from striatal terminals (P=0.016), and acetylcholine from striatal terminals (P=0.047); 4AP-evoked release of the latter four transmitters was inhibited equipotently. Release of all five neurotransmitters was inhibited within a clinical concentration range defined as <2 MAC (twice the ED50; equivalent to a solution concentration of 0.7 mM; Fig. 6). With the exception of glutamate, release evoked by the higher concentration of 4AP was less sensitive to inhibition by isoflurane. With the exception of dopamine, isoflurane inhibited KCl-evoked release by >20% of control release only at concentrations above the clinical concentration range (Fig. 6). Differences between neurotransmitters in isoflurane potency for inhibition of release were not related to the magnitude of the release stimulus (measured as FR), as shown by the more potent inhibition of 1 mM 4AP-evoked glutamate release compared with 100 µM 4AP-evoked GABA release (P=0.014), for example.
Fig 6.
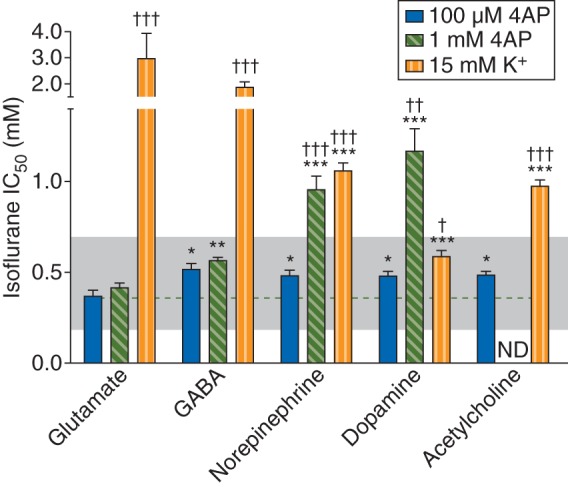
Inhibition by isoflurane of the release of various neurotransmitters evoked by 100 µM or 1 mM 4AP, or by 15 mM extracellular K+. Shaded area indicates the clinical concentration range of isoflurane for general anaesthesia in rat (0.5–2 times MAC). The dashed line shows the degree of inhibition of 100 μM 4AP-evoked glutamate release for comparison. *P<0.05, **P=0.0002, ***P<0.0001 vs glutamate release evoked by respective stimulus. †P<0.05, ††P=0.0003, †††P<0.0001 vs 100 µM 4AP-evoked release. ND, not determined.
Discussion
The relative contributions of pre- and postsynaptic effects of anaesthetics on synaptic transmission are difficult to resolve using standard brain slice and microdialysis approaches. Here, we show that the volatile anaesthetic isoflurane inhibits the release of five of the major transmitters implicated in the actions of general anaesthetics on the CNS.19 Differences in anaesthetic sensitivity between these neurotransmitter systems indicate neurotransmitter-specific specializations in presynaptic physiology that lead to pharmacological selectivity in anaesthetic effects on the release process. Importantly, isoflurane inhibits the release of the neurochemically distinct neurotransmitters GABA, acetylcholine, norepinephrine, and dopamine from isolated nerve terminals at clinically relevant concentrations, but with reduced potency and by distinct mechanisms compared with the release of the principal excitatory transmitter glutamate.
Neurotransmitters are selectively packaged into synaptic vesicles for storage and release by neurotransmitter- and nerve terminal-specific mechanisms that are poorly understood at a molecular level.9–11 This renders detailed determination of the mechanisms responsible for these differences difficult to resolve. The fundamental molecular mechanisms and machinery involved are common to all transmitters,20 but terminal-specific specializations contribute to distinct functional properties. These specializations include differences in presynaptic ion channels and their regulatory mechanisms,21 such as expression of specific voltage-gated Ca2+ channel subtypes22 and K+ channels,23 synaptic vesicle release machinery,24,25 and isoforms of the principal presynaptic Ca2+ sensor synaptotagmin.26,27 Such biochemical differences provide potential substrates for important pharmacological differences. Pharmacological characterization of presynaptic drug targets using currently available approaches is limited by the small size of typical CNS nerve terminals, but indirect approaches suggest that differential expression of presynaptic ion channels are likely involved.28
Neurotransmitter release is determined by the amount of Ca2+ entering the nerve terminal, coupling of Ca2+ entry to synaptic vesicle exocytosis, and the number of docked and primed vesicles.20 The amount of Ca2+ entry is determined by presynaptic Na+, Ca2+, and K+ channels that regulate excitability.29 The markedly greater sensitivity to isoflurane of 4AP-evoked release (Na+ channel-dependent) compared with KCl-evoked release (Na+ channel-independent) for all five transmitters studied likely indicates that targets upstream of Ca2+ entry and Ca2+-release coupling, such as Nav, are highly sensitive to volatile anaesthetics relative to downstream targets such as Cav and the synaptic vesicle fusion machinery, with the notable exception of dopamine release. Alternatively, the greater sensitivity of 4AP-evoked release could reflect synergistic interaction between effects of isoflurane on both upstream Na+ channel-dependent and the relatively insensitive downstream Ca2+-channel dependent mechanisms. The greater isoflurane sensitivity of glutamate release is possibly due to a relatively greater role or anaesthetic sensitivity of the specific Na+ channel subtype(s) expressed in glutamatergic terminals30 and/or to transmitter-specific differences in efficacy of presynaptic excitation/secretion coupling as proposed for other Na+ channel blockers.31 Other nerve terminal specializations that potentially influence the pharmacology of synaptic transmission include differences in expression of other presynaptic ion channels and other regulators of synaptic vesicle release. Presynaptic BK K+ channels, for example, selectively control glutamate compared with GABA release,23 and differential expression of SNAP-25 correlates with differences in Ca2+ responsiveness of release.24 Glutamate release is unique in its greater sensitivity to isoflurane compared with GABA release,5 and to the release of acetylcholine, norepinephrine, or dopamine, as we now show.
In contrast to the other transmitters studied, KCl-evoked dopamine release was inhibited by isoflurane with an IC50 only slightly higher than that for 100 μM 4AP-evoked release. Interestingly, this was not observed for the other catecholamine tested, norepinephrine, which indicates a greater anaesthetic sensitivity of Cav coupled to dopamine release and/or of mechanisms downstream of Ca2+ entry. The latter interpretation is consistent with findings that isoflurane inhibits exocytosis from dopamine-releasing PC12 rat pheochromocytoma cells stimulated with the Ca2+ ionophore ionomycin to bypass Ca2+ channels.32 Another unique feature of dopamine release is the much lower Ca2+ dependence and isoflurane IC50 for dopamine release evoked by 1 mM 4AP, possibly indicating effects on other targets affected at higher 4AP concentrations. Interestingly, different forms of stimulation evoke release from PC12 cells that requires distinct synaptotagmin isoforms,28 which might also determine the sensitivity to isoflurane. Certain types of glutamatergic terminals might also exhibit greater anaesthetic sensitivity of targets downstream of Na+ channel activation as suggested by electrophysiological evidence from the hippocampal CA1 synapse.33
Depolarization-evoked neurotransmitter release can occur by at least two major mechanisms: Ca2+-dependent vesicular release and reverse transport through membrane transporters.13 The latter mechanism is more significant with elevated extracellular K+-evoked release due to the sustained reversal of the resting membrane potential and K+ gradient, as evident in the reduced Ca2+ dependence of KCl-evoked glutamate and GABA release. Isoflurane at clinical concentrations does not significantly affect native glutamate or GABA transporters in isolated cortical nerve terminals34 or heterologously expressed dopamine or norepinephrine transporters,35 so it is likely that the effects of lower concentrations of isoflurane reported here result primarily from effects on vesicular release and ion channels that affect presynaptic excitation rather than directly on reverse transport. At higher concentrations, isoflurane inhibits both Ca2+-dependent and -independent KCl-evoked release by multiple mechanisms.30
Potential limitations of studies involving isolated nerve terminals include phenotypic heterogeneity in their neurones of origin, which could obscure pharmacological differences between terminals releasing the same transmitter, and the need to chemically evoke release. Supporting evidence includes the findings that volatile anaesthetics also inhibit action-potential-evoked synaptic vesicle exocytosis in single cultured neonatal hippocampal neurones36 and glutamate release in rat brain slice preparations.7,37 These limitations must be balanced by the advantages isolated terminals provide in analysing the presynaptic mechanisms of drug effects on transmitter release from mature adult neurones releasing various transmitters without interference from intrinsic neuronal networks or postsynaptic effects, as occurs in vivo or in brain slices and neurone cultures in vitro.13 While this approach allows selective investigation of the presynaptic effects of drugs in isolation of postsynaptic effects, postsynaptic actions also contribute to the net effect of drugs on synaptic transmission in vivo.
Neurochemical and electrophysiological evidence suggests that volatile anaesthetics inhibit glutamate release by blocking presynaptic Na+ channels, thereby inhibiting nerve terminal depolarization.15,28,29,37 Supportive electrophysiological evidence shows that isoflurane inhibits nerve terminal Na+ currents and action potential amplitude via Na+ channel block in isolated neurohypophysial nerve terminals38,39 and Na+ channel-mediated action potential amplitude in the giant calyx of Held.37 Evidence in vivo further supports a role for Na+ channel inhibition in immobilization by volatile anaesthetics.40,41
In summary, isoflurane at clinical concentrations directly inhibits the release of multiple neurotransmitters from isolated nerve terminals by transmitter-specific presynaptic mechanisms. These findings have important implications for the mechanisms of inhaled anaesthetics on neurotransmission in specific neuronal networks involving different neurotransmitters, and also general relevance to presynaptic neuropharmacology. The greater anaesthetic sensitivity of glutamate release implies a unique feature of glutamatergic terminals, possibly a greater dependence on Na+ channel function15,31 and/or contributions of additional inhibitory mechanisms.23 Differential anaesthetic effects on the release of the five major classical CNS neurotransmitters support terminal-specific expression of ion channel subtypes, signal transduction pathways, and/or Ca2+-exocytosis coupling mechanisms with distinct anaesthetic sensitivities. These differences are relevant to selective effects of anaesthetics on functionally specialized neuronal networks and pathways integral to the multiple dose-dependent neurological endpoints characteristic of general anaesthesia.19,42
Declaration of interest
H.C.H. is an Editor of the British Journal of Anaesthesia and a Senior Editor of Anesthesiology.
Funding
This study was supported by National Institutes of Health (grant GM 58055).
Acknowledgement
The authors wish to acknowledge helpful comments from members of the Hemmings laboratory.
References
- 1.Hemmings HC, Jr, Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL. Emerging molecular mechanisms of general anesthetic action. Trends Pharmacol Sci. 2005;26:503–10. doi: 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Sonner JM, Antognini JF, Dutton RC, et al. Inhaled anesthetics and immobility: mechanisms, mysteries, and minimum alveolar anesthetic concentration. Anesth Analg. 2003;97:718–40. doi: 10.1213/01.ANE.0000081063.76651.33. [DOI] [PubMed] [Google Scholar]
- 3.Miao N, Frazer MJ, Lynch C., III Volatile anesthetics depress Ca2+ transients and glutamate release in isolated cerebral synaptosomes. Anesthesiology. 1995;83:593–603. doi: 10.1097/00000542-199509000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Schlame M, Hemmings HC., Jr Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiology. 1995;82:1406–16. doi: 10.1097/00000542-199506000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Westphalen RI, Hemmings HC., Jr Selective depression by general anesthetics of glutamate versus GABA release from isolated cortical nerve terminals. J Pharmacol Exp Ther. 2003;304:1188–96. doi: 10.1124/jpet.102.044685. [DOI] [PubMed] [Google Scholar]
- 6.Westphalen RI, Hemmings HC., Jr Volatile anesthetic effects on glutamate versus GABA release from isolated rat cortical nerve terminals: four-aminopyridine evoked release. J Pharmacol Exp Ther. 2006;316:216–23. doi: 10.1124/jpet.105.090662. [DOI] [PubMed] [Google Scholar]
- 7.Kirson ED, Yaari Y, Perouansky M. Presynaptic and postsynaptic actions of halothane at glutamatergic synapses in the mouse hippocampus. Br J Pharmacol. 1998;124:1607–14. doi: 10.1038/sj.bjp.0701996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maclver MB, Mikulec AA, Amagasu SM, Monroe FA. Volatile anesthetics depress glutamate transmission via presynaptic actions. Anesthesiology. 1996;85:823–34. doi: 10.1097/00000542-199610000-00018. [DOI] [PubMed] [Google Scholar]
- 9.Ghijsen WE, Leenders AG, Lopes da Silva FH. Regulation of vesicle traffic and neurotransmitter release in isolated nerve terminals. Neurochem Res. 2003;28:1443–52. doi: 10.1023/a:1025606021867. [DOI] [PubMed] [Google Scholar]
- 10.Südhof TC. Neurotransmitter release. Handb Exp Pharmacol. 2008;184:1–21. doi: 10.1007/978-3-540-74805-2_1. [DOI] [PubMed] [Google Scholar]
- 11.Verhage M, McMahon HT, Ghijsen WE, et al. Differential release of amino acids, neuropeptides, and catecholamines from isolated nerve terminals. Neuron. 1991;6:517–24. doi: 10.1016/0896-6273(91)90054-4. [DOI] [PubMed] [Google Scholar]
- 12.Tibbs GR, Barrie AP, Van Mieghem FJE, McMahon HT, Nicholls DG. Repetitive action potentials in isolated nerve terminals in the presence of 4-aminopyridine: effects on cytosolic free Ca2+ and glutamate release. J Neurochem. 1989;53:1693–9. doi: 10.1111/j.1471-4159.1989.tb09232.x. [DOI] [PubMed] [Google Scholar]
- 13.Nicholls DG. Bioenergetics and transmitter release in the isolated nerve terminal. Neurochem Res. 2003;28:1433–41. doi: 10.1023/a:1025653805029. [DOI] [PubMed] [Google Scholar]
- 14.Westphalen RI, Hemmings HC., Jr Volatile anesthetic effects on glutamate versus GABA release from isolated rat cortical nerve terminals: basal release. J Pharmacol Exp Ther. 2006;316:208–15. doi: 10.1124/jpet.105.090647. [DOI] [PubMed] [Google Scholar]
- 15.Westphalen RI, Yu J, Krivitski M, Jih T-Y, Hemmings HC., Jr Regional differences in nerve terminal Na+ channel subtype expression and Na+ channel-dependent glutamate and GABA release in rat central nervous system. J Neurochem. 2010;113:1611–20. doi: 10.1111/j.1471-4159.2010.06722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamura HI, Snyder SH. High affinity transport of choline into synaptosomes of rat brain. J Neurochem. 1973;21:1355–74. doi: 10.1111/j.1471-4159.1973.tb06022.x. [DOI] [PubMed] [Google Scholar]
- 17.Taheri S, Halsey MJ, Liu J, Eger EI, II, Koblin DD, Laster MJ. What solvent best represents the site of action on inhaled anesthetics in humans, rats, and dogs. Anesth Analg. 1991;72:627–34. doi: 10.1213/00000539-199105000-00010. [DOI] [PubMed] [Google Scholar]
- 18.Galván E, Sitges M. Characterization of the participation of sodium channels on the rise in Na+ induced by 4-aminopyridine (4-AP) in synaptosomes. Neurochem Res. 2004;29:347–55. doi: 10.1023/b:nere.0000013737.17288.ce. [DOI] [PubMed] [Google Scholar]
- 19.Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–20. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- 20.Dittman J, Ryan TA. Molecular circuitry of endocytosis at nerve terminals. Annu Rev Cell Dev Biol. 2009;25:133–60. doi: 10.1146/annurev.cellbio.042308.113302. [DOI] [PubMed] [Google Scholar]
- 21.Meir A, Ginsburg S, Butkevich A, et al. Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol Rev. 1999;79:1019–88. doi: 10.1152/physrev.1999.79.3.1019. [DOI] [PubMed] [Google Scholar]
- 22.Reid CA, Clements JD, Bekkers JM. Nonuniform distribution of Ca2+ channel subtypes on presynaptic terminals of excitatory synapses in hippocampal cultures. J Neurosci. 1997;17:2738–45. doi: 10.1523/JNEUROSCI.17-08-02738.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martire M, Barrese V, D'Amico M, et al. Pre-synaptic BK channels selectively control glutamate versus GABA release from cortical and hippocampal nerve terminals. J Neurochem. 2010;115:411–22. doi: 10.1111/j.1471-4159.2010.06938.x. [DOI] [PubMed] [Google Scholar]
- 24.Verderio C, Pozzi D, Pravettoni E, et al. SNAP-25 modulation of calcium dynamics underlies differences in GABAergic and glutamatergic responsiveness to depolarization. Neuron. 2004;41:599–610. doi: 10.1016/s0896-6273(04)00077-7. [DOI] [PubMed] [Google Scholar]
- 25.Bragina L, Giovedì S, Barbaresi P, Benfenati F, Conti F. Heterogeneity of glutamatergic and GABAergic release machinery in cerebral cortex: analysis of synaptogyrin, vesicle-associated membrane protein, and syntaxin. Neuroscience. 2010;165:934–43. doi: 10.1016/j.neuroscience.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Mashimo T, Südhof TC. Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–81. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z, Wu Y, Wang Z, et al. Release mode of large and small dense-core vesicles specified by different synaptotagmin isoforms in PC12 cells. Mol Biol Cell. 2011;22:2324–36. doi: 10.1091/mbc.E11-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemmings HC., Jr Sodium channels and the synaptic mechanisms of inhaled anaesthetics. Br J Anaesth. 2009;103:61–9. doi: 10.1093/bja/aep144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Craig AM, Boudin H. Molecular heterogeneity of central synapses: afferent and target regulation. Nat Neurosci. 2001;4:569–78. doi: 10.1038/88388. [DOI] [PubMed] [Google Scholar]
- 30.Westphalen RI, Kwak N-B, Daniels K, Hemmings HC., Jr Regional differences in the effects of isoflurane on neurotransmitter release. Neuropharmacology. 2011;61:699–706. doi: 10.1016/j.neuropharm.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prakriya M, Mennerick S. Selective depression of low-release probability excitatory synapses by sodium channel blockers. Neuron. 2000;26:671–82. doi: 10.1016/s0896-6273(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 32.Herring BE, Xie Z, Marks J, Fox AP. Isoflurane inhibits the neurotransmitter release machinery. J Neurophysiol. 2009;102:1265–73. doi: 10.1152/jn.00252.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winegar BD, MacIver MB. Isoflurane depresses hippocampal CA1 glutamate nerve terminals without inhibiting fiber volleys. BMC Neurosci. 2006;7:5. doi: 10.1186/1471-2202-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westphalen RI, Hemmings HC., Jr Effects of isoflurane and propofol on glutamate and GABA transporters in isolated cortical nerve terminals. Anesthesiology. 2003;98:364–72. doi: 10.1097/00000542-200302000-00016. [DOI] [PubMed] [Google Scholar]
- 35.Shahani SK, Lingamaneni R, Hemmings HC., Jr General anesthetic actions on norepinephrine, dopamine, and gamma-aminobutyric acid transporters in stably transfected cells. Anesth Analg. 2002;95:893–9. doi: 10.1097/00000539-200210000-00019. [DOI] [PubMed] [Google Scholar]
- 36.Hemmings HC, Jr, Yan W, Westphalen RI, Ryan TA. The general anesthetic isoflurane depresses synaptic vesicle exocytosis. Mol Pharmacol. 2005;67:1591–9. doi: 10.1124/mol.104.003210. [DOI] [PubMed] [Google Scholar]
- 37.Wu XS, Sun JY, Evers AS, Crowder M, Wu LG. Isoflurane inhibits transmitter release and the presynaptic action potential. Anesthesiology. 2004;100:663–70. doi: 10.1097/00000542-200403000-00029. [DOI] [PubMed] [Google Scholar]
- 38.OuYang W, Wang G, Hemmings HC., Jr Isoflurane and propofol inhibit presynaptic Na+ channels in isolated rat neurohypophysial nerve terminals. Mol Pharmacol. 2003;64:373–81. doi: 10.1124/mol.64.2.373. [DOI] [PubMed] [Google Scholar]
- 39.OuYang W, Hemmings HC., Jr Depression by isoflurane of the action potential and underlying voltage-gated ion currents in isolated rat neurohypophysial nerve terminals. J Pharmacol Exp Ther. 2005;312:801–8. doi: 10.1124/jpet.104.074609. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Sharma M, Eger EI, II, Laster MJ, Hemmings HC, Jr, Harris RA. Intrathecal veratridine administration increases minimum alveolar concentration in rats. Anesth Analg. 2008;107:875–8. doi: 10.1213/ane.0b013e3181815fbc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Guzinski M, Eger EI, II, et al. Bidirectional modulation of isoflurane potency by intrathecal tetrodotoxin and veratridine in rats. Br J Pharmacol. 2010;159:872–8. doi: 10.1111/j.1476-5381.2009.00583.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Dort CJ, Baghdoyan HA, Lydic R. Neurochemical modulators of sleep and anesthetic states. Int Anesthesiol Clin. 2008;46:75–104. doi: 10.1097/AIA.0b013e318181a8ca. [DOI] [PMC free article] [PubMed] [Google Scholar]