

Abstract
Purpose
This study aims to investigate μ-calpain expression profiles in the anterior temporal neocortex in patients with intractable epilepsy, and to determine whether its pattern of expression is related to pathological changes seen in these patients.
Methods
The study subjects consisted of 30 patients with intractable epilepsy and a control group of 10 patients with brain trauma who underwent resection of the anterior temporal lobe. μ-Calpain expression in surgically resected anterior temporal cortices of patients with intractable epilepsy were analyzed using the RT-PCR, Western blot, immnohistochemistry and immunofluorescence staining. GFAP expression was detected by immunohistochemical staining. The related pro-inflammatory cytokines were quantified by elisa. Clinicopathological characteristics were evaluated by HE staining.
Results
Analysis by Western blot and RT-PCR revealed that inactive μ-calpain expression and the calpain-cleaved spectrin fragment in surgically resected anterior temporal cortices of patients with intractable epilepsy were significantly increased compared to the tissues from corresponding regions of the control group. Immunohistological staining demonstrated that μ-calpain was overexpressed in the cell cytoplasm of neurons and glial cells in patients with intractable epilepsy and GFAP was overexpressed in the cell cytoplasm of glial cells in patients with intractable epilepsy. The level of pro-inflammatory cytokines, such as IL-1β, IL-6 and TGF-β1 were significantly increased in patients with intractable epilepsy. HE staining indicated μ-calpain overexpression is an independent prognostic factor for pathological changes such as neuronal loss, neuronal degeneration, gliosis and astrocytosis.
Conclusion
These data suggest that overexpression of μ-calpain is relationship with intractable epilepsy as well as the clinicopathological characteristics in these patients.
Keywords: Intractable epilepsy, μ-Calpain, Clinicopathological characteristics
1. Introduction
Epilepsy is a common devastating neurological condition. Although 80% of epileptic patients are seizure-free with rational anti-epileptic drug (AED) therapy, approximately 20% of patients with epilepsy have a medically intractable condition even when treated with various AEDs at maximal dosages, either alone or in combination.1 Such epilepsy cases are, thus, referred to as intractable epilepsy (IE).
Recurrent seizures may cause necrosis within certain cell populations.2,3 Studies have also identified upregulation of bcl-2 family genes, including Bcl-2, Bcl-XL and activated caspase 3 in temporal neocortices from patients with temporal lobe epilepsy (TLE).4,5 Therefore, the bcl-2 and caspase families, which regulate cell death, may play a role in ongoing neuropathologic processes in human epilepsy.6 Further, more recent works have shown that increased Bax expression and activated caspases contribute to neuronal loss through apoptosis.7
Calpains, a class of Ca2+-activated cysteine proteases, are major mediators for Ca2+ signals in many biological systems. There are two subtypes of calpains: I or μ-calpain and II or m-calpain; μ-calpain is the predominant type found in tissues, including neocortex.8 Calpains are proposed to participate in the turnover of cytoskeletal proteins and regulate kinases, cytokine, transcription factors and receptors.9
Pathological activation of calpain results in the cleavage of numerous neuronal substrates that negatively affect neuronal structure and function, thereby, leading to inhibition of essential neuronal survival mechanisms. Indeed, many proteins have been found to be calpain substrates, such as caspase 3, caspase 9, Bax, and Bcl-xL.10 Moreover, activated calpain mediates the degradation of calpain substrates in the course of neuronal death and contributes to the pathophysiology of neurological disorders, such as ischemia, trauma, Alzheimer’s disease and demyelination.11–14
Bi et al. (1997) first reported the effect of calpain activation in animal models of epilepsy and, essentially, found a high degree of calpain expression.15 Further evidence for increased calpain activity has been described in the hippocampus of rodent models of temporal lobe epilepsy.16 Later, Araujo et al. investigated the activation of calpains and caspases in degenerating areas of the hippocampus, as well as the proteolysis of known substrates [spectrin, for calpains, and poly-ADP ribose polymerase (PARP) for caspase 3] in the hippocampus of rats given kainic acid (KA) systemically. They found that early calpain activation, but not caspase activation, is involved in neurotoxicity in the hippocampus after status epilepticus; calpain inhibition was strongly correlated with neurodegeneration in the hippocampus following seizures.16 Calpain-mediated mechanisms may be responsible for seizure offset, probably through AMPA glutamate receptor internalization and further degradation.17 Little information, however, is currently available concerning the role of calpains in intractable epilepsy. Therefore, we investigated the expression of μ-calpain in patients with intractable epilepsy and its correlation with clinicopathological characteristics.
2. Materials and methods
2.1. Patients selection
Intractable seizures were defined as those not controlled in all patients by maximal tolerated doses of at least two AEDs, including phenytoin, valproic acid, carbamazepine, phenobarbital, topiramate, oxcarbazepine, clonazepam, gabapentin and lamotrigine. A total of 30 patients with intractable epilepsy were recruited from the files of the department of neurosurgery of the following hospitals: The First Affiliated Hospital of Chongqing Medical University and Xinqiao Hospital of the Third Military Medical University. All of these patients underwent resection of the anterior temporal lobe. The diagnosis of epilepsy was determined according to the 1981 International Classification of Epileptic Seizures by the International League Against Epilepsy. Prior to surgery, the lesion was localized in all patients via several methods, including brain magnetic resonance imaging, a 24-h electroencephalogram or video electroencephalogram, sphenoidal electrode monitoring, and intraoperative electrocorticography. Table 1 summarizes the clinical features of the patients.
Table 1.
Patient characteristics.
| Total (n = 30) | |
|---|---|
| Age (years) | 24.06 ±9.42 |
| Gender (n) | |
| Male | 16 |
| Female | 14 |
| Onset of seizure (n) | |
| <18 years | 20 |
| ≥18 years | 10 |
| Course of epilepsy (n) | |
| <10 years | 18 |
| ≥10 years | 12 |
| Frequency of seizures per month (n) | |
| 3–4 | 5 |
| 4–8 | 15 |
| >8 | 10 |
| Family history of epilepsy (n) | |
| Yes | 3 |
| No | 27 |
| MRIa (n) | |
| Abnormal | 13 |
| Normal | 17 |
| Patterns of seizures (n) | |
| Complex focal seizures | 10 |
| Generalized tonic–clonic seizure | 4 |
| Tonic spasm | 3 |
| Secondary generalized seizure | 7 |
| Multiple seizure patterns | 6 |
Values are means ± SEM, or number.
Normal means no structural abnormality of brain, and abnormal MRI means hippocampal sclerosis, focal cortical dysplasia and temporal lobe malacia, multiple seizure patterns include simplex partial seizure, complex partial seizure and secondarily generalized seizure.
The control group consisted of temporal lobe samples obtained from 10 patients who underwent neurosurgical intervention due to brain trauma in the Neurosurgical Department of The First Affiliated Hospital of Chongqing Medical University. All controls were diagnosed as brain trauma, and neuropathologists found no abnormality in the specimen slides of these control patients. Table 2 shows the clinical features of the controls.
Table 2.
Patient characteristics.
| Patient no. | Gender/age (Y) | Clinical diagnosis | Source of tissues | Region of tissues | Adjacent tissue pathology |
|---|---|---|---|---|---|
| 1 | 15, M | Trauma | Surgical specimen | TNr, PCr | No particular findings |
| 2 | 26, F | Trauma | Surgical specimen | TNI | No particular findings |
| 3 | 42, M | Brain tumor | Surgical specimen | TNI | No particular findings |
| 4 | 20, M | Trauma | Surgical specimen | TNr, FCr | No particular findings |
| 5 | 22, F | Trauma | Surgical specimen | TNr | No particular findings |
| 6 | 16, M | Trauma | Surgical specimen | TNI, HI | No particular findings |
| 7 | 27, M | Trauma | Surgical specimen | TNr, PCr | No particular findings |
| 8 | 31, F | Trauma | Surgical specimen | TNI | No particular findings |
| 9 | 17, M | Trauma | Surgical specimen | TNr, Hr | No particular findings |
| 10 | 33, F | Brain tumor | Surgical specimen | TNr | No particular findings |
The study was approved by the ethics committee on human research at Chongqing Medical University. Informed consent was obtained from the patients or their relatives for the use of any data and tissues for research, which was performed in accordance with the Helsinki Declaration.
2.2. RT-PCR
cDNA equivalent to 20 ng of total RNA was subjected to subsequent semi-quantified PCR analysis using the following primers: CAPN1 (sense, TCG TGC TCG CCC TTA TGC; antisense, CTT GTC CAG GTC AAA CTT CC), and GAPDH (sense, ATC TGG CAC CAC ACC TTC TAC A; anti-sense, GTT TGG TGG ATG CCA CAG GAC T). In preliminary experiments, optimal cycling conditions were established for amplification of each cDNA. PCR products were separated on 1.5% agarose gel containing 10 g/ml ethidium bromide, photographed using the UVsolo system (Whatman Biometra) and densitometric analysis was performed with the BioDocAnalyze software (Whatman Biometra). Results were calculated according to levels of target mRNAs in relation to those of the housekeeping gene, GAPDH (three samples from each group were analyzed by PCR).
2.3. Western blotting
Tissues were homogenized and lysed in Laemmli buffer. The protein concentration of the lysates was determined using a Coomassie blue G-250 kit (Sigma, St. Louis, USA). The protein extracts (50 μg) were resolved by 10% SDS polyacrylamide gel electrophoresis and electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Dupont, Wilmington, USA). The membranes were blocked with 3% BSA (Sigma) and then incubated for 2 h at room temperature in PBS. After extensive washing with phosphate buffered saline (PBS), the membranes were incubated with the following primary antibodies: rabbit anti-human μ-calpain (1:200; Santa Cruz Biotechnology), 145 kDa α-spectrin specific antibody (1:200; Santa Cruz Biotechnology), and monoclonal anti-GAPDH (Santa Cruz Biotechnology) at a dilution of 1:1000 for 1 h. Goat anti-rabbit HPRIgG (1:5000; Sigma) was used for detection. Subsequently, the protein bands were visualized after exposure to film and the pixel density of the scanned film images was quantified using Labworks Analysis Software (UVP, Upland, USA). Western blot data were normalized relative to the density of the GAPDH bands.
2.4. Cytokine quantification
The overall cytokine levels in the anterior temporal lobe were measured by the Multi-Analyte ELISA array Kit (SA Biosciences, Frederick, MD, USA). Protein homogenates were extracted from the anterior temporal lobe using the Halt Protease Inhibitor Cocktail Kit (Pierce), centrifuged at 78,500 × g for one 1 h at 4 °C, and were collected and stored at −70 °C until assayed. According to the kit’s manual instructions, the levels of 12 cytokines and chemokines (IL-1β, IL-6, IL-10, IL-12, IL-17a, IFN-γ, TNF-α and TGF-β1) were detected simultaneously at 1:20 dilutions. Negative and positive controls supplied by the kits were also included. The reactions were analyzed at a wavelength of 450 nm using a 96-well microplate reader, Model 680 (Bio-Rad, Hercules, CA, USA).
2.5. Hematoxylin-eosin
The resected brain tissue was immediatedly fixed in 10% buffered formalin. After fixation in formalin for 48 h, the paraffin-embedded tissue was sectioned into 10 μm slices for immunohistochemistry and immunofluorescence analysis, and then mounted onto polylysine-coated slides. One section of each specimen was processed for hematoxylin-eosin staining. Control samples were prepared in the same way. Surgical specimens, sectioned with a microtome and stained with hematoxylin-eosin, were submitted for neuropathological evalution..
2.6. Immunohistochemistry
The procedure was performed according to the manufacturer’s protocol. First, paraffin sections were deparaffinized, hydrated through a graded ethanol series and incubated in 0.3% H2O2 for 15 min. The sections were heated in a microwave oven for 10 min at 98 °C in citrate buffer (pH 6) for antigen retrieval, and blocked in normal goat serum (1:10; Zhongshan Golden Bridge Inc, Beijing, China) for 10 min. The sections were incubated in the following primary antibodies: primary rabbit anti-human μ-calpain (1:150; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at 37 °C, primary rabbit anti-human GFAP (1:200; Zhong shan Golden Bridge Inc, China). Anti-rabbit secondary antibody was used for detection for 30 min at 37 °C. Immunoreactivity was observed with 3,30-diaminobenzidine (Zhongshan Golden Bridge Inc.). The OLYMPUS PM20 automatic microscope (Olympus, Osaka, Japan) and TC-FY-2050 pathology system (Yuancheng Inc., Beijing, China) were used to collect the images. Primary antibodies were replaced with PBS (pH 7.2) and served as negative controls.
2.7. Immunofluorescence
Sections were deparaffinized and antigen was recovered as described in the previous section. Tissues were permeabilized with 0.5% (v/v) Triton X-100. After blocking with 10% goat serum in PBS for 30 min, followed by incubation with the following primary antibodies: rabbit anti-human μ-calpain (1:150) in PBS at 4 °C overnight, rabbit antibody GFAP(1:200). Sections containing μ-calpain antibody were incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (H + L) (1:200, 10 μg/ml; green; Santa Cruz Biotechnology) in PBS in the dark for 60 min at room temperature. Sections containing GFAP antibody were incubated with tetra-methylrhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit IgG (1:200, 10 μg/ml; red; Zhong shan Golden Bridge Inc., Beijing, China). Fluorescence was detected by laser scanning confocal microscopy (Leica Microsystems Heidelberg GmbH, Heidelberg, Germany) on an Olympus IX 70 inverted microscope equipped with a Fluoview FVX confocal scanhead. The optical densities for every sample were analyzed automatically and semi-quantitatively using the Motic Med 6.0 CMIAS pathology image analysis system (Beihang Motic, Inc., Beijing, China).
2.8. Statistical analysis
Data are represented as means ± standard error of the mean (SEM). Student’s t-test was used for statistical analysis with SPSS 11.0 between epileptic tissues and the control tissues. p Value of <0.05 was used as the minimum criteria for statistical significance.
3. Results
3.1. Demographic and clinical characteristics of the epilepsy subjects
The mean age of the epilepsy patients was 24.06 ± 9.42 years, with 16 men and 14 women in the experimental group. The mean duration of seizure recurrence was 8.93 ± 4.06 years. The control group consisted of 10 men and 9 women, with an average age of 24.90 ± 8.65 years. Statistical analysis showed that there were no significant differences in age or sex between the IE patients and controls (p > 0.05). 50% of patients had neuronal loss while 86.7% had experienced seizure recurrence for at least 5 years, with 43.3% having recurrences for more than 10 years.
3.2. μ-Calpain was overexpressed in the anterior temporal neocortex of patients with intractable epilepsy
The μ-calpain gene was analyzed in epileptic tissues and control tissues using semi-quantification of RT-PCR products. The expression of the μ-calpain gene in the control group was significantly lower compared to the epilepsy group (Fig. 1A). mRNA expression for μ-calpain was measured by densitometric evaluation of PCR results. GAPDH served as the internal control. Values are expressed as mean ± SEM, with p < 0.05 considered statistically significant.
Fig. 1.

Calpain was overexpressed in the anterior temporal neocortex of patients with intractable epilepsy. (A) Identification of the expression patterns of μ-calpain in the control and the epileptic human samples by semi-quantitative RT-PCR of mRNA levels. Total RNA was isolated from the anterior temporal neocortex. GAPDH was used as an internal control. Densitometry analysis was performed using FluorChem software. The intensity of bands was quantified and normalized by that of GAPDH. The data are expressed as mean ± SD. The value of *p < 0.05 is indicated as significantly different. (B) Western blot analysis was used to validate the differential displays for inactive μ-calpain and α-spectrin (μ-calpain-mediated breakdown of spectrin) in the control and the epileptic human samples. Total proteins (10 μg/lane) were extracted from human samples by SDS-PAGE for each sample and probed with the primary antibody of μ-calpain. Densitometry analysis was performed using FluorChem software. Representative Western blots qualitatively visualizing μ-calpain expression levels. Histograms of the normalized μ-calpain band intensities for the epileptic group and the control group. The data are expressed as mean ± SD. The value of *p < 0.05 is indicated as significantly different.
Protein expression of μ-calpain in the controls as well as the temporal lobes of IE patients, via Western blotting, indicate that relatively strong expression of the inactive form of 80-kDa μ-calpain and a calpain-cleaved spectrin fragment of 145 kDa was observed in the epileptic tissues, whereas faintly stained bands were present in the control group samples (Fig. 1B). μ-Calpain expression was normalized by calculating the ratio of the optical density of the bands for μ-calpain and GAPDH. The optical density ratio was 0.5146 ± 0.131 for epileptic tissues and 0.2756 ± 0. 0.070 for control tissues; these values were significantly different (p < 0.05). Analysis of the calpain-cleaved 145 kDa spectrin fragment expression in the control and the temporal lobe of IE patients by Western blotting indicated that relatively strong expression of 145 kDa α-spectrin was observed in the epileptic tissues, whereas faintly stained bands were present in the control group samples. The optical density ratio was 0.4746 ± 0.080 for epileptic tissues and 0.2056 ± 0.0467 for control tissues; these values were significantly different (p < 0.05).
3.3. Immunohistochemical study of μ-calpain expression and localization in normal tissues and intractable epilepsy
Prominent μ-calpain staining in the anterior temporal neocortex of patients with IE correlates with positive expression of μ-calpain (buffy particles in the cytoplasm of neurons and glial cells), and was observed in the temporal lobe cortices in both IE patients and the control group using immunohistochemistry and immunofluorescence staining (Fig. 2A and B). Positive staining for μ-calpain in this section is brown. There was significantly stronger μ-calpain staining in epileptic temporal lobe tissues compared to control. The mean optical densities for the IE group and control group were 0.4124 ± 0.039 and 0.2521 ± 0.042, respectively. There was significantly higher expression of μ-calpain in the temporal lobe cortex of the IE group compared to the control group (p < 0.05). Immunofluorescence shows positive staining for μ-calpain in this section as green. There was significantly stronger μ-calpain staining in epileptic temporal lobe tissues compared to control. The mean optical densities for the IE group and control group were 0.8236 ± 0.102 and 0.4046 ± 0.0910, respectively. There was significantly higher expression of μ-calpain in the temporal lobe cortex of the IE group compared to the control group (p <0.05).
Fig. 2.

Calpain immunoreactivity in the temporal lobe of IE patients by immunohistochemical staining. (A) Faint positive staining in the control illustrates decreased μ-calpain expression. Strongly positive staining in the cortex of the temporal lobe of a patient with IE reflects increased μ-calpain expression (buffy particles in the cytoplasm of neurons and glial cells). Scale bars = 100 μm. (B) Immunofluorescence of μ-calpain expression in the control and patients with temporal lobe epilepsy (TLE). The white arrows indicate positive cells (a: neuron; b: glial cell) representing increased μ-calpain expression. Scale bars = 75 μm. The data are expressed as mean ± SD, and the value of *p < 0.05 is indicated as significantly different.
3.4. GFAP was overexpressed in the cell cytoplasm of glial cells in patients with intractable epilepsy
Prominent GFAP staining in the anterior temporal neocortex of patients with IE correlates with positive expression of GFAP (buffy particles in the cytoplasm of glial cells), and was observed in the temporal lobe cortices in both IE patients and the control group using immunohistochemistry and immunofluorescence staining (Fig. 3). Positive staining for GFAP in this section is brown. The mean optical densities for the IE group and control group were 0.5482 ± 0.042 and 0.3942 ± 0.038, respectively. There was significantly higher expression of GFAP in the temporal lobe cortex of the IE group compared to the control group (p < 0.05). Immunofluorescence staining shows positive staining for GFAP in this section as red. The mean optical densities for the IE group and control group were 0.7338 ± 0.0972 and 0.5923 ± 0.0945, respectively. There was significantly higher expression of GFAP in the temporal lobe cortex of the IE group compared to the control group (p < 0.05).
Fig. 3.
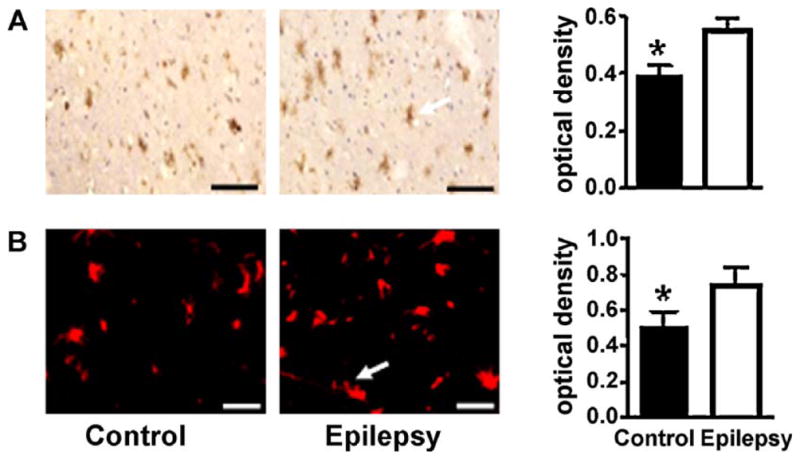
GFAP immunoreactivity in the temporal lobe of IE patients by immunohistochemical staining. (A) Faint positive staining in the control illustrates decreased GFAP expression. Strongly positive staining in the cortex of the temporal lobe of a patient with IE reflects increased GFAP expression (buffy particles in the cytoplasm of glial cells). (B) Immunofluorescence of μ-calpain expression in the control and patients with temporal lobe epilepsy. The white arrows indicate positive cells (glial cell) representing increased μ-calpain expression. Scale bars = 75 μm. The data are expressed as mean ± SD, and the value of *p < 0.05 is indicated as significantly different.
3.5. IL-1β, IL-6 and TGF-β1 were upregulated in patients with intractable epilepsy
There were no differences in protein homogenates levels of IL-10, IL-12, IL-17a, IFN-γ, TNF-α levels between the study groups. Protein homogenates levels of IL-1β, IL-6 and TGF-β1 were higher and statistically significant in the IE group than in control group (Fig. 4).
Fig. 4.

The levels of IL-1β, IL-6 and TGF-β1 were increased in IE patients. Protein homogenates were extracted from the anterior temporal lobe and the levels of IL-1β, IL-6 and TGF-β1 were detected simultaneously at 1:20 dilutions. The concentrations of IL-1β and TGF-β1 in epileptic tissues were significantly greater compared to controls (p < 0.05). The concentration of IL-6 in epileptic tissues was especially significant (p < 0.01). The data are expressed as mean ± SD, and the value of *p < 0.05, **p < 0.01 is indicated as significantly different.
3.6. Clinicopathological characteristics of the anterior temporal lobe in intractable epilepsy
Temporal lobe tissues taken from the control group and from patients with intractable epilepsy were sliced into 10 μm sections using a microtome and stained with hematoxylin-eosin. We found the following four types of pathological changes: neuronal loss, neuronal degeneration, gliosis and astrocytosis. Fig. 5B, D, and F are representative of specimens showing neuronal loss, neuronal degeneration and gliosis. These findings are consistent with previous studies of intractable epilepsy revealing neuronal degeneration and necrosis.18–20
Fig. 5.
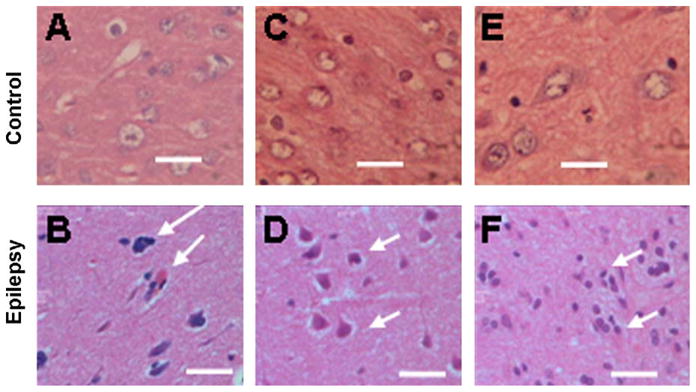
Hematoxylin-eosin staining of brain tissue from control and patient group. The control group consisted of temporal lobe samples from 10 patients who underwent neurosurgical intervention due to brain trauma. Thirty anterior temporal lobe samples were taken from patients with intractable epilepsy. The tissue was sliced into 10 μm sections using a microtome and stained with hematoxylin-eosin. The controls are represented by A, C, E. The epileptic anterior temporal lobe samples are represented by B, D, and F. The white arrows are pointing at neuronal loss (B) and neuronal degeneration (D) and gliosis (F). Scale bars = 100 μm.
4. Discussion
Since seizures can lead to the impairment of neurons, it can be surmised that the most important finding in the pathophysiology of intractable epilepsy is apoptosis and necrosis of neurons21. Previous studies have reported that neuronal loss existed in patients with epilepsy, especially in those with recurrent or status epilepsy18–20. In our study, we found the following pathological changes: neuron loss, neuronal degeneration, gliosis and astrocytosis. Neuron loss and neuronal degeneration are two, distinct separate features. Neuronal degeneration is due to hypoxia and hypoperfusion, but can be reversed if hypoxia and hypoperfusion are improved. However, neuron loss occurs as a result of deterioration of neuronal degeneration, and the injured neuron is swallowed by microglia. Gliosis and astrocytosis have been confirmed by overexpression of GFAP. Our study results show that 50% of patients had neuronal loss; therefore, we inferred that neuronal loss, a hallmark of neurodegenerative diseases, plays an important pathogenic role in the progression of IE.
To our knowledge, this is the first time the expression of μ-calpain in the anterior temporal neocortex of patients with IE has been reported. In our study, we found that the level of μ-calpain and the 145 kDa SBDP corresponds to μ-calpain-mediated cleavage expression in IE brain tissues were up-regulated phenomenally.
μ-calpain, the main subtype of the calpain family which was found in the central nervous system by Guroff more than 50 years ago,22 was required in concentrations of 2–80 μM Ca2+ for half-maximal activity. The process of μ-calpain activation occurs by an established progression of events beginning with calcium binding to the EF-hand structures. Once bound, μ-calpain becomes autocatalytic, cleaving itself on both large and small subunits, and leaving truncated N-terminals of 76 and 18 kDa, respectively.23 Thus, this process could catalyze the proteolysis involved in cytoskeletal remodeling, cytokine, cell-cycle regulation, signal transduction, cell differentiation, embryonic development and vesicular trafficking.24,25 However, pathological overactivation of μ-calpain mediates abnormal degradation of many proteins and, eventually, causes cell death.26
Spectrin is widely distributed throughout the cytoplasm and is easily accessible for cleavage by different proteases, giving rise to stable spectrin breakdown products (SBDPs) that can be identied by immunostaining. It is now known that μ-calpain-mediated breakdown of αII-spectrin creates several αII-spectrin breakdown products (SBDPs). Caspase-3-mediated cleavage creates 150 and 120 kDa SBDPs, whereas μ-calpain creates 150 and 145 SBDPs. In this study, the cleavage product of μ-calpain (including the 150 kDa and 145 kDa spectrin) and the cleavage product of caspase 3 (including the 150 kDa and 120 kDa spectrin) were regulated by μ-calpain. Therefore, this study selected 145 kDa SBDP, instead of 150 kDa, as a marker of increased μ-calpain expression. The 145 kDa SBDP corresponds to μ-calpain-mediated cleavage was found to be increased in the patient specimens.
The previous study by Araujo et al.16 had shown that seizures lead to calpain activation and proteolysis of spectrin in the hippocampus as a result of KA injection. This clinical finding shows that temporal neocortex from intractable epilepsy patients express raised levels of Bcl-2, Bcl-XL and activated caspase 3.6 Moreover, during focal cerebral ischemia-reperfusion, μ-calpain is an upstream regulator of caspase 3,27 and has been suggested to be involved in apoptosis by activating caspase-3 in IE.
Certainly, μ-calpain can also injure neurons by other pathways. μ-calpain could regulate cytokines.28–30 While cytokines are associated with pathological changes in intractable epilepsy.31,32 This clinical finding shows that marked activation of microglia and astrocytes and diffuse cell death were observed in epileptogenic tissue. IL-1beta, IL-8, IL-12p70 and MIP-1beta were significantly increased in the epileptogenic cortex. IL-6 and MCP-1 were significantly higher in patients with family history of epilepsy.31 Their results suggest that active neuroinflammation and marked cellular injury occur in pediatric epilepsy and may play a common pathogenic role or consequences in childhood epilepsy of diverse etiologies. In our study, we found that upregulated expression of IL-1β, IL-6 and TGF-β1 belongs to inflammation markers. Therefore, we theorized that μ-calpain can cause these pathological results through inflammation.
However, several issues need to be considered when interpreting the results of this study. Firstly, because of the practical difficulty in obtaining normal human brain specimens, we had to use structurally normal brain tissue obtained from temporal lobectomies performed for the treatment of traumatic brain injuries as controls. We made a preliminary evaluation of the control tissues by Western blot analysis to determine whether expression of μ-calpain in brain trauma specimens were accordant. The results show that there is no difference in the expression of μ-calpain among the five control anterior temporal neocortex samples which were collected. Recent studies have shown that structurally normal brain tissue obtained from traumatic brain injury can be used as controls.33,34 Secondly, μ-calpain’s involvement in the pathological process leading to causative or consequential epileptic seizures remains unclear. Based on the potential effects of μ-calpain on neuronal apoptosis, we hypothesize that its upregulation may play an important pathogenic role in the progression of IE. However, due to the limitations of the human study, the abovementioned hypothesis should be further investigated in animal models and in vitro experiments.
In conclusion, μ-calpain is upregulated in the anterior temporal neocortex of patients with IE, which may correlate with pathological changes in these regions. The biological significance of overexpression of μ-calpain warrants further investigation.
Acknowledgments
We thank the patients and their families for their participation in this study. The authors sincerely thank Xinqiao Hospital for their support in brain tissue procurement, and the National Institutes of Health of China and the Ethics Committee on Human Research of the Chongqing Medical University.
Footnotes
Conflict of interest
The authors do not have any conflicts of interest to disclose.
References
- 1.Aylward RL. Epilepsy: a review of reports, guidelines, recommendations and models for the provision of care for patients with epilepsy. Clinical Medicine (London England) 2008;8:433–8. doi: 10.7861/clinmedicine.8-4-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujikawa DG, Zhao S, Ke X, Shinmei SS, Allen SG. Mild as well as severe insults produce necrotic, not apoptotic, cells: evidence from 60-min seizures. Neuroscience Letters. 2010;469:333–7. doi: 10.1016/j.neulet.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 3.Tian FF, et al. Mossy fiber sprouting, hippocampal damage and spontaneous recurrent seizures in pentylenetetrazole kindling rat model. Acta Neurologica Belgica. 2009;109:298–304. [PubMed] [Google Scholar]
- 4.Pollard H, et al. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 5.Graham S, Chen J, Stetler RA, Zhu RL, Jin KL, Simon RP. Expression of the proto-oncogene bcl-2 is increased in the rat brain following kainate-induced seizures. Restorative of Neurology and Neuroscience. 1996;9:243–50. doi: 10.3233/RNN-1996-9407. [DOI] [PubMed] [Google Scholar]
- 6.Henshall DC, et al. Alterations in bcl-2 and caspase gene family protein expression in human temporal lobe epilepsy. Neurology. 2000;55:250–7. doi: 10.1212/wnl.55.2.250. [DOI] [PubMed] [Google Scholar]
- 7.Uysal H, et al. Is the cell death in mesial temporal sclerosis apoptotic? Epilepsia. 2003;44:778–84. doi: 10.1046/j.1528-1157.2003.37402.x. [DOI] [PubMed] [Google Scholar]
- 8.Persson H, Kawashima S, Karlsson JO. Immunohistochemical localization of calpains and calpastatin in the rabbit eye. Brain Research. 1993;611:272–8. doi: 10.1016/0006-8993(93)90513-m. [DOI] [PubMed] [Google Scholar]
- 9.Croall DE, Ersfeld K. The calpains: modular designs and functional diversity. Genome Biology. 2007;8:218. doi: 10.1186/gb-2007-8-6-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Molecular Neurobiology. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Das A, Guyton MK, Butler JT, Ray SK, Banik NL. Activation of calpain and caspase pathways in demyelination and neurodegeneration in animal model of multiple sclerosis. CNS and Neurological Disorders Drug Targets. 2008;7:313–20. doi: 10.2174/187152708784936699. [DOI] [PubMed] [Google Scholar]
- 12.Nicholson AM, Ferreira A. Increased membrane cholesterol might render mature hippocampal neurons more susceptible to beta-amyloid-induced calpain activation and tau toxicity. Journal of Neuroscience. 2009;29:4640–51. doi: 10.1523/JNEUROSCI.0862-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang YN, et al. Calpain-mediated N-cadherin proteolytic processing in brain injury. Journal of Neuroscience. 2009;29:5974–84. doi: 10.1523/JNEUROSCI.6178-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun M, Zhao Y, Xu C. Cross-talk between calpain and caspase-3 in penumbra and core during focal cerebral ischemia-reperfusion. Cellular and Molecular Neurobiology. 2008;28:71–85. doi: 10.1007/s10571-007-9250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi X, Chen J, Baudry M. Developmental changes in calpain activity, GluR1 receptors and in the effect of kainic acid treatment in rat brain. Neuroscience. 1997;81:1123–35. doi: 10.1016/s0306-4522(97)00218-2. [DOI] [PubMed] [Google Scholar]
- 16.Araujo IM, et al. Calpain activation is involved in early caspase-independent neurodegeneration in the hippocampus following status epilepticus. Journal of Neurochemistry. 2008;105:666–76. doi: 10.1111/j.1471-4159.2007.05181.x. [DOI] [PubMed] [Google Scholar]
- 17.Sierra-Paredes G, Cornes JM, Sierra-Marcuno G. Calpain inhibitor I retards seizure offset in the hippocampus of freely moving rats. Neuroscience Letters. 1999;263:165–8. doi: 10.1016/s0304-3940(99)00136-6. [DOI] [PubMed] [Google Scholar]
- 18.Dawodu S, Thom M. Quantitative neuropathology of the entorhinal cortex region in patients with hippocampal sclerosis and temporal lobe epilepsy. Epilepsia. 2005;46:23–30. doi: 10.1111/j.0013-9580.2005.21804.x. [DOI] [PubMed] [Google Scholar]
- 19.Ozbas-Gerceker F, et al. Neurotrophin receptor immunoreactivity in the hippocampus of patients with mesial temporal lobe epilepsy. Neuropathology and Applied Neurobiology. 2004;30:651–64. doi: 10.1111/j.1365-2990.2004.00582.x. [DOI] [PubMed] [Google Scholar]
- 20.Santhakumar V, Aradi I, Soltesz I. Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: a network model of the dentate gyrus incorporating cell types and axonal topography. Journal of Neurophysiology. 2005;93:437–53. doi: 10.1152/jn.00777.2004. [DOI] [PubMed] [Google Scholar]
- 21.Holmes GL. Clinical evidence that epilepsy is a progressive disorder with special emphasis on epilepsy syndromes that do progress. Advances in Neurology. 2006;97:323–31. [PubMed] [Google Scholar]
- 22.Guroff G, Neutral A. Calcium-activated proteinase from the soluble fraction of rat brain. The Journal of Biological Chemistry. 1964;239:149–55. [PubMed] [Google Scholar]
- 23.Elce JS, Hegadorn C, Arthur JS. Autolysis Ca2+ requirement, and heterodimer stability in m-calpain. The Journal of Biological Chemistry. 1997;272:11268–75. doi: 10.1074/jbc.272.17.11268. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Liu MC, Wang KK. Calpain in the CNS: from synaptic function to neurotoxicity. Science Signaling. 2008;1:re1. doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- 25.Liu J, Liu MC, Wang KK. Physiological and pathological actions of calpains in glutamatergic neurons. Science Signaling. 2008;1:tr3. doi: 10.1126/scisignal.123tr3. [DOI] [PubMed] [Google Scholar]
- 26.Ray SK, et al. Oxidative stress and Ca2+ influx upregulate calpain and induce apoptosis in PC12 cells. Brain Research. 2000;852:326–34. doi: 10.1016/s0006-8993(99)02148-4. [DOI] [PubMed] [Google Scholar]
- 27.McDonough JH, Jr, Shih TM. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neuroscience and Biobehavioral Reviews. 1997;21:559–79. doi: 10.1016/s0149-7634(96)00050-4. [DOI] [PubMed] [Google Scholar]
- 28.Imam SA, et al. Increased calpain correlates with Th1 cytokine profile in PBMCs from MS patients. Journal of Neuroimmunology. 2007;190:139–45. doi: 10.1016/j.jneuroim.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, Temple S, Roberts S, Price P. Complex effects of IL1A polymorphism and calpain inhibitors on interleukin 1 alpha (IL-1 alpha) mRNA levels and secretion of IL-1 alpha protein. Tissue Antigens. 2008;72:67–71. doi: 10.1111/j.1399-0039.2008.01052.x. [DOI] [PubMed] [Google Scholar]
- 30.Hayakawa M, et al. Mature interleukin-33 is produced by calpain-mediated cleavage in vivo. Biochemical and Biophysical Research Communications. 2009;387:218–22. doi: 10.1016/j.bbrc.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 31.Choi J, et al. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. Journal of Neuroinflammation. 2009;6:38. doi: 10.1186/1742-2094-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi J, Koh S. Role of brain inflammation in epileptogenesis. Yonsei Medical Journal. 2008;49:1–18. doi: 10.3349/ymj.2008.49.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li JM, et al. Decreased expression of thyroid receptor-associated protein 220 in temporal lobe tissue of patients with refractory epilepsy. Biochemical and Biophysical Research Communications. 2006;348:1389–97. doi: 10.1016/j.bbrc.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Xiao F, et al. Overexpression of N-WASP in the brain of human epilepsy. Brain Research. 2008;1233:168–75. doi: 10.1016/j.brainres.2008.07.101. [DOI] [PubMed] [Google Scholar]