

Abstract
The differentiation and maintenance of Th17 cells require a unique cytokine milieu and activation of lineage-specific transcription factors. This process appears to be antagonized by the transcription factor T-bet, which controls the differentiation of Th1 cells. Considering that T-bet-deficient (T-bet−/−) mice are largely devoid of natural killer (NK) cells due to a defect in the terminal maturation of these cells, and because NK cells can influence the differentiation of T helper cells, we investigated whether the absence of NK cells in T-bet-deficient mice contributes to the augmentation of autoreactive Th17 cell responses. We show that the loss of T-bet renders the transcription factors Rorc and STAT3 highly responsive to activation by stimuli provided by NK cells. Furthermore, reconstitution of T-bet−/− mice with wild-type NK cells inhibited the development of autoreactive Th17 cells through NK cell-derived production of IFN-γ. These results identify NK cells as critical regulators in the development of autoreactive Th17 cells and Th17-mediated pathology.
Introduction
T-bet (Tbx21), a member of the tissue-specific T-box transcription factor family, is critical for the lineage commitment of CD4+ T helper 1 (Th1) cells and for their maintenance of immune cell effector function and IFN-γ production. Recent studies have suggested that, in addition to its effects on Th1 cells, T-bet can also influence, either directly or indirectly, the phenotype of Th2 cells, B cells, dendritic cells (DCs) and macrophages [1, 2]. T-bet might also negatively regulate IL-17-secreting Th cells, as T-bet-deficient (T-bet−/−) mice have increased levels of Th17 cells, particularly in inflamed organs [3–8].
The development of Th17 cells requires the cytokines IL-6 and TGF-β for lineage commitment and IL-23 for maintenance of the Th17 phenotype [9, 10]. The transcription factor STAT3 plays a critical role in Th17 cell differentiation, as it regulates the expression of two orphan nuclear receptors, RORγt and RORα, in the developing Th17 cells [11]. T-bet-deficient mice display aberrant Th17 cell responses. Although it has been suggested that T-bet-deficiency impacts the expression of the IL-23 receptor, the mechanisms responsible for the presence of abnormal Th17 responses in T-bet-deficient mice remain elusive [5].
It is well established that the commitment of naïve T cells into distinct Th cell subsets is regulated by antigen-presenting cells (APCs) and by cytokines present in the local microenvironment during T cell receptor ligation [12]. As innate natural killer (NK) and natural killer T (NKT) cells produce large amounts of cytokines, both cell types might make a significant contribution to the cytokine milieu in which Th cell differentiation occurs [13]. Consistent with this possibility, both NK and NKT cells have been implicated in the differentiation of Th1 and Th2 cells [14, 15].
As T-bet-deficient mice exhibit a profound, stem cell-intrinsic defect in their capacity to generate NK cells and (Vα14i+) NKT cells [16], we tested whether these cell deficiencies might affect autoimmune Th17 cell responses in T-bet−/− mice. To this aim, we employed an experimental model for human autoimmune myasthenia gravis (MG) that is induced in C57BL/6 mice through immunization with acetylcholine receptor (AChR). The disease, called experimental autoimmune MG (EAMG), is T and B cell-mediated and is characterized by the development of AChR-reactive antibodies in the neuromuscular junctions [17]. Using this model, we show that the augmented, autoreactive Th17 responses in T-bet-deficient mice are inhibited by NK cells in a process that depends on T-bet expression in T cells and cytokine production by NK cells.
Materials and methods
Mice
T-bet-deficient (T-bet−/−) mice [2] and other mutant RAG1−/−, γc−/− and IFN-γ−/− mice were either available in our colony or purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and maintained on the C57BL/6 (B6) background after 11 generations of backcrossing. Wild-type (WT) littermates (T-bet+/+) were used as controls. Only female mice at 7–8 weeks of age (at the initiation of the experiments) were used. IFN-γ−/−RAG1−/− and γc−/− RAG1−/− mutant mice (B6 background) were generated by crossing the single mutants. All mice were bred and maintained in specific pathogen-free conditions in accordance with the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Barrow Neurological Institute, St. Joseph's Hospital and Medical Center (Phoenix, AZ, USA) or the Tianjin Neurological Institute (Tianjin, China).
Autoantigens and immunization protocol
Acetylcholine receptor was purified as previously described [18]. Mice were injected subcutaneously (s.c.) with 20 μg of AChR in complete Freund's adjuvant (CFA) (Difco, Detroit, MI, USA) in a total volume of 100 μl along the shoulders and back. Mice were boosted twice at day 30 and day 60 with 20 μg of AChR in incomplete Freund's adjuvant (IFA) s.c.
Reconstitution of T-bet−/− mice with NK cells or NKT cells
NK cells were isolated from splenocytes using CD49b (DX-5) beads [19]. NK1.1+CD3+ NKT cells were sorted from splenocytes of WT mice. In some experiments, FITC-labelled α-GalCer-tetramers (kindly provided by Dr. M. Kronenberg) [20] were used to sort invariant NKT (iNKT) cells. The purities of the isolated cells were >95% upon re-analysis by flow cytometry. The isolated NK cells were stimulated with lipopolysaccharide (LPS, 10 μg/ml; Sigma, St. Louis, MO, USA) and IL-15 (15 ng/ml; R&D, Minneapolis, MN, USA), and the isolated NKT cells were stimulated with α-GalCer (5 μg/ml; obtained from kirin Brewery Co., Takasaki, Gunma, Japan) for 24 h in culture medium (complete RPMI 1640 containing 50 IU/ml penicillin, 50 μg/ml streptomycin, 2 mm L-glutamine, 10 mm HEPES, 10% FBS, 10 μm β-mercaptoethanol and 1% non-essential amino acids). 2.5–5 × 105 cells were injected intravenously (i.v.) into recipient mice.
FACS analysis
For surface staining, single cell suspensions were prepared and stained with fluorescently labelled antibodies: NK1.1 (PK136), CD3 (145-2C11), CD4 (GK1.4) and CD8 (53–6.7) (BD Bioscience, San Diego, CA, USA). For intracellular cytokine staining, single cell suspensions were prepared and incubated at 37 °C for 4 days in round-bottom plates (2 × 106 cells/well) with or without antigens (AChR, 10 μg/ml; Con A, 2 μg/ml) and stimulated with phorbol myristate acetate (PMA, 20 ng/ml), ionomycin (1 μg/ml) and brefeldin A (5 μg/ml) for another 5 h at 37 °C. After harvesting, cells were stained for surface markers, fixed and permeabilized with reagents from the Cytofix/Cytoperm kit (BD Bioscience), then stained with fluorescent conjugated anti-IFN-γ (XMG1.2) and anti-IL-17 (TC11-18H10.1) monoclonal Abs (BD Bioscience), or anti-pSTAT3 (ABIN732648) polyclonal Ab (Antibodies-online Inc., Atlanta, GA, USA). Flow cyto-metric data were collected on a FACSAria™ flow cytometer (Becton Dickinson, Mountain View, CA, USA) and analysed with Diva™ software.
Th17 differentiation in vitro
CD4+ T cells were isolated from splenocytes using FACSAria™ flow cytometer and cultured for 3 ~4 days in medium with plate-bound anti-CD3 (145-2C11, 2 μm/ml) and anti-CD28 (PV-1, 2 μg/ml) plus irradiated syngeneic splenocytes (2000 rad) as antigen-presenting cells and stimulated with recombinant cytokines IL-6 (30 ng/ml), TGF-β1 (3 ng/ml) and IL-23 (20 ng/ml). IL-17 production in the supernatant was measured by commercially available ELISA kit. Intracellular analysis of IL-17 produced by CD4+ T cells was also evaluated by FACS analysis.
Transwell experiments
Transwell experiments were performed in 24-well plates. Isolated CD4+ T cells (2 × 105) from T-bet−/− mice were stimulated with anti-CD3 (2 μm/ml), anti-CD28 (2 μg/ml) and irradiated syngeneic splenocytes (1 × 106) under Th17 polarization condition with recombinant cytokines IL-6 (30 ng/ml), TGF-β1 (3 ng/ml) and IL-23 (20 ng/ml). In addition, isolated NK cells (2 × 105) from WT mice were placed in transwell chambers (Millicell, 0.4 μ; Millipore, Billerica, MA, USA). After 3–4 days of co-culture, NK cells were removed, IL-17 production from the supernatant of the bottom compartment was measured by ELISA kit and Rorc transcripts of the CD4+ T cells in the bottom compartment were determined by qRT-PCR.
ELISA
IL-17 ELISA was detected using the Quantikine mouse IL-17 immunoassay (R&D System). The limit of detection was 5 pg/ml. Briefly, the isolated CD4+ cells (2 × 105/well) from the spleens of T-bet−/− and B6 mice were co-cultured with the indicated cells (2 × 105/well) and stimulated with LPS (10 μg/ml) and IL-15 (15 ng/ml) for NK cells or α-Galcer (5 μg/ml) for NKT cells for 72 h and then supernatants were harvested and analysed for the presence of IL-17 by ELISA. Results were expressed as Mean ± SE.
qRT-PCR
Total RNA was isolated from cells with the RNAeasy Mini kit (QIAGEN GmbH, Valencia, CA, USA). cDNA was synthesized with SuperScript® III Reverse Transcriptase (Life Technologies Co., Grand Island, NY, USA) from 1 μg total RNA. Aliquots of cDNA were used as template for real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) using a Sybr-Green PCR master mix kit (Applied Biosystems, Carlsbad, CA, USA) containing either primers for Rorc or primers for internal control gene hypoxanthine-guanine phosphoribosyltransferase (HPRT). The primers for detection of Rorc transcript have been previously described [21] and are as follows: Rorc; forward, 5'- CCCCCTGCCCAGAAACACT-3', reverse, 5'-GGATGCCCCCATTCACTTACTTCT-3'. Each reaction contained cDNA derived from 10 ng total RNA. Triplicate reactions of each sample were performed and experiments were repeated twice. The relative amounts of mRNA were calculated by plotting the Ct (cycle number), and the fold change in the target gene Rorc normalized to HPRT and relative to the expression in control group T-bet+/+ was calculated for each sample using the 2−ΔΔCt comparative method.
NK cell cytotoxicity assay
NK cell cytotoxicity was assessed by the chromium release method. Freshly purified NK cells as effector cells were incubated with 51Cr-labelled YAC1 cells from murine lymphoma cell line or Th17-polarizing CD4+ T cells (5 × 103) as the target cells at various effector cells/target cell ratios. The culture was incubated for 4 h, and the supernatants were collected and 51Cr-release was measured by a γ-counter (PerkinElmer, Waltham, MA, USA). The percentage of specific lysis was calculated according to the following formula: (experimental release – spontaneous release)/(maximum release – spontaneous release) × 100. Cytotoxicity assay was performed in triplicate.
BrdU incorporation assay
For in vivo bromodeoxyuridine (BrdU) labelling assay, mice were injected with 100 μl of 10 mg/ml BrdU (BD Biosciences). After 18–24 h, BrdU-labelled cells harvested from the culture or single cell suspensions prepared from spleen were stained with mAb against CD4, IL-17 and BrdU. Samples were analysed on a FACSAria™ flow cytometer using Diva™ software.
Western blotting
Western blots were performed using anti-pSTAT3 (49/p-Stat3) and anti-STAT3 (84/Stat3) (BD Bioscience) antibodies followed by HRP-conjugated goat anti-mouse IgG and ECL detection system (Pierce, Rockford, IL, USA).
Statistics
Comparisons between groups were evaluated by the one-tailed unpaired Student's t-test for two groups, and anova for multiple comparisons. Values of P < 0.05 were considered significant.
Results
AChR-primed T-bet−/− mice generate strong autoreactive Th17 cell responses
We evaluated AChR-induced Th1 and Th17 responses in T-bet−/− mice. As expected, AChR-reactive, IFN-γ-producing CD4+ and CD8+ T cells were reduced in lymph nodes and spleen of T-bet-deficient mice compared with WT mice (Fig. 1A, B). In contrast, AChR-reactive, IL-17A (referred as IL-17 thereafter)-producing cells were pronouncedly increased 4- to 6-fold in the CD4+ T cells (Fig. 1A), while no significant change was observed in the CD8+ IL-17-producing T cells (Fig. 1B). Thus, we focused on CD4+ T cells in our subsequent investigations. Production of IL-17 from sorted T-bet−/− CD4+ T culture stimulated with AChR was significantly induced (Fig. 1C), and qRT-PCR of sorted CD4+ T cells from T-bet−/− mice cultured in the presence of AChR showed significant induction of Rorc, a gene encoding the Th17 lineage-specific transcription factor RORγ (Fig. 2A).
Figure 1.
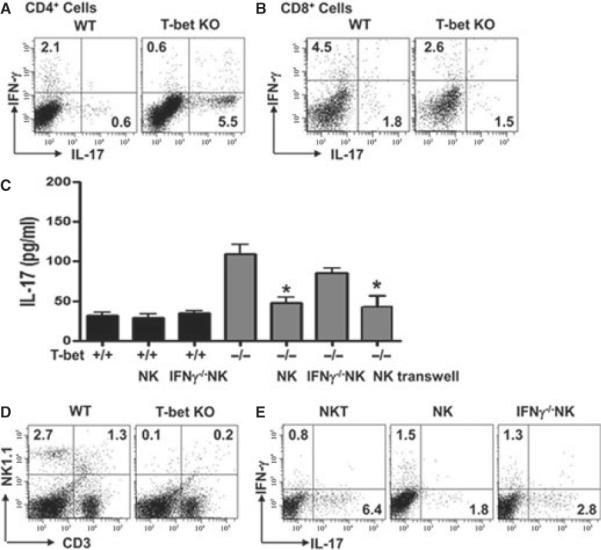
Reconstitution of T-bet−/− mice with NK cells, but not natural killer T (NKT) cells, reduces autoreactive Th17 cell responses. WT and T-bet−/− mice were immunized with AChR/CFA as described in Materials and methods. A portion of these mice were reconstituted with NK cells or NKT cells. (A,B) Lymphocytes from spleen were analysed for the production of IFN-γ and IL-17 in response to AChR (10 μg/ml) by intracellular cytokine staining; dot plots were gated on CD4+ cells (A) or CD8+ cells (B). (C) IL-17 production was determined by ELISA in sorted CD4+ T cells stimulated with NK cells from the indicated sources. Transwells were used in some NK cell-T cell co-cultures. Data shown are means ± SD and are representative of three independent experiments. (D) Frequencies of NK and NKT cells in WT and T-bet−/− mice were analysed by FACS. (E) Left panel: NKT cells were isolated from WT mice, activated by α-GalCer and transferred into T-bet−/− mice. AChR-reactive IFN-γ- and IL-17-producing T cells were analysed. Middle and right panels: NK cells of the indicated sources were isolated, activated with IL-15 and LPS and transferred into T-bet−/− mice, and AChR-reactive IFN-γ- and IL-17-producing cells were analysed. All dot plots show representative data of spleen from two to three separate experiments.
Figure 2.
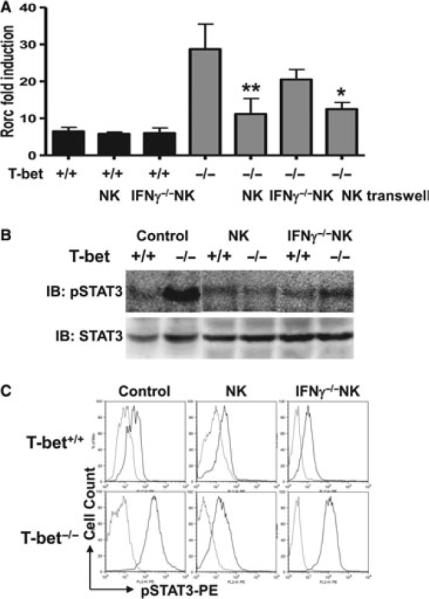
Loss of T-bet renders Rorc and STAT3 hyperresponsive to activation signals provided by NK cells. CD4+ T cells were sorted from pooled spleen and lymph node cells of acetylcholine receptor (AChR)-primed WT and T-bet−/− mice. A portion of these wells also received NK cells. Parallel co-cultures were set up using transwells. (A) Fold induction of Rorc transcripts at 3 days after stimulation measured by qRT-PCR. (B) STAT3 phosphorylation (pSTAT3) analysed by Western blotting. (C) pSTAT3 in AChR-stimulated CD4+ T cells measured by FACS by intracellular staining. Data represent two independent experiments.
Reconstitution of NK cells in T-bet−/− mice reverses the augmented Th17 response
Because both NK and NKT cells can influence the differentiation of T helper cells, we investigate the potential contribution of NK cell and NKT cell deficiency to the unabated Th17 phenotype observed in AChR-primed T-bet−/− mice. For this purpose, we quantified NK and NKT cells in AChR-primed T-bet−/− and WT mice. Consistent with an earlier report [16], both NK and NKT cells were largely absent from spleen (Fig. 1D), lymph nodes (T-bet−/− NK cells: 0.3 ± 0.2%, T-bet−/− NKT cells: 0.2 ± 0.2%) and peripheral blood (T-bet−/− NK cells: 0.4 ± 0.2%, T-bet−/− NKT cells: 0.2 ± 0.1%) in T-bet−/− mice compared with WT mice. When we transferred α-GalCer-activated NKT cells into T-bet−/− mice during immunization with AChR, we did not observe a significant change in AChR-reactive Th17 cells (Fig. 1E, left panel). Likewise, adoptive transfer of NKT cells did not significantly alter AChR-reactive Th1 and Th17 cell responses in WT mice (IFN-γ: 2.3 ± 0.5%; IL-17: 0.8 ± 0.1%). A recent study reported that NKT cells can suppress the Th17 lineage [22]. However, we did not find such an effect of NKT cells in EAMG using either T-bet+/+ or T-bet−/− Th17 cells.
Next, we isolated NK cells from RAG1−/− mice, avoiding contamination of these cells with NKT cells, and adoptively transferred these cells into AChR-primed T-bet−/− mice. Remarkably, NK cells activated with LPS and IL-15 prevented aberrant Th17 cell responses in T-bet−/− mice (Fig. 1E, middle panel), whereas the transfer of NK cells neither enhanced nor decreased IFN-γ and IL-17 production in WT mice (IFN-γ: 3.2 ± 0.4%; IL-17: 0.7 ± 0.2%).
The early and burst release of IFN-γ by NK cells prior to T cell priming and Th cell lineage differentiation has been shown to favour Th1 cell development in multiple experimental systems [23]. We reasoned that IFN-γ produced by NK cells could directly or indirectly influence Th17 cell differentiation in EAMG. To test this possibility, we isolated NK cells from IFN-γ−/− RAG1−/− mice and adoptively transferred these cells into AChR-primed T-bet−/− mice. Interestingly, these activated IFN-γ-deficient NK cells lost, at least in part, their capacity to suppress AChR-reactive T-bet−/− Th17 cells (Fig. 1E right panel; IFN-γ−/− NK cells: 3.5- to 4.8-fold increase; IFN-γ+/+ NK cells: 1.5- to 2.5-fold increase; no NK cells: 4- to 6-fold increase). Importantly, NK cells failed to alter the frequency of T-bet+/+ Th17 cells (0.7 ± 0.2%; and Fig. 1C). Clearly, our results indicated that the opposing effects of NK cells on Th17 and Th1 cells in peripheral lymphoid organs are dependent on the presence of T-bet.
NK cells inhibit STAT3 phosphorylation in T-bet-deficient T cells
We sought to understand the mechanisms responsible for the inhibitory effect of NK cells on the differentiation of T-bet-deficient Th17 cells. qRT-PCR results showed that reconstitution of T-bet−/− mice, but not WT mice, with activated NK cells enhanced the expression of Rorc in CD4+ T cells (Fig. 2A). As STAT3 is a downstream transcription factor that is critically important for Th17 lineage development, we investigated the capacity of NK cells to modulate STAT3 phosphorylation in CD4+ cells from WT and T-bet−/− mice. Western blot and FACS analysis showed that STAT3 phosphorylation was not significantly affected in WT mice receiving NK cells (Fig. 2B, C). In contrast, transfer of NK cells into T-bet−/− mice strongly inhibit STAT3 phosphorylation (Fig. 2).
Ex vivo effects of NK cells on Th17 cells and contributions of direct NK cell-mediated cytolytic activity
Our results thus far suggested that NK cells can suppress T-bet−/− autoreactive Th17 cells in a manner that involves, at least in part, IFN-γ production by NK cells and the enhancement of Rorc expression and STAT3 phosphorylation in CD4+ T cells. Ex vivo co-culture experiments of enriched NK cells and AChR-reactive Th17 cells showed that NK cells suppressed T-bet−/− but not T-bet+/+ Th17 cells, and these effects were partially dependent on IFN-γ production by NK cells (Fig. 3A).
Figure 3.
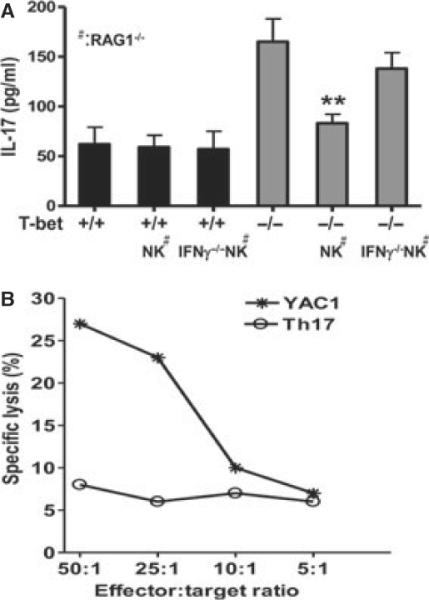
Ex vivo effects of NK cells on Th17 cells and contribution of direct NK cell-mediated cytotoxic activity. (A) NK cells were sorted from lymphocytes pooled from spleen and lymph nodes of IFN-γ+/+RAG1−/− or IFN-γ−/−RAG1−/− mice and stimulated with LPS (10 μg/ml) and IL-15 (15 ng/ml). CD4+ T cells were sorted from acetylcholine receptor (AChR)/CFA-primed T-bet+/+ or T-bet−/− mice. 2 × 105 sorted NK cells and CD4+ T cells were cultured at a ratio of 1:1 in the presence of plain T cell medium or medium supplemented with acetylcholine receptor (10 μg/ml) for 48 h. NK cells were subsequently depleted using anti-NK1.1 mAb (20 μg/ml). IL-17 production in culture supernatants was quantified by ELISA. Eight mice in each group were used to generate cells for co-culture. (B) Th17-polarizing CD4+ T cells and YAC1 cells were incubated with 51Cr. The effector (NK) and target cells (Th17 cells or YAC1 cells, 5 × 104) were incubated at various ratios, and cytolytic effects were measured in a 4-h standard Cr51-release assay.
A recent study has shown that NK cells can directly lyse autoreactive T cells via the NKG2A-CD94/Qa1 pathway [24]. To test whether NK cells inhibited T-bet−/− autoreactive Th17 cells via direct cytolytic effects, we quantified the frequency and numbers of CD4+ T cells in co-cultures of NK cells and CD4+ T cells after 3 days. We were unable to detect a reduction in CD4+ T cells under any of the conditions tested (data not shown). Further, NK cells retained their capacity to suppress Th17 cell responses when separated from these cells in transwells. Additionally, NK cells efficiently lysed YAC1 target cells but not Th17 cells in a dose-dependent manner (Fig. 3B). Collectively, these experiments confirm the direct inhibitory effect of IFN-γ-producing NK cells on T-bet−/− autoreactive Th17 cells and exclude direct cytolysis of Th17 cells as a possible mechanism.
NK cells inhibit expansion of T-bet−/− Th17 cells in vivo
γc−/− RAG1−/− mice are devoid of T, NKT, B and NK cells, but have intact APCs. This model allowed us to investigate the capacity of NK cells to suppress T-bet−/− Th17 cells without interfering with endogenous cells. We pooled lymphocytes from AChR-immunized WT and T-bet−/− mice, sorted CD4+ T cells by FACS for Th17 polarization. These cells were then co-injected with NK cells into the γc−/− RAG1−/− hosts. Compared with WT Th17 cells, a larger proportion of T-bet−/− Th17 cells expanded in the host (Fig. 4A). These T-bet−/− T cells also maintained their capacity to produce IL-17 in response to AChR stimulation. Co-transfer of IFN-γ+/+ but not IFN-γ−/− NK cells inhibited the expansion of T-bet−/− Th17 cells and IL-17 production (Fig. 4). Co-transfer of neither IFN-γ+/+ nor IFN-γ−/− NK cells affected the expansion of T-bet+/+ Th17 cells or IL-17 production (Fig. 4).
Figure 4.
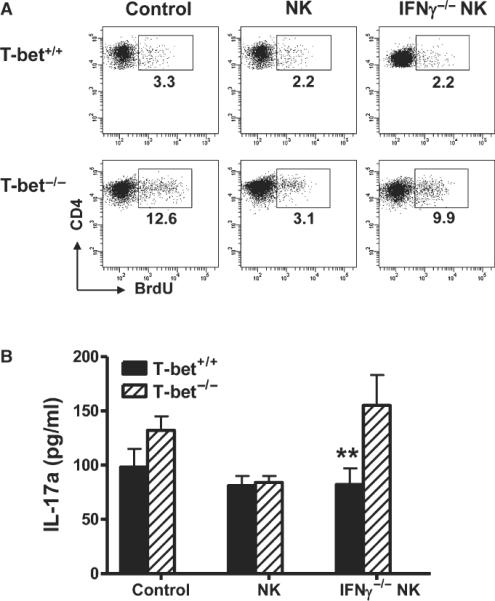
NK cells inhibit the in vivo expansion of Th17 cells in T-bet−/− mice.NK cells were sorted from lymphocytes pooled from spleens and lymph nodes of IFN-γ+/+RAG1−/− or IFN-γ−/−RAG1−/− mice and stimulated with LPS (10 μg/ml) and IL-15 (15 ng/ml). CD4+ T cells were sorted from AChR/CFA-primed T-bet+/+ or T-bet−/− mice and cultured under Th17 polarizing condition. 2 × 105 sorted NK cells and Th17-polarizing CD4+ T cells were passively transferred into γc−/− RAG1−/− mice at a ratio of 1:1. (A) Percentages of proliferation of CD4+ T cells were analysed by FACS at day 15 after cell transfer (Mice were injected with bromodeoxyuridine at day 14 after cell transfer). Dot plots are representative of two separate experiments. (B) 2 × 105 of CD4+ cells/well were cultured with AChR (10 μg/ml) in the presence of anti-NK1.1 (20 μg/ml) for 3 days, and IL-17 was measured by ELISA. Data shown are representative of two separate experiments (n = 3 per experiment).
Effect of NK cells on developing Th17 cells
Next, we investigated the effects of NK cells on the differentiation of naive CD4+ (CD25−CD62LhighCD44low) effector cells towards the Th17 lineage. Using a standard Th17 differentiation protocol, we found that naive T-bet+/+ and T-bet−/− CD4+ T cells became readily polarized towards the Th17 phenotype (Fig. 5, upper panels). When activated NK cells were added simultaneously with the cytokine cocktail used to induce Th17 differentiation, Th17 cell polarization was largely maintained in both T-bet+/+ and T-bet−/− CD4+ T cells (Fig. 5, lower panels). These findings suggested that the impact of NK cells on developing Th17 cells is much less pronounced than on fully differentiated Th17 cells.
Figure 5.
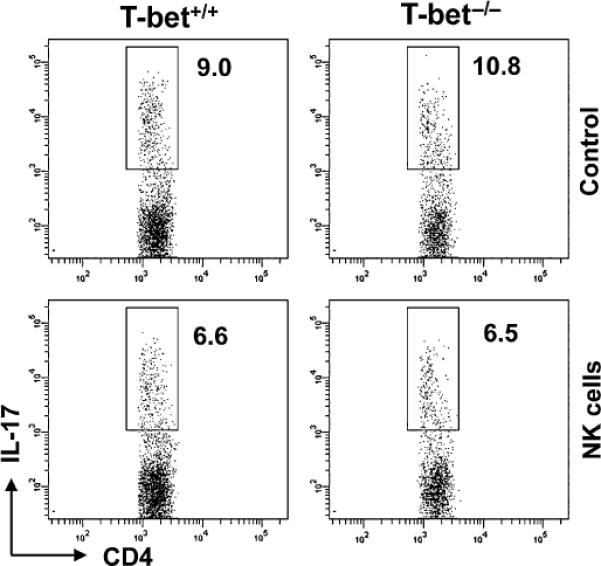
Effect of NK cells on developing Th17 cells. 2 × 105 (CD25−CD62LhighCD44lo) CD4+ T cells were sorted and cultured under Th17 polarizing conditions. Simultaneously, isolated NK cells were added to cultures. Production of IL-17 after 3 days of culture was analysed by intracellular cytokine staining. Dot plots shown are representative of four separate experiments.
Discussion
There are several possible explanations for the augmented autoreactive Th17 cell response in T-bet-deficient mice. First, the absence of Th1 cells in T-bet−/− mice might indirectly promote Th17 cell development, in part by the absence of Th1 cytokines that suppress Th17 cell differentiation, or by the antagonistic activity between T-bet and RORγ. Second, as suggested by Gocke et al. [5], T-bet expression promotes IL-23R induction, which, in turn, could influence the maintenance of the Th17 lineage [5]. Third, the paucity of NK and NKT cells in T-bet−/− mice [16] might contribute to a highly polarized Th17 phenotype. Because NK and NKT cells can influence the differentiation of T helper cells, we focused the last possibility in the current investigation.
Evidence derived from the ex vivo and in vivo experiments indicates that NK cells inhibit T-bet−/− autoreactive Th17 cells. Such inhibition is not achieved by direct killing of Th17 cells (Fig. 3B), rather production of IFN-γ by NK cells appear to play a major role. The transwell experiment suggests that in addition to release of IFN-γ, physical contact between NK cells and Th17 cells, and/or participation of other lymphocyte, that is, antigen-presenting cells, may also mediate suppression of Th17 cells.
A question remains to be answered is that whether NK cell inhibit Th17 cells non-specifically via release of IFN-γ. In other words, how does IFN-γ derived from NK cells differ those from Th1 cells, which has been suggested to have an antagonist effect on Th17 cells [10]. Additional investigation is needed to clarify this issue. The timing of IFN-γ production by NK cells differs from those produced by Th1 cells. NK cells are capable of release IFN-γ quickly and at high amount prior to T helper cell differentiation, whereas IFN-γ is produced by Th1 cells once antigen-specific immunity develops [25]. The early burst of cytokines by NK cells may be more important in determining the directions of T helpers.
Overall, our results provide new insight regarding the capacity of T-bet to negatively regulate Th17 cell development in the EAMG model of autoimmunity. We have shown that NK cells can inhibit the generation of Th17 cells and, specifically the expansion of polarized Th17 cells, in a process that is dependent, in large part, on IFN-γ production by NK cells. These findings imply that NK cells can downregulate Th17 cell responses to avoid pathologic autoimmunity, and that STAT3 activation in T-bet−/− T cells can be reversed by the addition of NK cells. Our findings provide the first demonstration that NK cells can inhibit T-bet-deficient, autoreactive Th17 cells in the peripheral immune system. These findings have important implications for the development of Th17 cell-mediated autoimmune pathology and suggest NK cells as novel targets for curbing Th17 cell-mediated autoimmunity.
Acknowledgment
The authors thank S. Rhodes for technical assistance and Y. Gan for her contribution to the present study. This work was supported in part by grants from the Muscular Dystrophy Association (FDS), ABRC (FDS 9-140) and the NIH (FDS AI083294, ALC AR53293, LVK AI070305 and HL089667).
Footnotes
Author statement WW and SS carried out the experiments. HGL, AC and LVK provided reagents and mutant mice. RL and FDS participated in the design of the study and wrote the paper. All authors read and approved the final manuscript.
Conflict of interest statement The authors have no financial conflict of interest.
References
- 1.Glimcher LH, Townsend MJ, Sullivan BM, Lord GM. Recent developments in the transcriptional regulation of cytolytic effector cells. Nat Rev Immunol. 2004;4:900–11. doi: 10.1038/nri1490. [DOI] [PubMed] [Google Scholar]
- 2.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 3.Mathur AN, Chang HC, Zisoulis DG, et al. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood. 2006;108:1595–601. doi: 10.1182/blood-2006-04-015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rangachari M, Mauermann N, Marty RR, et al. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–19. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gocke AR, Cravens PD, Ben LH, et al. T-bet regulates the fate of Th1 and Th17 lymphocytes in autoimmunity. J Immunol. 2007;178:1341–8. doi: 10.4049/jimmunol.178.3.1341. [DOI] [PubMed] [Google Scholar]
- 6.Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK. CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J Immunol. 2008;181:3906–14. doi: 10.4049/jimmunol.181.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Weiner J, Liu Y, et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206:1549–64. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazarevic V, Chen X, Shim JH, et al. T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol. 2011;12:96–104. doi: 10.1038/ni.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350–2. doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–48. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 11.Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 12.O'Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8:275–83. doi: 10.1016/s1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- 13.Shi FD, Ljunggren HG, Sarvetnick N. Innate immunity and autoimmunity: from self-protection to self-destruction. Trends Immunol. 2001;22:97–101. doi: 10.1016/s1471-4906(00)01821-4. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Fontecha A, Thomsen LL, Brett S, et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol. 2004;5:1260–5. doi: 10.1038/ni1138. [DOI] [PubMed] [Google Scholar]
- 15.Hong S, Wilson MT, Serizawa I, et al. The natural killer T cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med. 2001;7:1052–6. doi: 10.1038/nm0901-1052. [DOI] [PubMed] [Google Scholar]
- 16.Townsend MJ, Weinmann AS, Matsuda JL, et al. T-bet regulates the terminal maturation and homeostasis of NK and Valpha14i NKT cells. Immunity. 2004;20:477–94. doi: 10.1016/s1074-7613(04)00076-7. [DOI] [PubMed] [Google Scholar]
- 17.Shi FD, Wang HB, Li H, et al. Natural killer cells determine the outcome of B cell-mediated autoimmunity. Nat Immunol. 2000;1:245–51. doi: 10.1038/79792. [DOI] [PubMed] [Google Scholar]
- 18.Liu R, La Cava A, Bai XF, et al. Cooperation of invariant NKT cells and CD4 + CD25 + T regulatory cells in the prevention of autoimmune myasthenia. J Immunol. 2005;175:7898–904. doi: 10.4049/jimmunol.175.12.7898. [DOI] [PubMed] [Google Scholar]
- 19.Liu R, Van Kaer L, La Cava A, et al. Autoreactive T cells mediate NK cell degeneration in autoimmune disease. J Immunol. 2006;176:5247–54. doi: 10.4049/jimmunol.176.9.5247. [DOI] [PubMed] [Google Scholar]
- 20.Matsuda JL, Naidenko OV, Gapin L, et al. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med. 2000;192:741–54. doi: 10.1084/jem.192.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–87. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mars LT, Araujo L, Kerschen P, et al. Invariant NKT cells inhibit development of the Th17 lineage. Proc Natl Acad Sci U S A. 2009;106:6238–43. doi: 10.1073/pnas.0809317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi FD, Van Kaer L. Reciprocal regulation between natural killer cells and autoreactive T cells. Nat Rev Immunol. 2006;6:751–60. doi: 10.1038/nri1935. [DOI] [PubMed] [Google Scholar]
- 24.Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity. 2007;26:593–604. doi: 10.1016/j.immuni.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoenborn JR, Wilson CB. Regulation of interferon-γ during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]