

Abstract
Silica nanoparticles (SiO2 NPs) have been shown to exert cytotoxic effects in hepato-cytes and to cause liver injury. In the liver, Kupffer cells (KCs), as the resident macrophages, play an important role in the normal physiology and homeostasis of the liver. Nevertheless, few studies have attempted to clarify the role of KCs in hepatic injury induced by SiO2 NPs. In this study, we treated Buffalo rat liver (BRL) cells with the supernatants of SiO2 NP-stimulated KCs to determine KC-mediated hepatotoxicity and its underlying preliminary mechanism. We also examined the response of KCs and liver injury in vivo after the administration of SiO2 NPs. The results showed that KCs stimulated by SiO2 NPs release large amounts of reactive oxygen species, tumor necrosis factor-α and nitric oxide. After BRL cells were cultured with the supernatants of SiO2 NP-stimulated KCs, the viability of BRL cells was reduced, and increases in aspartate aminotransferase and lactate dehydrogenase leakage were observed. Exposure to SiO2 NPs in vivo caused KC hyperplasia, hepatic inflammation, and oxidative stress, which led to changes in the biochemical composition of the liver. These data suggest that SiO2 NPs activate KCs to mediate hepatic injury and that the preliminary mechanism involves the release of bioactive substances from KCs.
Keywords: silica nanoparticles, Kupffer cells, coculture, oxidative stress, metabolomics, hepatic injury
Introduction
Silica nanoparticles (SiO2 NPs), as one of the most popular nanomaterials, have been intensively investigated for their application in biomedical fields, for example as drug carriers,1 for gene therapy,2 and in molecular imaging.3 Together with these investigations, their potential side effects on human health have drawn increasing attention. Recent studies have focused on the toxicology of SiO2 NPs and have found that SiO2 NPs can cause harmful biological responses in diverse organs.4–9 The liver, as the major organ for the biotransformation of toxins, is a primary target organ for nanoparticles.10–12 Evidence has shown that SiO2 NPs induce cytotoxicity in hepatocytes13,14 and lead to liver injury, including particle accumulation,11 disturbances in metabolism,15 and fibrogenesis.16 However, in addition to the direct hepatotoxicity of SiO2 NPs, are there other factors that mediate hepatic injury induced by SiO2 NPs? Kupffer cells (KCs), the resident macrophages in the liver, play an important role in the normal physiology and homeostasis of the liver as well as participate in the acute and chronic responses of the liver to toxic compounds.17–20 Thus, it will be valuable to determine whether KCs mediate SiO2 NP-induced hepatic injury.
KCs are phagocytic and ingest substances to provide the first line of defense against invading particles.21 We previously found that SiO2 NPs were taken up mainly by KCs in the liver after intravenous injection.22 In addition to phagocytosis of particles, KCs are reported to contribute to the formation of silicotic nodules in the liver.23 These studies indicate that KCs may play a certain role in hepatic injury induced by SiO2 NPs, but the mechanism is still unclear. However, KCs are located in the hepatic sinusoids and lie in between or on top of endothelial cells, so how do they affect hepatocytes? The pathogenesis of hepatic injury resulting from acetaminophen/lipopolysaccharide treatment provides us with some insight into this question. A study has demonstrated that acetaminophen activates KCs to form reactive oxygen species (ROS) and nitric oxide (NO), which contribute to hepatotoxicity.24 Furthermore, a previous study has shown that KCs participate in monocrotaline/ lipopolysaccharide-induced liver injury through the release of inflammatory cytokines, such as tumor necrosis factor (TNF)-α.25 These studies suggest that KCs are activated by foreign stimuli and release a variety of bioactive mediators, which fulfill a crucial function in the response to liver injury. However, there have been few relevant reports addressing whether KCs can be activated to release these bioactive substances and mediate hepatic injury after phagocytosis of SiO2 NPs, which is essential to understanding the mechanism underlying hepatotoxicity induced by SiO2 NPs.
To explore the role of KCs in hepatic injury induced by SiO2 NPs, we first analyzed the release of ROS, NO, and TNF-α by SiO2 NP-stimulated KCs to investigate whether SiO2 NPs can activate these cells. To demonstrate KC-mediated hepatotoxicity and to determine the underlying preliminary mechanism, we established a noncontact coculture model by treating Buffalo rat liver (BRL) cells with particle free supernatants of SiO2 NP-stimulated KCs. An in vivo study was then carried out to examine the response of KCs after exposure to SiO2 NPs. Finally, clinical chemistry analysis and a 1H nuclear magnetic resonance (NMR) partial least squares discriminant analysis (PLS-DA)-based integrated metabolomics approach were used to assess liver injury induced by SiO2 NPs.
Materials and methods
Materials, characterization, and preparation
SiO2 NPs at ≥99% purity were purchased from Sigma-Aldrich (St Louis, MO, USA). The average size of SiO2 NPs was determined by transmission electron microscopy ([TEM] JEM-2010; JEOL Ltd, Tokyo, Japan). The characterization of size and zeta potential in RPMI 1640 medium (Life Technologies, Carlsbad, CA, USA) and physiological saline were performed using a Zetasizer 3000HS (Malvern Instruments Ltd, Malvern, UK) and laser diffraction particle size analyzer (LS230; Beckman Coulter, Inc, Brea, CA, USA), respectively.
SiO2 NPs were sterilized using ethylene oxide. In brief, after being weighed in clear tubes, SiO2 NPs were sent to the disinfection department for sterilization using an ethylene oxide sterilizer (3M; St Paul, MN, USA). The ethylene oxide sterilization line involved three different stages: preconditioning, sterilizing, and degassing. The temperature for sterilization was 55°C. The relative humidity was 40%–80%. The concentration range of ethylene oxide was 450–1200 mg/L. The sterilization lasted for 18 hours.
For in vitro studies, SiO2 NPs were suspended at concentrations of 50, 100, 200, 400, and 800 μg/mL in RPMI 1640 medium containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (Life Technologies, Carlsbad CA, USA). For in vivo studies, SiO2 NPs were prepared at a concentration of 5 mg/mL in physiological saline. To produce a less aggregated and uniform suspension, all samples were sonicated for at least 20 minutes before use.
KCs preparation and treatment
KCs were isolated according to Tukov et al.26 Briefly, the liver of a male Sprague Dawley (SD) rat was perfused in situ through the portal vein with liver perfusion medium (calcium-and magnesium-free Hank’s buffer). After digestion, the filtrate was centrifuged to pellet hepatocytes. This centrifugation resulted in a hepatocyte enriched pellet and a nonparenchymal cell enriched supernatant. A Percoll gradient was prepared by carefully layering 12 mL of 25% Percoll solution on to 10 mL of 50% Percoll solution. The nonparenchymal cell fraction was layered onto the Percoll gradient. This assembly was centrifuged to separate the nonparenchymal cell fraction into distinct zones in the gradient. The KC-enriched fraction (the 50% Percoll layer) was aspirated into a clean tube and washed and the pellet was resuspended in RPMI 1640 medium without serum. The viability of the isolated KCs was determined by trypan blue exclusion and was generally >95%. The cell concentration was adjusted to 1 ×106 viable cells/mL in a plastic culture flask for 20 minutes at 37°C in a humidified incubator. After this time, nonadherent cells were removed by replacing the culture medium with fresh complete culture medium. The purity of the KCs was determined by CD68 staining (AbD Serotec, Oxford UK) with a flow cytometer (Becton Dickinson, San Jose, CA, USA). For the in vitro study, KCs were plated into a 6-well culture plate at a density of 1 × 105 cells/ mL and allowed to attach for 24 hours. Different concentrations of SiO2 NPs suspensions were then applied to KCs. The cells without SiO2 NPs were used as the control.
Measurement of ROS and H2O2
The level of intracellular ROS in KCs was measured using the fluorescent probe 20,70-dichlorofluorescein diacetate (DCFH-DA). Briefly, a DCFH-DA stock solution (10 mM in methanol; Sigma-Aldrich) was diluted 1000-fold in RPMI 1640 without serum to yield a 10 μM working solution. After 24 hours of exposure to SiO2 NPs, KCs were washed twice with phosphate-buffered solution (PBS) and incubated in 2 mL of a working solution of DCFH-DA at 37°C for 30 minutes before the fluorescence was determined in a flow cytometer (Accuri C6™; BD Biosciences, San Jose, CA, USA).
H2O2, a type of ROS, was also measured. The H2O2 level in the supernatants of KCs stimulated with SiO2 NPs for 24 hours was analyzed using a kit (Beyotime Biotech Ltd Haimen, People’s Republic of China), and the absorbance was measured at 540 nm using a microplate reader (Wellscan MK3; Labsystems Dragon, Helsinki, Finland).
Measurement of TNF-α and NO
After KCs were treated with SiO2 NPs for 24 hours, the supernatants of KCs were collected. The levels of TNF-α were quantified using an ELISA kit (ExCell Bio, Shanghai, People’s Republic of China) according to the manufacturer’s instructions. The production of NO by KCs was measured using the Griess reagent (Beyotime Biotech Ltd), and the absorbance at 540 nm was recorded using a microplate reader.
Treatment of BRL cells
The BRL cell line was obtained from the Cell Bank of Type Culture Collection of the Chinese Academy of Sciences in Shanghai, People’s Republic of China. BRL cells were cultured in 96-well plates for cell viability assays or in 6-well plates for enzymatic analysis at a density of 5 × 104 cells/mL and allowed to attach for 24 hours. KCs were treated with suspensions of SiO2 NPs at different concentrations for 24 hours, and then the supernatant was harvested and centrifuged at 10,000 rpm for 10 minutes. For the coculture study, the supernatant of SiO2 NP- treated KCs was used to stimulate the BRL cells for 24 hours. BRL cells treated with the supernatant of KCs were used as the control.
BRL cell toxicity assay
BRL cell viability was determined using a Cell Counting Kit-8 assay (CCK-8; Beyotime Biotech Ltd), which assesses mitochondrial function by measuring the ability of viable cells to reduce CCK-8 into an orange formazan product. In brief, after BRL cells were cocultured with the supernatant of KCs, the cells were incubated with CCK-8 for 2 hours, and the absorbance of the plate was read at 450 nm using a microplate reader.
Aspartate aminotransferase (AST) leakage in the cultured medium of BRL cells was measured using an automatic analyzer (COBAS INTEGRA 400 plus; Roche, Basel, Switzerland). Lactate dehydrogenase (LDH) in the medium was determined using a commercially available LDH ELISA kit (Jiancheng, Nanjing, People’s Republic of China) according to the manufacturer’s protocols, and the absorbance was measured at 450 nm using a microplate reader.
Qualitative observation of BRL cell morphology
After BRL cells were cocultured with the supernatant of KCs for 24 hours, BRL cells were washed with PBS and the cellular morphology observed by phase contrast inverted microscopy (200× magnification).
Animal procedures
The animal study was approved by the Animal Ethics Committee of Shanghai Jiaotong University (People’s Republic of China). Twelve male SD rats weighing approximately 200 grams were purchased from Shanghai Laboratory Animal Center, Chinese Academy of Sciences ([SLACCAS] Shanghai, People’s Republic of China). One week prior to the experiment, the rats were housed in pairs, under controlled environmental conditions (23°C ± 0.5°C, 50% ± 5% humidity, illumination from 7 am to 7 pm). After an overnight fast, an intravenous injection (into the tail vein) of SiO2 NPs was administered as a single dose of nanoparticle suspension at 50 mg/kg (n = 6), whereas the controls were given physiological saline (n = 6). The concentration we used was similar to the concentration used in a previously published study11 At 48 hours postinjection, blood samples from the rats were collected and used for clinical biochemical analysis and hematology studies. Meanwhile, the left lateral lobe of the liver was removed and cut into two parts immediately after termination. One part was collected and used for histopathology and immunohistochemistry. The other part was snap frozen in liquid nitrogen and stored at −80°C until oxidative biomarkers and metabolomic analyses were performed.
Histopathology examination and immunohistochemistry
The liver samples were harvested and immediately fixed in a 10% formalin solution. Histopathology tests were performed using standard laboratory procedures. Briefly, the livers were embedded in paraffin blocks, sectioned into 5 μm slices, and then mounted onto glass slides. After hematoxylin–eosin staining, the histopathological reaction of the liver was evaluated using an optical microscope.
CD68 was detected in paraffin-embedded liver sections by immunohistochemistry For immunohistochemistry sections were mounted on silanized slides, deparaffinized and rehydrated. After the endogenous peroxidase activity was quenched the slides were placed in citrate buffer solution for antigen retrieval, blocked with normal goat serum, and incubated overnight at 4°C with a primary antibody (AbD Serotec). Immunoreactive complexes were detected using an avidin-biotin affinity system and visualized with the chromogen 3,30-diaminobenzidine tetrahydrochloride. Sections were counterstained with Mayer’s hematoxylin and examined microscopically. CD68 positive KCs were counted in ten randomly selected fields for each stained liver section using a light microscope (500× magnification).
Oxidative stress related biomarker assay
The livers from each group were weighed and cold protein lysis buffer was added at a ratio of 1:9 (w/v). The mixtures were homogenized at 4°C using an ultrasonic cell disruptor (Sonics Vibra Cell VCX105, Sonics Vibra Cell VCX105, Branson, MO, USA). The solution was then centrifuged at 12,000 rpm at 4°C for 15 minutes. The supernatants were collected for oxidative biomarker analysis. The activities of glutathione (GSH) and superoxide dismutase (SOD), and the levels of hydrogen peroxide (H2O2) and malondialdehyde (MDA) in the liver extracts were examined. All commercial colorimetric assay kits were purchased from Beyotime Biotech Ltd and the assays were performed according to manufacturers’ instructions. Protein concentrations were determined using the bicinchoninic acid method.
Clinical chemistry and hematology analysis
Standard spectrophotometric methods of a Cobas Integra 400 plus Automatic Biochemistry Analyzer (Roche) were used for the measurement of the following serum parameters: alanine aminotransferase, AST, alkaline phosphatase, total bile acid blood urea nitrogen, cholesterol, triglyceride, uric acid creatine kinase, high-density lipoprotein, and low-density lipoprotein. Hematological parameters consisting of white blood cell count (WBC), neutrophils (NEU), lymphocytes (LYM), monocytes (MONO), erythrocytes, hemoglobin, and platelet count were determined using a hematological autoanalyzer (Coulter T540 hematology system; Beckman Coulter, Inc). The TNF-α level in sera was measured using an ELISA kit.
1H NMR spectroscopic analysis of liver tissue extracts
Samples of liver tissue (250 mg) were homogenized in 2 mL of 50% acetonitrile in an ice water bath. The homogenates were centrifuged at 5070 g for 15 minutes at 4°C. The supernatants were removed and lyophilized before being reconstituted in 500 μL of D2O containing 1 mM3-(trimethylsilyl)[2,2,3,3–2H4] propionate. 1H NMR spectra were acquired for each sample at 500 MHz on a Bruker Avance III spectrometer (Bruker Corporation, Billerica, MA, USA) at ambient probe temperature. To explore the metabolic information embedded in NMR spectra, all 1D FIDs were multiplied by an exponential function of a 0.3 Hz line-broadening factor prior to Fourier transformation. The chemical shifts were referenced to the methyl group of sodium trimethylsilylpropionate (TSP) at δ 0.000. The integrals were normalized by the sum of the spectral intensity to compensate for differences in sample concentrations. PLS-DA was applied for the classification of NMR data.
Statistical analysis
Data were expressed as the mean ± standard deviation. Statistical analyses were performed using SPSS software (v 12.0; IBM Corporation, Armonk, NY, USA), and statistical comparisons were analyzed using the t-test and one-way ANOVA followed by Tukey’s Honestly Significant Difference (HSD) post hoc test. Differences were considered statistically significant when P < 0.05.
Results
Characterization of SiO2 NPs
SiO2 NPs used in this study were near-spherical, nonporous, and unmodified (Figure 1). The average size of SiO2 NPs measured by TEM was 15 nm. Since nanoparticles often agglomerates in solution, the sizes of SiO2 NPs and their agglomerates in RPMI 1640 medium and physiological saline were estimated using dynamic light scattering. The results showed that the agglomeration of SiO2 NPs was 92 nm in cell culture medium and 156 nm in physiological saline, which was nearly 6 to 10 times larger than the primary particle size. The values of the zeta potential were −8.5 ± 1.1 mV and −11.1 ± 1.3 mV in cell culture medium and physiological saline, respectively.
Figure 1.

Transmission electron microscopy image for silica nanoparticles.
Effects of SiO2 NPs on KCs
The ability of SiO2 NPs to induce intracellular oxidant production in KCs was assessed using 20,70-dichlorofluo rescein (DCF) fluorescence as a reporter of ROS generation. Figure 2A shows that SiO2 NPs resulted in a dose-dependent increase in intracellular ROS in the concentration range of 100–800 μg/mL (P < 0.05). In addition, H2O2 in the supernatant was measured as extracellular oxidant production. As shown in Figure 2B, SiO2 NPs induced a dose-dependent release of H2O2 into the supernatant of KCs.
Figure 2.

Reactive oxygen species, H2O2, tumor necrosis factor-α, and nitric oxide produced by SiO2 NP-stimulated Kupffer cells. (A) Intracellular reactive oxygen species (ROS) levels of Kupffer cells (KCs) incubated with different concentrations of silica nanoparticles (SiO2 NPs) for 24 hours; (B) H2O2 levels in the supernatant of KCs; (C) tumor necrosis factor (TNF)-α levels in the supernatant of KCs; and (D) the level of nitric oxide (NO) in the supernatant of KCs after treatment with SiO2 NPs for 24 hours.
Notes: Untreated KCs served as the negative control. Data represent mean ± SD, n = 3. *P < 0.05 when compared with control; **P < 0.01 when compared with control.
KCs were stimulated with SiO2 NPs for 24 hours, and secreted TNF-α was measured in cell culture supernatants. The ELISA results (Figure 2C) demonstrated that the secretion of TNF-α by KCs significantly increased at the concentrations of 400 µg/mL and 800 µg/mL (P < 0.01). NO released into the supernatant of KCs was measured by the Griess reaction. As shown in Figure 2D, SiO2 NP-stimulated KCs produced a dose-dependent increase in NO secretion in the concentration range of 200–800 μg/mL (P < 0.05).
KC-mediated cytotoxicity in BRL cells
After coculture in the supernatant of KCs, the viability of BRL cells was determined by CCK-8. As shown in Figure 3A, the supernatants of the KCs stimulated by SiO2 NPs in the concentration range of 100–800 μg/mL reduced the cellular viability of BRL cells (P < 0.05). In addition, the LDH and AST levels in the culture medium of BRL cells were measured. LDH levels significantly increased when BRL cells were treated with the supernatants of SiO2 NP-stimulated KCs (Figure 3B). AST levels were enhanced only when BRL cells were exposed to the supernatants of KCs that were treated with SiO2 NPs at the concentrations of 400 μg/mL and 800 μg/mL (P < 0.05) (Figure 3C).
Figure 3.

Kupffer cell-mediated cytotoxicity in Buffalo rat liver (BRL) cells. (A) The viability of BRL cells was measured by CCK-8 assay after coculture with the supernatants of silica nanaoparticles (SiO2 NPs) stimulated Kupffer cells (KCs) for 24 hours; (B) lactate dehydrogenase (LDH) levels in the medium of BRL cells after coculture with the supernatants of SiO2 NP-stimulated KCs for 24 hours; and (C) aspartate aminotransferase (AST) levels in the medium of BRL cells after coculture with the supernatants of SiO2 NP-stimulated KCs for 24 hours.
Notes: Data represent mean ± SD, n = 3. *P < 0.05 when compared with control; **P < 0.01 when compared with control.
Morphological characterization of BRL cells
After coculture in the supernatant of KCs, morphological changes of BRL cells were examined using phase contrast inverted microscopy. Figure 4A shows the morphology of control cells. At the low concentration of SiO2 NPs (50 μg/mL), the cells appear similar to control cells (Figure 4B). With increasing condensation of SiO2 NPs (100–800 μg/mL), BRL cells started to shrink and became irregular in shape after coculture in the supernatant of KCs (Figure 4C–F).
Figure 4.
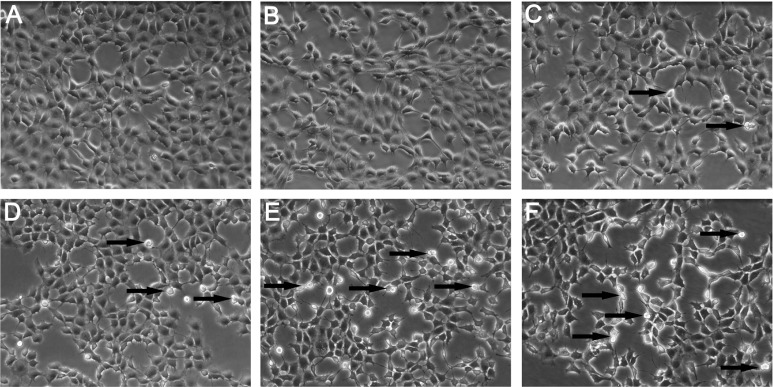
Morphological characterization of Buffalo rat liver (BRL) cells. BRL cells were treated with the supernatant of Kupffer cells stimulated by different concentrations of silica nanoparticles (SiO2 NPs). At the end of the 24-hour exposure, BRL cells were washed with phosphate-buffered solution and the cells were visualized by inverted microscopy (200× magnification). (A) Control; (B) 50 μg/mL; (C) 100 μg/mL; (D) 200 μg/mL; (E) 400 μg/mL; and (F) 800 μg/mL of SiO2 NPs.
Note: Arrows indicate cells with nuclear condensation and irregular shapes.
Histopathology examination and immunohistochemistry of the liver
The liver histopathological pictures are illustrated in Figure 5A-C. Compared with the control (Figure 5A), SiO2 NPs induced inflammatory cell infiltration at the portal area in the liver (Figure 5B and C). In addition, CD68, a KC-specific marker, was used to monitor KC activation. Compared with the control (Figure 5D), CD68-positive KCs became prominent and increased in number after the administration of SiO2 NPs (Figure 5E and F). KCs were counted in the liver sections (Figure 5G). In the control rat, the mean number of KCs was approximately seven cells/field. After exposure to SiO2 NPs for 48 hours, the number of KCs increased to 15 cells/field.
Figure 5.

Histological analyses and immunohistochemical stain of rat livers following an intravenous injection of silica nanoparticles (50 mg/kg body weight) at 48 hours. (A-C) Hematoxylin-eosin stain of liver sections (200× magnification) taken from (A) control and (B) silica nanoparticle (SiO2 NP)-treated group. The boxed area is magnified 500× and shown in (C). The liver of the SiO2 NP group showed inflammatory cell infiltrate (arrow). (D–F) Immunohistochemical stain for CD68 in liver sections (200× magnification) taken from (D) control and (E) SiO2 NP group. The boxed area is magnified 500× and shown in (F). The liver of the SiO2 NPs group showed strong positive signals for CD68 (arrow). (G) The mean number of Kupffer cells in the sections of livers per high-power field.
Notes: Data represent mean ± SD, n = 6. *P < 0.05 when compared with control.
Oxidative damage in the liver
The measurements of H2O2, GSH, SOD, and MDA levels in the liver are presented in Figure 6. When compared with the control, the liver exhibited a decrease in GSH activity and an elevation in MDA levels after the administration of SiO2 NPs (P < 0.05), while there were no obvious changes in the H2O2 and SOD levels.
Figure 6.

Changes in H2O2, GSH, SOD, and MDA levels of rat livers following an intravenous injection of SiO2 NPs (50 mg/kg body weight) at 48h. Levels of (A) H2O2; (B) GSH (C) SOD; and (D) MDA.
Notes: Data represent mean ± SD, n = 6. *P < 0.05 when compared with control.
Abbreviations: H2O2, hydrogen peroxide; GSH, glutathione; SOD, superoxide dismutase; MDA, malondialdehyde; prot, protein.
Clinical chemistry and hematology analysis
Significant changes in the serum clinical biochemistry parameters were observed after the administration of SiO2 NPs (Table 1). When compared with the control group, AST, total bile acid, blood urea nitrogen, cholesterol, and low-density lipoprotein levels were increased in the SiO2 NP group, whereas the triglyceride level was reduced. The hematology analysis (Table 2) showed that WBC, LYM, MONO, and NEU were increased, whereas the platelet count level was reduced after the administration of SiO2 NPs. The level of TNF-α released into the serum was also elevated after the administration of SiO2 NPs.
Table 1.
Effect of SiO2 NPs on selected clinical chemistry parameters
| Parameters | Control | SiO2 NPs |
|---|---|---|
| ALT (U/L) | 81 ± 8.22 | 83.33 ± 16.8 |
| AST (U/L) | 150.83 ± 14.19 | 186.33 ± 17.17* |
| ALP (U/L) | 264.16 ± 30.92 | 273.33 ± 28.34 |
| TBA (μM) | 21.1 ± 4.55 | 53.05 ± 15.6* |
| BUN (mM) | 5.78 ± 0.35 | 6.77 ± 1.26* |
| CHOL (mM) | 1.87 ± 0.07 | 2.36 ± 0.43* |
| TG (mM) | 0.6 ± 0.2 | 0.38 ± 0.07* |
| UA (μM) | 68.5 ± 5.4 | 57.83 ± 10.05 |
| CK (U/L) | 733.41 ± 133.37 | 786.08 ± 76.28 |
| HDL (mM) | 0.77 ± 0.04 | 0.86 ± 0.11 |
| LDL (mM) | 0.31 ± 0.02 | 0.47 ± 0.11* |
| TNF-α (pg/mL) | 46.02 ± 8.45 | 71.86 ± 12.27* |
Notes: Data are presented as mean ± SD, n = 6.
P < 0.05 when compared with vehicle control group.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CHOL, cholesterol; CK, creatine kinase; HDL, high density lipoprotein; LDL, low density lipoprotein; SiO2 NPs, silica nanoparticles; TBA, total bile acid; TG, triglyceride; TNF-α, tumor necrosis factor-α; UA, uric acid.
Table 2.
Effect of SiO2 NPs on selected clinical hematology parameters
| Parameters | Control | SiO2 NPs |
|---|---|---|
| WBC (109/L) | 4.47 ± 0.63 | 6.87 ± 1.34* |
| LYM (109/L) | 3.18 ± 0.56 | 4.18 ± 0.6* |
| MONO (109/L) | 0.07 ± 0.05 | 0.13 ± 0.05* |
| NEU (109/L) | 1.27 ± 0.35 | 2.33 ± 0.96* |
| RBC (109/L) | 5.77 ± 0.17 | 5.91 ± 0.3 |
| HGB (g/L) | 118.5 ± 5.63 | 119.67 ± 2.8 |
| PLT (109/L) | 481.67 ± 47.4 | 313.17 ± 32.31* |
Notes: Data are presented as mean ± SD, n = 6.
P < 0.05 when compared with vehicle control group.
Abbreviations: HGB, hemoglobin; LYM, lymphocytes; MONO, monocytes; NEU, neutrophils; PLT, platelet count; RBC, erythrocytes; SiO2 NPs, silica nanoparticles; WBC, white blood cell count.
1H NMR spectroscopic measurement and PLS-DA of liver tissue
1H NMR spectra of the liver tissue extracts after SiO2 NPs administration are shown in Figure 7A. PLS-DA was applied for the classification of the NMR data. The score plots on the first two latent variables (LVs) of the 1H NMR spectra from the control samples and the SiO2 NP-treated samples are shown in Figure 7B. The corresponding loading plot (Figure 7C) revealed the metabolites contributing to these differences. Following treatment with SiO2 NPs, increases in the levels of lactate, phosphorylcholine, sn-glycero-3-phosphocholine, tyrosine, phenylalanine, and lysine were detected together with decreases in the levels in succinate, glucose, and glycine. Detailed analysis of the loading plot indicated variations in the endogenous metabolites, which are summarized in Table 3.
Figure 7.

1H NMR spectra and PLS-DA of liver tissue extracts from rats following an intravenous injection of silica nanoparticles (50 mg/kg body weight) at 48 hours. (A) 1H NMR spectra; (B) PLS-DA score plots derived from 1H NMR spectra; and (C) coefficient plots derived from 1H NMR spectra.
Notes: (a) Control and (b) silica nanoparticle (SiO2 NP)-treated group.
Abbreviations: Glu, glucose; Gly, glycine; GPC, sn-glycero-3-phosphocholine; Lac, lactate; Lys, lysine; NMR, nuclear magnetic resonance; PC, phosphorylcholine; Phe, phenylalanine; ppm, parts per million; Suc, succinate; Thr, threonine; Tyr, tyrosine; Var ID, variable ID.
Table 3.
Summary of variations of liver metabolites
| Metabolite | Chemical shift | Variation following SiO2 NP treatment |
|---|---|---|
| Suc | 2.41(s) | ↓ |
| Lac | 4.13(q), 1.34(d) | ↑ |
| Glu | 4.64(d), 3.24 (dd), 3.49(t), 3.38(t), 3.45(m), 3.89(dd) | ↓ |
| PC | 4.18(m), 3.60(t), 3.22(s) | ↑ |
| GPC | 4.33(m), 3.68(m), 3.23(s), 3.60(dd), 3.68(dd), 3.90(m), 3.87(m), 3.94(m) | ↑ |
| Tyr | 7.19(d), 6.92(d), 3.05(dd), 3.19(dd), 3.93(dd) | ↑ |
| Phe | 7.33(d), 7.43(t), 7.37(t), 3.98(dd), 3.27(dd), 3.12(dd) | ↑ |
| Gly | 3.57(s) | ↓ |
| Lys | 3.75(t), 1.90(m), 1.452(m), 1.50(m), 1.72(m), 3.02(t) | ↑ |
Notes: Chemical shift in 1H NMR spectra, indicated by (s) singlet, (d) doublet, (dd) double-doublet, (t) triplet, and (m) multiplet peaks. Variations compared to control samples: ↑ indicates relative increase in signal; ↓ indicates relative decrease in signal.
Abbreviations: Glu, glucose; Gly, glycine; GPC, sn-glycero-3-phosphocholine; Lac, lactate; Lys, lysine; PC, phosphorylcholine; Phe, phenylalanine; SiO2 NP, silica nanoparticle; Suc, succinate; Tyr, tyrosine.
Discussion
Recent studies have shown that SiO2 NPs can induce cytotoxicity and oxidative stress in hepatocytes.13,14 In addition to hepatocytes, there are nonparenchymal cells in the liver, such as KCs. KCs are resident macrophages and play an important role in the defense against invading particles via phagocytosis.26,27 Some studies have described KCs as the primary responders to a toxicant where the released molecules are considered mediators of subsequent hepatic damage.28,29 However, few relevant reports have addressed the role of KCs in hepatic toxicity induced by SiO2 NPs. In this study, we demonstrated that SiO2 NPs activate KCs to mediate hepatic injury in vitro and in vivo, which might be essential to understanding the mechanisms of SiO2 NP-induced hepatotoxicity.
KCs can be activated by foreign stimuli, and the activated KCs play a major role in the initiation and maintenance of liver damage via the production of bioactive substances. ROS and proinflammatory cytokines are thought to be the immediate responses of KCs to a stimulus.30,31 To determine whether SiO2 NPs could activate KCs to release these substances, we treated KCs with SiO2 NPs. The results showed that SiO2 NPs induced increases in ROS formation in KCs (Figure 2A), which indicated that KCs were activated. Whether activated KCs can affect hepatocytes partly depends on whether the increased ROS can be released from these cells. Furthermore, we found that the level of H2O2 in the supernatants of KCs was significantly increased after exposure to SiO2 NPs (Figure 2B), which demonstrated that intracellular ROS are able to diffuse throughout these cells. In addition, our results revealed that KCs were stimulated by SiO2 NPs to release TNF-α and NO (Figure 2C and D). These bioactive mediators may have harmful effects on hepatocytes.
To examine the hypothesis that activated KCs mediate hepatic injury, we established a noncontact coculture model using the supernatants of SiO2 NP-stimulated KCs to culture BRL cells. In addition to the morphological changes (Figure 4), we also observed that the viability of BRL cells was reduced, and leakage of LDH and AST increased (Figure 3). LDH and AST are relatively stable enzymes in BRL cells, thus their leakage from the intracellular compartment of hepatocytes into the extracellular space indicates that plasma membrane disruption and hepatocyte damage have occurred.32,33 In this study, the elevated levels of these enzymes in the supernatants of BRL cells may be correlated with the increased release of ROS, TNF-α, and NO by activated KCs, as previously mentioned. Studies have demonstrated that ROS are capable of causing oxidative damage to major cellular structures, in particular the mitochondria and the plasma membrane.34,35 NO has been reported to downregulate cytochrome P450 and to suppress liver protein and DNA synthesis, and these activities may contribute to hepatotoxicity.36 Additionally, TNF-α, a potent inflammatory cytokine produced by activated KCs, can induce multiple mechanisms to initiate apoptosis in hepatocytes that leads to hepatic injury.37 These data suggest that KCs activated by SiO2 NPs mediate hepatotoxicity and that the preliminary mechanism might occur through the release of ROS, TNF-α, and NO.
An in vivo analysis was carried out to determine whether SiO2 NPs activate KCs and induce liver injury. The results showed that the number of sinusoidal KCs increased after administration of SiO2 NPs (Figure 5E and F), which indicated that SiO2 NPs activated the phagocytic activity of sinusoidal cells by increasing the number of KCs to help remove accumulating nanoparticles.38 The observed hyperplasia and activation of KCs could also lead to the increased formation of bioactive mediators contributing to hepatic injury, such as ROS, TNF-α, and NO, which we demonstrated in the in vitro study. Additionally, we observed infiltration of inflammatory cells into the liver and increases in the serum levels of WBC, LYM, MONO, NEU, and TNF-α, which suggests that SiO2 NPs may induce inflammation. Aderem39 reported that inflammation may result from the internalization of nanoparticles into macrophages and the subsequent activation of these macrophages. Because KCs can phagocytose SiO2 NPs and become activated, we deduced that KCs played a critical role in the inflammation that we observed. There is a link between inflammation and oxidative stress where recruited inflammatory cells can generate oxidative stress by activating oxidative stress responsive transcription factors.40 In this study, we found that SiO2 NPs caused oxidative stress in the liver, with a decrease in GSH activity and an elevation in MDA levels. Oxidative stress has been suggested to play an important role in the mechanisms of toxicity for a number of nanoparticles.43,44 In addition to the direct oxidative stress in hepatocytes induced by SiO2 NPs,13,14 the activated KCs and recruited inflammatory cells mediated oxidative damage.
We also investigated the biochemical variation in the serum and liver to determine the hepatic injury induced by SiO2 NPs. In agreement with our in vitro results, the level of AST in the serum was raised after the administration of SiO2 NPs. In the liver, the decrease in glucose levels and increase in lactate levels coupled with the alteration in succinate levels might reflect the effect of SiO2 NPs on glycolysis and the mitochondrial Krebs cycle.41 Phosphorylcholine and sn-glycero-3-phosphocholine are constituents of cell membranes and, thus, increased levels might be associated with the disruption of cellular membranes.42 Another metabolic consequence of liver injury was that treatment with SiO2 NPs resulted in the perturbation of amino acid metabolism, such as increased levels of threonine, phenylalanine, and lysine and a decrease in the level of glycine.
Conclusion
Our results demonstrated that KCs can be activated by SiO2 NPs and release bioactive mediators, such as ROS, TNF-α, and NO, which subsequently contributes to hepatotoxicity. Our in vivo study indicated that SiO2 NPs cause KC hyperplasia, hepatic inflammation, and oxidative stress, which lead to changes in the biochemical composition of the liver. These data suggest that activated KCs mediate the hepatic injury induced by SiO2 NPs. In the future, additional studies are needed to clarify whether other cell types in the liver, such as endothelial cells or hepatic stellate cells, are involved in hepatic injury induced by nanoparticles.
Acknowledgments
This work was supported by grants from the Natural Science Foundation of China (no 30870680), Major Program of the National Natural Science Foundation of China (no 81190132), the National Key Technology R&D Program (no 2012BAI22B01), and the Shanghai Sci-Tech Committee Foundation (no 11DZ2291700).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Huo Q, Liu J, Wang LQ, Jiang Y, Lambert TN, Fang E. A new class of silica cross-linked micellar core-shell nanoparticles. J Am Chem Soc. 2006;128:6447–6453. doi: 10.1021/ja060367p. [DOI] [PubMed] [Google Scholar]
- 2.Roy I, Ohulchanskyy TY, Bharali DJ, et al. Optical tracking of organically modified silica nanoparticles as DNA carriers: a nonviral, nanomedicine approach for gene delivery. Proc Natl Acad Sci U S A. 2005;102:279–284. doi: 10.1073/pnas.0408039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao X, Hilliard LR, Mechery SJ, et al. A rapid bioassay for single bacterial cell quantitation using bioconjugated nanoparticles. Proc Natl Acad Sci U S A. 2004;101:15027–15032. doi: 10.1073/pnas.0404806101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaewamatawong T, Shimada A, Okajima M, et al. Acute and subacute pulmonary toxicity of low dose of ultrafine colloidal silica particles in mice after intratracheal instillation. Toxicol Pathol. 2006;34:958–965. doi: 10.1080/01926230601094552. [DOI] [PubMed] [Google Scholar]
- 5.Guzman E, Liggieri L, Santini E, Ferrari M, Ravera F. Effect of hydrophilic and hydrophobic nanoparticles on the surface pressure response of DPPC Monolayers. J Phys Chem C. 2011;115:21715–21722. [Google Scholar]
- 6.Guzman E, Liggieri L, Santini E, Ferrari M, Ravera F. Influence of silica nanoparticles on dilational rheology of DPPC–palmitic acid Langmuir monolayers. Soft Matter. 2012;8:3938–3948. [Google Scholar]
- 7.Wu J, Wang C, Sun J, Xue Y. Neurotoxicity of silica nanoparticles: brain localization and dopaminergic neurons damage pathways. ACS Nano. 2011;5:4476–4489. doi: 10.1021/nn103530b. [DOI] [PubMed] [Google Scholar]
- 8.Hasezaki T, Isoda K, Kondoh M, Tsutsumi Y, Yagi K. Hepatotoxicity of silica nanoparticles with a diameter of 100 nm. Pharmazie. 2011;66:698–703. [PubMed] [Google Scholar]
- 9.Steenland K, Rosenman K, Socie E, Valiante D. Silicosis and end-stage renal disease. Scand J Work Environ Health. 2002;28:439–442. doi: 10.5271/sjweh.696. [DOI] [PubMed] [Google Scholar]
- 10.Xie G, Wang C, Sun J, Zhong G. Tissue distribution and excretion of intravenously administered titanium dioxide nanoparticles. Toxicol Lett. 2011;205:55–61. doi: 10.1016/j.toxlet.2011.04.034. [DOI] [PubMed] [Google Scholar]
- 11.Cho M, Cho WS, Choi M, et al. The impact of size on tissue distribution and elimination by single intravenous injection of silica nanoparticles. Toxicol Lett. 2009;189:177–183. doi: 10.1016/j.toxlet.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 12.Nishimori H, Kondoh M, Isoda K, Tsunoda S, Tsutsumi Y, Yagi K. Histological analysis of 70-nm silica particles-induced chronic toxicity in mice. Eur J Pharm Biopharm. 2009;72:626–629. doi: 10.1016/j.ejpb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad J, Ahamed M, Akhtar MJ, et al. Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell line HepG2. Toxicol Appl Pharmacol. 2012;259:160–168. doi: 10.1016/j.taap.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 14.Ye Y, Liu J, Xu J, Sun L, Chen M, Lan M. Nano-SiO2 induces apoptosis via activation of p53 and Bax mediated by oxidative stress in human hepatic cell line. Toxicol In Vitro. 2010;24:751–758. doi: 10.1016/j.tiv.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Lu X, Tian Y, Zhao Q, Jin T, Xiao S, Fan X. Integrated metabonomics analysis of the size-response relationship of silica nanoparticles-induced toxicity in mice. Nanotechnology. 2011;22:055101. doi: 10.1088/0957-4484/22/5/055101. [DOI] [PubMed] [Google Scholar]
- 16.Liu T, Li L, Teng X, et al. Single and repeated dose toxicity of mesoporous hollow silica nanoparticles in intravenously exposed mice. Biomaterials. 2011;32:1657–1668. doi: 10.1016/j.biomaterials.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 17.Winwood PJ, Arthur MJ. Kupffer cells: their activation and role in animal models of liver injury and human liver disease. Semin Liver Dis. 1993;13:50–59. doi: 10.1055/s-2007-1007337. [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Tao Q, Sun M, et al. Kupffer cells are associated with apoptosis, inflammation and fibrotic effects in hepatic fibrosis in rats. Lab Invest. 2010;90:1805–1816. doi: 10.1038/labinvest.2010.123. [DOI] [PubMed] [Google Scholar]
- 19.Thakur V, McMullen MR, Pritchard MT, Nagy LE. Regulation of macrophage activation in alcoholic liver disease. J Gastroenterol Hepatol. 2007;22( Suppl 1):S53–S56. doi: 10.1111/j.1440-1746.2006.04650.x. [DOI] [PubMed] [Google Scholar]
- 20.West MA, Billiar TR, Curran RD, Hyland BJ, Simmons RL. Evidence that rat Kupffer cells stimulate and inhibit hepatocyte protein synthesis in vitro by different mechanisms. Gastroenterology. 1989;96:1572–1582. doi: 10.1016/0016-5085(89)90529-5. [DOI] [PubMed] [Google Scholar]
- 21.Nishimori H, Kondoh M, Isoda K, Tsunoda S, Tsutsumi Y, Yagi K. Silica nanoparticles as hepatotoxicants. Eur J Pharm Biopharm. 2009;72:496–501. doi: 10.1016/j.ejpb.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Xie G, Sun J, Zhong G, Shi L, Zhang D. Biodistribution and toxicity of intravenously administered silica nanoparticles in mice. Arch Toxicol. 2010;84:183–190. doi: 10.1007/s00204-009-0488-x. [DOI] [PubMed] [Google Scholar]
- 23.Liu T, Li L, Fu C, Liu H, Chen D, Tang F. Pathological mechanisms of liver injury caused by continuous intraperitoneal injection of silica nanoparticles. Biomaterials. 2012;33:2399–2407. doi: 10.1016/j.biomaterials.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Michael SL, Pumford NR, Mayeux PR, Niesman MR, Hinson JA. Pretreatment of mice with macrophage inactivators decreases acetaminophen hepatotoxicity and the formation of reactive oxygen and nitrogen species. Hepatology. 1999;30:186–195. doi: 10.1002/hep.510300104. [DOI] [PubMed] [Google Scholar]
- 25.Yee SB, Ganey PE, Roth RA. The role of Kupffer cells and TNF-alpha in monocrotaline and bacterial lipopolysaccharide-induced liver injury. Toxicol Sci. 2003;71:124–132. doi: 10.1093/toxsci/71.1.124. [DOI] [PubMed] [Google Scholar]
- 26.Tukov FF, Maddox JF, Amacher DE, Bobrowski WF, Roth RA, Ganey PE. Modeling inflammation-drug interactions in vitro: a rat Kupffer cell-hepatocyte coculture system. Toxicol In Vitro. 2006;20:1488–1499. doi: 10.1016/j.tiv.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Tseng MT, Lu X, Duan X, et al. Alteration of hepatic structure and oxidative stress induced by intravenous nanoceria. Toxicol Appl Pharmacol. 2012;260:173–182. doi: 10.1016/j.taap.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Laskin DL, Laskin JD. Role of macrophages and inflammatory mediators in chemically induced toxicity. Toxicology. 2001;160:111–118. doi: 10.1016/s0300-483x(00)00437-6. [DOI] [PubMed] [Google Scholar]
- 29.Labib R, Turkall R, Abdel-Rahman MS. Endotoxin potentiates cocaine-mediated hepatotoxicity by nitric oxide and reactive oxygen species. Int J Toxicol. 2003;22:305–316. doi: 10.1080/10915810305117. [DOI] [PubMed] [Google Scholar]
- 30.Vrba J, Modriansky M. Oxidative burst of Kupffer cells: target for liver injury treatment. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2002;146:15–20. doi: 10.5507/bp.2002.003. [DOI] [PubMed] [Google Scholar]
- 31.Valatas V, Kolios G, Manousou P, et al. Secretion of inflammatory mediators by isolated rat Kupffer cells: the effect of octreotide. Regul Pept. 2004;120:215–225. doi: 10.1016/j.regpep.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 32.El-Tawil OS, Abou-Hadeed AH, El-Bab MF, Shalaby AA. d-Amphetamine-induced cytotoxicity and oxidative stress in isolated rat hepatocytes. Pathophysiology. 2011;18:279–285. doi: 10.1016/j.pathophys.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Hussain SM, Hess KL, Gearhart JM, Geiss KT, Schlager JJ. In vitro toxicity of nanoparticles in BRL 3A rat liver cells. Toxicol In Vitro. 2005;19:975–983. doi: 10.1016/j.tiv.2005.06.034. [DOI] [PubMed] [Google Scholar]
- 34.Xia T, Kovochich M, Nel AE. Impairment of mitochondrial function by particulate matter (PM) and their toxic components: implications for PM-induced cardiovascular and lung disease. Front Biosci. 2007;12:1238–1246. doi: 10.2741/2142. [DOI] [PubMed] [Google Scholar]
- 35.Sayes CM, Gobin AM, Ausman KD, Mendez J, West JL, Colvin VL. Nano-C60 cytotoxicity is due to lipid peroxidation. Biomaterials. 2005;26:7587–7595. doi: 10.1016/j.biomaterials.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 36.Gardner CR, Heck DE, Yang CS, et al. Role of nitric oxide in acetaminophen-induced hepatotoxicity in the rat. Hepatology. 1998;27:748–754. doi: 10.1002/hep.510270316. [DOI] [PubMed] [Google Scholar]
- 37.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdelhalim MA, Jarrar BM. Gold nanoparticles administration induced prominent inflammatory, central vein intima disruption, fatty change and Kupffer cells hyperplasia. Lipids Health Dis. 2011;10:133. doi: 10.1186/1476-511X-10-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aderem A. Phagocytosis and the inflammatory response. J Infect Dis. 2003;187( Suppl 2):S340–S345. doi: 10.1086/374747. [DOI] [PubMed] [Google Scholar]
- 40.Donaldson K, Stone V, Seaton A, MacNee W. Ambient particle inhalation and the cardiovascular system: potential mechanisms. Environ Health Perspect. 2001;109( Suppl 4):523–527. doi: 10.1289/ehp.01109s4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lei R, Wu C, Yang B, et al. Integrated metabolomic analysis of the nano-sized copper particle-induced hepatotoxicity and nephrotoxicity in rats: a rapid in vivo screening method for nanotoxicity. Toxicol Appl Pharmacol. 2008;232:292–301. doi: 10.1016/j.taap.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 42.Wei L, Liao P, Wu H, et al. Metabolic profiling studies on the toxicological effects of realgar in rats by (1)H NMR spectroscopy. Toxicol Appl Pharmacol. 2009;234:314–325. doi: 10.1016/j.taap.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Manna P, Ghosh M, Ghosh J, Sil PC. Contribution of nano-copper particles to in vivo liver dysfunction and cellular damage: Role of IkBa/NF-kB, MAPKs and mitochondrial signal. HYPERLINK Nanotoxicology. 2012;6:1–21. doi: 10.3109/17435390.2011.552124. [DOI] [PubMed] [Google Scholar]
- 44.Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2) Apoptosis. 2012;17:852–870. doi: 10.1007/s10495-012-0705-6. [DOI] [PubMed] [Google Scholar]