

Abstract
Background
The oncogenic potential of colony stimulating factor 1 receptor (CSF-1R) has been well described, while its relevance for human acute myelogenous leukemia (AML) is still undetermined. In a recent clinical trial for AML, sunitinib was found to hold potential therapeutic benefit, however, the mechanism for this remains unknown.
Materials and Methods
In this study, we treated three myeloid cell lines, Mono-Mac 1, THP-1, and U937, with sunitinib, and a small-molecule CSF-1R inhibitor (cFMS-I) to test the anticancer effect of such treatment.
Results
Mono-Mac 1 cells had inhibited proliferation and extracellular-signal regulated kinase activity as a result of CSF-1R inhibition and a dose-dependent increase in CSF-1R expression with both sunitinib and cFMS-I.
Conclusion
Our results suggest potential for CSF-1R as an important target of sunitinib or other similar drugs. Future study of CSF-1R may produce more targeted therapeutic approaches and aid in the development of personalized medicine for AML.
Keywords: AML, M-CSF, CSF-1, CSF-1R, cFMS, leukemia, tyrosine kinase, individualized medicine
Colony-stimulating factor 1 (CSF-1), previously known as macrophage colony-stimulating factor (MCSF), is a hematopoietic growth factor, which synergizes with other factors to regulate the survival, proliferation, differentiation and motility of cells of the monocyte lineage (1,2). In light of the mitogenic response that results from CSF-1 signaling cascade, it is not surprising that the receptor for CSF-1, colony stimulating factor 1 receptor (CSF-1R) or cFMS, has been identified as an oncogene. Originally, v-FMS was identified as a transforming retroviral product contained in the genome of Susan McDonough strain of feline sarcoma virus (SM-FeSV), a retrovirus which causes fibrosarcoma in cats (3). Additionally, CSF-1R is also up-regulated and subsequently mutated in one fifth of cases of Friend murine leukemia virus (F-MuLV)-derived leukemia (4). Studies of the v-FMS gene have identified sites of mutation that promote transformation which causes one of two phenotypic outcomes: inactivation of regulation and ligand independent activation. A 50 amino acid truncation in the carboxyl-terminus along with 11 other residues of an unknown source (5) was identified in v-FMS that inactivates the regulatory domain (6). This deletion, however, is only responsible for partial transformation (5,7). Additional mutations in the extracellular domain promote autophosphorylation and full transformation, with additive effects observed when both carboxyl-terminus and extracellular domain mutation are combined (5,8). Similar findings have been observed with carboxyl-terminus and extracellular mutation of a humanized v-FMS (5)
Expression of v-FMS in myeloid cells causes transformation, supporting the belief that this oncogene may be involved in acute myelogenous leukemia (AML) (9,10). Consequently, several studies in humans have also implicated CSF-1R in AML. It was reported that CSF-1R expression is increased in later stages of AML (11) and mutant FMS was identified in 23% of human AML M4 (12). If CSF-1R is indeed important in AML, the increased likelihood of CSF-1R involvement in differentiated AML may imply either that these cells rely on CSF-1R signaling to a greater extent, that these tumors are more differentiated due to CSF-1R activity (which promotes differentiation), or both.
Current therapeutics for AML, among other targets, are designed to inhibit specific tyrosine kinase receptors, but may have cross-reactivity to many targets. sunitinib, for example, inhibits vascular endothelial growth factor, platelet-derived growth factor, tyrosine protein kinase, and FMS-related tyrosine kinase 3 (FLT3) (13,14). However, sunitinib can also inhibit CSF-1R (15,16). Considering that genes for all of these may be involved in cancer, it is difficult to ascertain exactly which receptors are being affected in achieving therapeutic results. In a recent phase 1 clinical trial for AML, sunitinib was shown to promote partial remission (6/14 patients) (17). Although the doses used (0.125–0.25 μM: 50 ng/ml–100 ng/ml) were sufficient for both FLT3 and CSF-1R inhibition (15,16), it was asserted that the clinical response was due to FLT3, as 4/4 patients with FLT3 mutations were partial responders. However, other studies suggest that the effect of sunitinib on AML is unlikely to be solely due to FLT3 inhibition. Whereas sunitinib was found to promote mitogen-activated protein kinase/extracellular-signal regulated kinase (MAPK/ERK) inhibition (18), FLT3 inhibition alone did not reduce constitutive ERK activation in AML cells, including those with FLT3 mutations (19). Further, where sunitinib induces differentiation in both FLT3 mutant and wild type AML cells (20), suggesting that that at least, additional receptors are involved in these effects. Considering that CSF-1R is important in differentiation and MAPK/ERK signaling, and the crossreactivity of sunitinib to CSF-1R kinase, our study was designed to investigate the possible role of CSF-1 receptor signaling in AML.
Materials and Methods
Cell culture
As a model for AML, three myeloid cell lines were used. U937 cells are derived from pleural effusion from a patient with histiocytic lymphoma (21) and resemble promonocytic cells (22). THP-1 cells are derived from acute monocytic leukemia (M5b) (23) and resemble monocytes (24). Mono-Mac 1 cells are derived from peripheral monoblastic leukemia (M5a) (25) and resemble mature monocytes (24,26). THP-1 and U937 cells were grown in RPMI 1640 supplemented with 10% FBS, 2mM L-glutamine, and penicillin (50 U/ml)/streptomycin (50 μg/ml). Mono-Mac 1 cells were grown in the same media but supplemented with 1X non-essential amino acids and 1 mM sodium pyruvate (Invitrogen, Carlsbad, CA, USA).
The compounds used for the treatment of cells included sunitinib (0.01–0.1μM) (LC laboratories, Woburn, MA, USA), U0126 (1–10 μM) (Merck KGaA, Darmstadt, Germany), staurosporine (0.5 μM) (Sigma-Aldrich, St. Louis, MO, USA), and a small molecule (8-indan-5-yl-2-[4-(1-methylpiperidin-4-yl) phenylamino]-5-oxo-5,8-dihydropyrido[2,3-d]pyrimidine-6-carboxylic acid methoxyamide), an inhibitor of CSF-1R, designated herein as cFMS-I. The optimization of the latter compound for CSF-1R inhibition has been described previously (27). The cFMS-I inhibitor was a gift from Johnson & Johnson Pharmaceutical Research & Development.
Western blot and immunoprecipitation
For cell signaling experiments, all three AML cell lines were serum starved (0.1% FBS) for 48 hours and treated with CSF-1 (10 ng/ml) for 20 minutes at which point the samples were harvested. Samples were then analysed by western blot using Tris/Glycine buffer and transferred onto a hybond-P membrane (Amersham, GE, Fairfield, CT, USA). All protein samples were quantified by using a BCA assay to ensure similar protein quantities in all lanes of the western gel (Thermo Scientific Inc.). Antibodies used in western blot experiments were CSF-1R (sc692, 1:1000, Santa Cruz, Santa Cruz, CA, USA), phospho-tyrosine (sc-508, 1:1000, Santa Cruz,), phospho-ERK (sc-7383, 1:1000, Santa Cruz), total ERK (sc-94, 1:1000, Santa Cruz), and β-actin (A2228, 1:10,000, Sigma-Aldrich,). All antibodies were incubated with the blot overnight at 4°C in 5% BSA TTBS. The secondary antibodies mouse IgG-HPR (sc-2061, 1:10,000, Santa Cruz,) or rabbit IgG-HPR (sc-2030, 1:10,000, Santa Cruz,) were incubated for 1 hour at room temperature in 5% milk TTBS. The signal was detected using Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific Inc., Waltham, MA, USA).
Immunoprecipitation (IP) was conducted using CSF-1R antibody followed by western blot for phospho-tyrosine and then cFMS. Mono-Mac 1 cells or macrophages were treated overnight with inhibitor (cFMS-I, sunitinib or anti-CSF-1) and stimulated with CSF-1 (10 ng/ml) for 5 minutes and then harvested in lysis buffer. Samples were harvested in lysis buffer (150 mM NaCl, 40 mM Tris HCl pH 7.4, 1% NP40, 1 mM Dithiothreitol, 1 mM EDTA). Protein lysate (1000 μg) from Mono-Mac 1 cells was used for IP with 6 μg of CSF-1R or rabbit IgG control (Santa Cruz). IP was done over night at 4°C using Dynabeads conjugated to protein G (Invitrogen) in conjunction with the recommended BS3 cross-linking protocol (Thermo Fisher Scientific Inc.). The total precipitate was then analyzed by western blot as noted above.
Cell proliferation and toxicity assays
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was conducted following 3 days of treatment with inhibitor for all three cell lines (Roche, Basel, Switzerland). All cell lines were initially plated at 25 × 103 cells/96 well. Student’s t-test was conducted to compare the effect of no treatment to that with the highest drug concentration for each cell line. The optical density was read at 4 hours (test: 550 nm, reference: 630 nm). Propidium iodide assay was performed after 24 hours of treatment of Mono-Mac 1 cells using standard protocol (Sigma-Aldrich). An annexin/7-Aminoactinomycin D (7AAD) assay was carried out after 48 hours of treatment with cFMS-I, sunitinib, or U0126 using the manufacturer protocol (BD Biosciences, Franklin Lakes, NJ, USA). All flow cytometry was conducted on a Guava Easy Cyte mini machine (Guava Technologies, Billerica, MA, USA). The annexin/7AAD data was analyzed using FlowJo 9.2 software (Ashland, OR, USA).
Results
In order to assess the relevance of CSF-1R in AML, three myeloid cell lines, Mono-Mac 1, THP-1 and U937, were treated with compounds that affect the CSF-1R and other pathways. Firstly, a specific CSF-1R inhibitor (cFMS-I) was used at concentrations from .01 to 1 μM (Figure 1A). After allowing the cells to grow for three days, only Mono-Mac 1 cells demonstrated reduced proliferation with cFMS-I treatment as determined by MTT assay. As sunitinib has been used in clinical trials for AML, and can also inhibit cFMS, all three cell lines were treated with sunitinib at concentrations from 0.01 to 0.1 μM (Figure 1B). At these concentrations, sunitinib can inhibit CSF-1R (15,16) and these are similar to those used in a recent clinical trial (17). As with the treatment with cFMS-I, sunitinib caused differences in proliferation of the Mono-Mac 1 cells. In order to confirm that the downstream signaling for CSF-1R is similarly important for the growth of these cells, all three cell lines were treated with a MEK1/2 inhibitor, U0126 at concentrations from 1 to 10 μM (Figure 1C). Surprisingly, all three cell lines showed some inhibition of growth with 10 μM concentration of U0126. However, U0126 treatment produced a smaller degree of inhibition than did cFMS-I and sunitinib treatment.
Figure 1. The effect of colony stimulating factor 1 receptor inhibitor, sunitinib, and U0126 on acute myelogenous leukemia cell line proliferation.
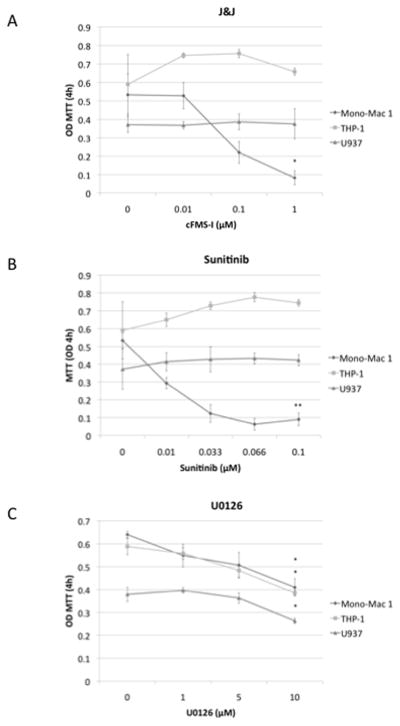
Mono-Mac 1, THP-1 and U937 cells were treated for 3 days with cFMS-I (A), sunitinib (B), and U0126 (C) * P≤ 0.05 for comparison of treatment at the highest concentration to that of no treatment.
To determine the nature of the toxicity of these three treatments, the three cell lines were cultured with cFMS-I (1 μM), sunitinib (0.1 μM), U0126 (10 μM), or stuarosporine (0.5 μM) for 24 hours and a propidium iodide cell-cycle assay was conducted. In Mono-Mac 1 cells, cFMS-I increased the G1 population from 57.92% to 64.04% (Figure 2B), Sunitinitb increased G1 cells from 57.92% to 63.4% (Figure 2C), and U0126 increased the G1 population from 57.92% to 62.6% (Figure 2D). Staurosporine, as expected, caused DNA fragmentation due to cell death (data not shown). cFMS-I treatment, however, caused very small changes in cell-cycle of THP-1 or U937 cells, altering the percentage of cells in the G1 phase of the cell cycle from 61.14% to 61.64% and 45.52% to 46.52% respectively (data not shown). In U937 cells, sunitinib increased the percentage of cells in G1 from 45.52% to 48.04% and U0126 increased the population from 45.52% to 53.9% (data not shown). In order to assess the possible role of apoptosis in the toxicity of these treatments, an annexin/7AAD assay was conducted (data not shown). No apoptosis was observed in any of these cell lines, except on treatment with stuarospoine (0.5 μM).
Figure 2. The effect of colony stimulating factor 1 receptor inhibitor, sunitinib, and U0126 on Mono-Mac 1 cell cycle distribution.
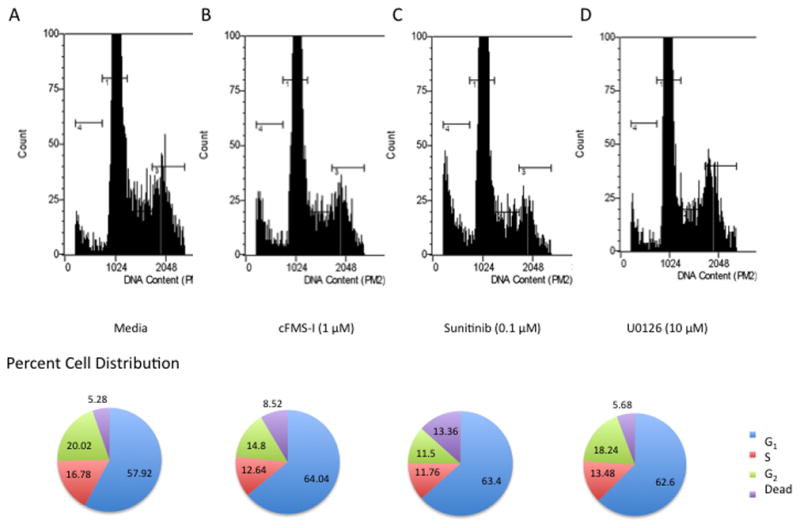
Mono-Mac 1 cells were treated for 24 hours with medium alone (A), cFMS-I (B), sunitinib (C), and U0126 (D).
We next evaluated the effect of these drug treatments on the cell signaling of the three AML cell lines. We found that of the three cell lines, Mono-Mac 1 cells were the only cell line to have constitutive ERK activity (Figure 3A). Furthermore, this activity was slightly responsive to CSF-1 and was inhibited by cFMS-I (1 μM). THP-1 cells did not have ERK activity with CSF-1 and therefore, we did not observe any inhibition with cFMS-I treatment. U937 cells also did not have ERK activation with CSF-1, but interestingly, did have slight ERK activation with cFMS-I. This may have been due to off-target activity. Next, we compared the effects of cFMS-I (1 μM), sunitinib (0.1 μM), and U0126 (10 μM) on ERK activity in Mono-Mac 1 cells with and without CSF-1 treatment (Figure 3B). As shown in Figure 3A, the Mono-Mac 1cells had high ERK activity and CSF-1 induced slight ERK activation. cFMS-I, sunitinib, and U0126 all inhibited ERK activity. sunitinib caused the greatest inhibition of ERK, followed by cFMS-I, and U0126 had the smallest effect on ERK. Interestingly, the degree of inhibition of each compound closely corresponds to the efficacy and potency, according to the MTT assay (Figure 1A–C). Further, even though inhibition by cFMS-I is not competitive with regards to CSF-1, we did observe an effect of CSF-1 treatment on ERK activity, implying that the inhibition by cFMS-I is not complete in these conditions. Additionally, some CSF-1 ERK activation was noted in the U0126 treated Mono-Mac 1 cells, also suggesting that the inhibition of ERK is incomplete with 10 μM of U0126 treatment. We then compared the level of CSF-1R expression in the three AML cell lines by western blot. We were only able to detect CSF-1R in Mono-Mac 1 cells (Figure 3C). The observation that CSF-1R is not expressed in THP-1 and U937 cells has been reported in previously (11). Interestingly, we found that the CSF-1R was down-regulated after 5 minutes of treatment with CSF-1 (Figure 3D).
Figure 3. Cell signaling and colony stimulating factor 1 receptor (CSF-1R) receptor function with drug treatment in acute myelogenous leukemia cell lines.
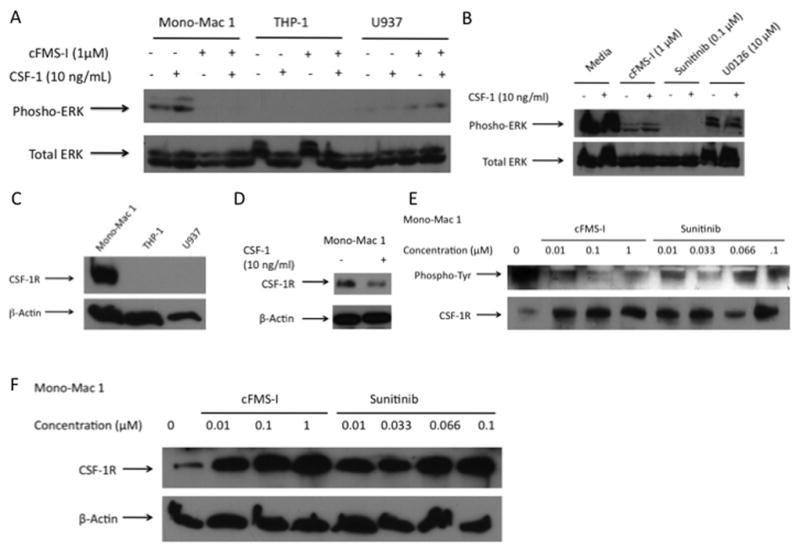
A: Mono-Mac 1, THP-1 and U937 cells were treated for 24 hours with medium alone or cFMS-I and were re-stimulated with and without colony-stimulating factor 1 (CSF-1) for 20 minutes. Phospho-extracellular-signal regulated kinase (ERK) and total ERK are shown. B: Mono-Mac 1 cells were treated with medium alone, cFMS-I, sunitinib, or U0126 for 24 hours and were re-stimulated with and without CSF-1 for 20 minutes. Phospho-ERK and total ERK are shown. C: Western blot of CSF-1R expression in Mono-Mac 1, THP-1, and U937 cells. D: The expression of CSF-1R in Mono-Mac 1 with and without 5 minutes of CSF-1 treatment cells. E: Mono-Mac 1 cells were pre-treated with cFMS-I or sunitinib for 24 hours and were then treated with CSF-1 for 5 minutes. Immunoprecipitation for CSF-1R followed by western blot or phospho-tyrosine and CSF-1R and F: for total cFMS.
To determine if effect of cFMS-I on CSF-1R phosphorylation occurred at similar doses as the inhibition of growth observed from the MTT assay, We next treated Mono-Mac 1 cells were treated with cFMS-I (0.01–1 μM) and sunitinib (0.01–1μM) (Figure 3E). Following IP for cFMS, we observed inhibition of phosphorylation in a similar dose dependent manner for cFMS-I. sunitinib also had inhibitory effects on CSF-1R activation, but these were not clearly dose dependent. Both treatments appeared to increase the amount of total CSF-1R by IP. By western blot, both treatments did indeed show a profound and dose dependent increase in the amount of total CSF-1R receptor in Mono-Mac 1 cells (Figure 3F).
Discussion
Our findings show that Mono-Mac 1 cells alone are inhibited by cFMS-I and sunitinib treatment, while U0126 treatment is somewhat toxic to all three cell lines. The nature of the inhibitory effect was primarily G1 arrest, as shown by cell-cycle analysis and annexin/7AAD staining. We found that Mono-Mac 1 cells had constitutive ERK activation that was inhibited with toxic treatments and was partly responsive to CSF-1. Mono-Mac 1 cells were the only cell line to express CSF-1R and demonstrated down-regulation of CSF-1R with CSF-1 treatment. We found that cFMS-I inhibited the receptor activity at similar concentrations that caused growth arrest in these cells. In agreement with previous publications, we also showed that sunitinib inhibits CSF-1R phosphorylation, supporting the possibility that CSF-1R may be one target of the drug. Finally, both drugs were observed to increase the expression of CSF-1R receptor in Mono-Mac 1 cells in a dose-dependent manner.
While we have not conclusively proven that CSF-1R is required for sunitinib efficacy in AML, we provide ample evidence that it is a very likely target. Two compounds that have activity against the CSF-1R kinase inhibited the growth of the only cell line, of the three tested found to have CSF-1R expression. We cannot exclude, however, the possibility that another target common to both drugs is responsible for this effect. Alternatively, the two drugs may have separate targets in Mono-Mac 1 cells other than cFMS. Another possibility is that CSF-1R is involved in the toxicity of both compounds, but is not sufficient by itself as a target. Consistent with the notion that CSF-1R is indeed inhibited, ERK inhibition was observed with both treatments, as well as with U0126, a known MEK1/2 and ERK inhibitor. This inhibition also explains the cell-cycle arrest that we report, as the ERK pathway is involved in the G1/S transition (28). While ERK is activated by CSF-1 in myeloid cells (29,30), ERK activation is a common pathway to many extracellular signals and does not specifically implicate CSF-1R (31). Finally, the up-regulation of CSF-1R in response to drug treatment of Mono-Mac 1 cells shows that the expression of the receptor in these cells is linked to it’s phosphorylation status and likely indicates that this receptor function is important for cell division and is highly regulated by these cells. It is, however, also possible that CSF-1R up-regulation is a non-specific event in response to growth arrest. In both cases, the induction of CSF-1R by tyrosine kinase inhibitors may be an important marker to study and correlate to patient outcomes.
The nature of CSF-1R signaling in Mono-Mac 1 cells is likely not the same as for those with the wild-type protein. While we did not sequence the gene in these cells, our results show that the receptor has different epitopes than that of donor derived-macrophages. The CSF-1R antibody used detected two expected bands in primary cells (data not shown), but only one band in Mono-Mac 1 cells under similar electroporation conditions (Figure 3C–F). This implies that the peptide detected by the antibody is mutated or differentially modified post-transcriptionally by phosphorylation of glycosylation. It is possible that such modification may lead to ligand-independent activation, however, it is also likely that the regulatory domain is not affected as there appears to be a down-regulation of the signaling by CSF-1 treatment in Mono-Mac 1 cells (Figure 3D). Furthermore, if CSF-1R function is indeed needed for the growth of Mono-Mac 1 cells, it is unlikely that CSF-1 produced by these cells activates the pathway as this cell line produces extremely low quantities of CSF-1 (data not shown). Future studies will be needed with this cell line to determine the exact nature of CSF-1 signaling.
We treated three cell lines that represent different stages of AML with inhibitors for both extracellular tyrosine kinase receptors and for downstream ERK signaling. Of the three cell lines used, Mono-Mac 1 cells display the most mature phenotype (mature monocyte) (24,26) and are the only one of the three to express CSF-1R (Figure 3C). This is consistent with previous literature that reports more probable up-regulation and mutation of CSF-1R in well-differentiated forms of AML (11,12). This observation further suggests that CSF-1R is a driving factor for proliferation of some AML cell types. However, it is unclear why sunitinib, which can affect multiple extracellular receptors, has minimal effects on U937 and THP-1 cells. These cell lines might have mutations downstream of receptors and do not rely on such signaling for growth. It may be an important future study to determine the need for surface receptor signaling in various types of AML, and correlate this data to drug responsiveness. Even though U937 and THP-1 cells were not observed to have high constitutive ERK activation (Figure 3A), U0126 treatment had some effect on the proliferation of these cells (Figure 1C). It may be that even undetectable levels of ERK activation have significant effects on cellular processes in these cells. This implies that ERK status in AML may be a deceptive finding, however, this needs to be evaluated further, as our results may be unspecific or due to inhibition of other pathways.
Our data suggests that the level of differentiation level of AML may be an important factor in the selection of personalized medicine. Furthermore, we propose that CSF-1R signaling may be more important for mature phenotypes of AML, or that the induction of cFMS, and possibly other receptors, might make a good marker for drug responsiveness. We believe our findings may encourage clinical trials testing these compounds to incorporate such criteria in the future or during retrospective analyses of past data. Likewise, Mono-Mac 1 cells may be an interesting model for the role of CSF-1R in future in vitro studies. Finally, this report emphasizes the understudied role of CSF-1R in AML and highlights the unique signaling of this pathway in transformed myeloid cells.
Acknowledgments
We would like to thank Dr. Wenhui Hu for helpful discussions. We would also like to thank Dr. Carl L. Manthey and Dr. Margery A. Chaikin from Johnson and Johnson for helpful advice in this project. This investigation was supported by the National Institutes of Health under a Ruth L. Kirschstein National Research Service Award (1T32MH079785), providing support to MK, as well as RO1 grant support to JR from NINDS and NIMH.
References
- 1.Haine V, Fischer-Smith T, Rappaport J. Macrophage Colony-Stimulating Factor in Pathogenesis of HIV Infection: Potential Target for Therapeutic Intervention. J Neuroimmune Pharmacol. 2006;1:1–19. doi: 10.1007/s11481-005-9003-1. [DOI] [PubMed] [Google Scholar]
- 2.Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol. 2004;14(11):628–638. doi: 10.1016/j.tcb.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 3.McDonough SK, Larsen S, Brodey RS, Stock ND, Hardy WD., Jr A transmissible feline fibrosarcoma of viral origin. Cancer Res. 1971;31(7):953–956. [PubMed] [Google Scholar]
- 4.Gisselbrecht S, Fichelson S, Sola B, Bordereaux D, Hampe A, Andre C, Galibert F, Tambourin P. Frequent c-fms activation by proviral insertion in mouse myeloblastic leukaemias. Nature. 1987;329(6136):259–261. doi: 10.1038/329259a0. [DOI] [PubMed] [Google Scholar]
- 5.Woolford J, McAuliffe A, Rohrschneider LR. Activation of the feline c-fms proto-oncogene: multiple alterations are required to generate a fully transformed phenotype. Cell. 1988;55(6):965–977. doi: 10.1016/0092-8674(88)90242-5. [DOI] [PubMed] [Google Scholar]
- 6.Roussel MF, Dull TJ, Rettenmier CW, Ralph P, Ullrich A, Sherr CJ. Transforming potential of the c-fms proto-oncogene (CSF-1 receptor) Nature. 1987;325(6104):549–552. doi: 10.1038/325549a0. [DOI] [PubMed] [Google Scholar]
- 7.Browning PJ, Bunn HF, Cline A, Shuman M, Nienhuis AW. “Replacement” of COOH-terminal truncation of v-fms with c-fms sequences markedly reduces transformation potential. Proc Natl Acad Sci U S A. 1986;83(20):7800–7804. doi: 10.1073/pnas.83.20.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roussel MF, Downing JR, Rettenmier CW, Sherr CJ. A point mutation in the extracellular domain of the human CSF-1 receptor (c-fms proto-oncogene product) activates its transforming potential. Cell. 1988;55(6):979–988. doi: 10.1016/0092-8674(88)90243-7. [DOI] [PubMed] [Google Scholar]
- 9.Wheeler EF, Askew D, May S, Ihle JN, Sherr CJ. The v-fms oncogene induces factor-independent growth and transformation of the interleukin-3-dependent myeloid cell line FDC-P1. Mol Cell Biol. 1987;7(5):1673–1680. doi: 10.1128/mcb.7.5.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheeler EF, Rettenmier CW, Look AT, Sherr CJ. The v-fms oncogene induces factor independence and tumorigenicity in CSF-1 dependent macrophage cell line. Nature. 1986;324(6095):377–380. doi: 10.1038/324377a0. [DOI] [PubMed] [Google Scholar]
- 11.Parwaresch MR, Kreipe H, Felgner J, Heidorn K, Jaquet K, Bodewadt-Radzun S, Radzun HJ. M-CSF and M-CSF-receptor gene expression in acute myelomonocytic leukemias. Leuk Res. 1990;14(1):27–37. doi: 10.1016/0145-2126(90)90143-w. [DOI] [PubMed] [Google Scholar]
- 12.Ridge SA, Worwood M, Oscier D, Jacobs A, Padua RA. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc Natl Acad Sci U S A. 1990;87(4):1377–1380. doi: 10.1073/pnas.87.4.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. [PubMed] [Google Scholar]
- 14.O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101(9):3597–3605. doi: 10.1182/blood-2002-07-2307. [DOI] [PubMed] [Google Scholar]
- 15.Mashkani B, Griffith R, Ashman LK. Colony stimulating factor-1 receptor as a target for small molecule inhibitors. Bioorg Med Chem. 2010;18(5):1789–1797. doi: 10.1016/j.bmc.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 16.Murray LJ, Abrams TJ, Long KR, Ngai TJ, Olson LM, Hong W, Keast PK, Brassard JA, O’Farrell AM, Cherrington JM, Pryer NK. SU11248 inhibits tumor growth and CSF-1R-dependent osteolysis in an experimental breast cancer bone metastasis model. Clin Exp Metastasis. 2003;20(8):757–766. doi: 10.1023/b:clin.0000006873.65590.68. [DOI] [PubMed] [Google Scholar]
- 17.Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O’Farrell AM, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–993. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 18.Nishioka C, Ikezoe T, Yang J, Takeshita A, Taniguchi A, Komatsu N, Togitani K, Koeffler HP, Yokoyama A. Blockade of MEK/ERK signaling enhances sunitinib-induced growth inhibition and apoptosis of leukemia cells possessing activating mutations of the FLT3 gene. Leuk Res. 2008;32(6):865–872. doi: 10.1016/j.leukres.2007.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Siendones E, Barbarroja N, Torres LA, Buendia P, Velasco F, Dorado G, Torres A, Lopez-Pedrera C. Inhibition of Flt3-activating mutations does not prevent constitutive activation of ERK/Akt/STAT pathways in some AML cells: a possible cause for the limited effectiveness ofmonotherapy with small-molecule inhibitors. Hematol Oncol. 2007;25(1):30–37. doi: 10.1002/hon.805. [DOI] [PubMed] [Google Scholar]
- 20.Nishioka C, Ikezoe T, Yang J, Yokoyama A. Sunitinib, an orally available receptor tyrosine kinase inhibitor, induces monocytic differentiation of acute myelogenous leukemia cells that is enhanced by 1,25-dihydroxyvitamin D(3) Leukemia. 2009;23(11):2171–2173. doi: 10.1038/leu.2009.152. [DOI] [PubMed] [Google Scholar]
- 21.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS, et al. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167(4):1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genois N, Robichaud GA, Tremblay MJ. Mono Mac 1: a new in vitro model system to study HIV-1 infection in human cells of the mononuclear phagocyte series. J Leukoc Biol. 2000;68(6):854–864. [PubMed] [Google Scholar]
- 23.Auwerx J. The human leukemia cell line, THP-1: a multifacetted model for the study of monocyte-macrophage differentiation. Experientia. 1991;47(1):22–31. doi: 10.1007/BF02041244. [DOI] [PubMed] [Google Scholar]
- 24.Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, Tada K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26(2):171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- 25.Sundstrom C, Nilsson K. Establishment and characterization of a human histiocytic lymphoma cell line (U-937) Int J Cancer. 1976;17(5):565–577. doi: 10.1002/ijc.2910170504. [DOI] [PubMed] [Google Scholar]
- 26.Collins SJ. The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood. 1987;70(5):1233–1244. [PubMed] [Google Scholar]
- 27.Huang H, Hutta DA, Rinker JM, Hu H, Parsons WH, Schubert C, DesJarlais RL, Crysler CS, Chaikin MA, Donatelli RR, Chen Y, Cheng D, Zhou Z, Yurkow E, Manthey CL, Player MR. Pyrido[2,3-d]pyrimidin-5-ones: a novel class of antiinflammatory macrophage colony-stimulating factor-1 receptor inhibitors. J Med Chem. 2009;52(4):1081–1099. doi: 10.1021/jm801406h. [DOI] [PubMed] [Google Scholar]
- 28.Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14–3–3 and cytoplasmic localization. J Biol Chem. 2003;278(49):49254–49260. doi: 10.1074/jbc.M306614200. [DOI] [PubMed] [Google Scholar]
- 29.Bhatt NY, Kelley TW, Khramtsov VV, Wang Y, Lam GK, Clanton TL, Marsh CB. Macrophage-colony-stimulating factor-induced activation of extracellular-regulated kinase involves phosphatidylinositol 3-kinase and reactive oxygen species in human monocytes. J Immunol. 2002;169(11):6427–6434. doi: 10.4049/jimmunol.169.11.6427. [DOI] [PubMed] [Google Scholar]
- 30.Kelley TW, Graham MM, Doseff AI, Pomerantz RW, Lau SM, Ostrowski MC, Franke TF, Marsh CB. Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J Biol Chem. 1999;274(37):26393–26398. doi: 10.1074/jbc.274.37.26393. [DOI] [PubMed] [Google Scholar]
- 31.Roux PP, Blenis J. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68(2):320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]