

Abstract
Increasing evidence indicates that high levels of serum erythropoietin (Epo) can lessen ischemia-reperfusion injury in the heart and multiple cardiac cell types have been suggested to play a role in this Epo effect. To clarify the mechanisms underlying this cardioprotection, we explored Epo treatment of coronary artery endothelial cells and Epo cardioprotection in a Mus musculus model with Epo receptor expression restricted to hematopoietic and endothelial cells (ΔEpoR). Epo stimulation of coronary artery endothelial cells upregulated endothelial nitric oxide synthase (eNOS) activity in vitro and in vivo, and enhanced nitric oxide (NO) production that was determined directly by real time measurements of gaseous NO release. Epo stimulated phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and mitogen-activated protein kinase kinase (MEK)/extracellular signal regulated kinase (ERK) signaling pathways, and inhibition of PI3K, but not MEK activity, blocked Epo-induced NO production. To verify the potential of this Epo effect in cardioprotection in vivo, ΔEpoR-mice with Epo response in heart restricted to endothelium were treated with Epo. These mice exhibited a similar increase in eNOS phosphorylation in coronary artery endothelium as that found in wild type (WT) mice. In addition, in both WT- and ΔEpoR-mice, exogenous Epo treatment prior to myocardial ischemia provided comparable protection. These data provide the first evidence that endothelial cell response to Epo is sufficient to achieve an acute cardioprotective effect. The immediate response of coronary artery endothelial cells to Epo stimulation by NO production may be a critical mechanism underlying this Epo cardioprotection.
Keywords: Endothelial cells, ischemia, myocardial infarction, nitric oxide, Endothelial nitric oxide synthase
Introduction
Erythropoietin (Epo), a cytokine required for erythrocyte production, provides protection against ischemic injury in heart, vascular endothelium and other tissues of rodents [9, 14, 39]. In animal models of cardiac ischemia-reperfusion injury, Epo administered 24 hr before or up to 24 hr following coronary artery ligation reduced infarct size [9, 12, 16, 24, 25]. Epo could directly protect against apoptosis induced by hypoxia in cardiomyocytes in culture studies [12, 28].
Epo has been reported to stimulate nitric oxide production by vascular endothelial cells mediated in part by induction and activation of endothelial nitric oxide synthase (eNOS/NOS3), particularly at reduced oxygen levels [3, 4]. However, Epo administered immediately prior to ischemia-reperfusion in the isolated rabbit heart model did not change nitrate and nitrite [34]. Subsequently, in the in vitro isolated rat heart ischemia-reperfusion model, it was shown that 24 hour pretreatment with Epo provided protection dependent on nitric oxide (NO) [7]. The differences in these studies indicate that the Epo-cardioprotective activity may proceed via mechanisms that are both dependent and independent of NO. The important role of eNOS in Epo cardioprotection was illustrated by ischemia-reperfusion injury where Epo cardioprotection was significantly diminished in eNOS-/--mice and appeared to be linked to phosphoinositide 3-kinases (PI3K) signaling [8, 29].
EpoR in endocardium links Epo to normal heart and vascular development. Although the bulk of cardiac tissue mass is cardiomyocytes, the endothelial cell number in heart is about 3 times greater [5]. Embryonic EpoR-/--mice have severely affected angiogenesis and decreased complexity of vascular networks by day E10.5, exhibit ventricular hypoplasia at day E12.5 and show increased myocardium and endocardium apoptosis prior death in utero due to severe anemia at day E13.5 [21, 42, 46]. Epo stimulation of endothelial cells induced eNOS activation and NO production [4], proliferation, chemotaxis and angiogenesis, and promoted endothelial progenitor cell migration [1, 39]. Mice overexpressing an Epo transgene exhibited elevated eNOS and plasma NO that contributes to prevention of cardiovascular disease such as hypertension and thromboembolism despite 80% hematocrit, and inhibition of NO synthase resulted in cardiovascular dysfunction and death [30]. The important role of eNOS in endothelial function suggests that Epo stimulation of endothelium contributes significantly to Epo cardioprotection. EpoR expression was also shown in cardiomyocytes and cardiac fibroblast cells [27, 41]. Epo stimulation of cardiomyocytes increased eNOS expression and NO production, which prevented cardiomyocytes from apoptosis [8]. Epo treatment in cardiac fibroblasts induced activation of JAK/STAT and ERK/MAPK signaling pathways [27]. Since cardiac fibroblasts contribute to post-ischemic remodeling and serve as a rich of source of cytokines, chemokines and growth factors, it was proposed that cardiac fibroblast cells contribute to Epo cardioprotection [27]. Hence, Epo-cardioprotective activity may proceed via multiple mechanisms involving diverse cell types.
To determine the contribution of cardiac endothelial response alone to Epo cardioprotection, mice were generated with endogenous EpoR expression restricted largely to hematopoietic and endothelial cells (ΔEpoR). ΔEpoR-mice that lack gross developmental defects show Epo cardioprotection in contrast to eNOS-/--mice that showed no Epo protection to myocardial ischemia-reperfusion injury. This suggests that the restoration of eNOS Epo response in endothelial cells via EpoR expression is sufficient to regain Epo cardioprotection as observed in wild type (WT) mice.
Methods
Animals
Mice with EpoR restricted to hematopoietic and endothelial cells (ΔEpoR) were generated by selective rescue of disrupted endogenous EpoR expression using a Tie2-Cre recombinase transgene [13]. TgEpoR-mice with EpoR expression restricted to erythroid cells were created using an EpoR transgene driven by the GATA-1-erythroid promoter on an EpoR-/- background [37]. C57Bl6/J WT-mice were purchased (Jackson Laboratory) and eNOS-/--mice were originally from Paul Huang (Massachusetts General Hospital). Only male mice were used in current study due to the concern of sexual dimorphism in Epo production. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). The NIDDK Animal Care and Use Committee approved all animal protocols.
Primary cell preparation
Ventricular cardiomyocytes, endothelial cells and fibroblasts were isolated from adult hearts from C57Bl/6 WT- and ΔEpoR-mice (n=3) (12 weeks old) by a modified perfusion method [36]. Briefly, After perfusion digestion, the ventricular tissue was minced, gently triturated and incubated in 5 ml of the same digestion buffer (the Tyrode’s buffer containing 0.1 mmol/L CaCl2 and Liberase Blendzyme 2) for 3 min at 37°C, the total dissociated cells were transferred into equal volume of the Tyrode’s buffer (pH 7.4 in mmol/L: NaCl 140, KCl 4, MgCl2 1, HEPES 5, D-glucose 10) containing 0.2 mmol/L CaCl2 and 10 mg/ml bovine serum albumin (Sigma) to stop the digestion. After gradually adding CaCl2 back to a final concentration of 1.2 mmol/L, Cardiac endothelial cells were sorted using CD146 (LSEC) MicroBeads, mouse [#130-092-007] and a MACS® Cell Separation system (Miltenyi Biotec, Inc, CA) following guidelines from the manufacture and then cultured in EGM®-2MV microvascular endothelial cell growth medium (Lonza). The flow-through containing cardiomyocytes and fibroblast cells were resuspended into modified insulin-transferrin-selenite MEM buffer containing 100 U/ml penicillin-streptomycin, 2 mmol/L L-glutamine, 0.1 mg/ml bovine serum albumin, 10 mmol/L 2,3-butanedione monoxime, 10 μg/ml insulin, 5.5 μg/ml transferrin, 5 ng/ml selenium, plated in petri dishes and incubated at 37°C for 60 min. The attached fibroblast cells were grown in 10 % CO2, in Dulbecco’s Modified Eagle’s Medium (GIBCO) supplemented with 10% FCS (Hyclone) and 1% pen/strep (GIBCO). The cardiomyocytes were plated into laminin coated 6-well plate. Cells were cultured at 37°C for 48 hours before they were processed for immunostaining and total RNA extraction. Neonatal cardiomyocytes from mice (2 day old) were isolated by collagenase/pancreatin digestion for 8-10 minutes at 37°C, gentle pipetting and transferring cell suspension to 50% bovine fetal serum (FBS), repeated three times. Cells were filtered and purified by selective sedimentation. Human coronary artery endothelial cells (HCAEC) (Lonza, Walkersville, MD) were cultured for 2-3 passages in EGM®-2MV microvascular endothelial cell growth medium (Lonza).
NO measurement
Culture media nitrite levels for HCAEC, neonatal cardiomyocytes and HL-1 cells were performed using a modified Griess reaction. Nitrite and nitrate content were determined using the HPLC-based NOx analyzer ENO-20 (Eicom, San Diego, CA); nitrite and nitrate are separated on a HPLC column and then a Cd reducing column is used to reduce nitrate to nitrite. Free (NO) gas production from confluent cultures of HCAEC in a T75 flask was measured using chemiluminescence NOx analyzer (ECO Physics) with 21% O2 (balance N2) as carrier gas. Area under the peak indicating NO production was determined using Origin software. Cells were also pre-incubated (30-60 minutes) with inhibitors for nitric oxide synthase (NG-nitro-L-arginine methyl ester), mitogen-activated protein kinase kinase (MEK) (U0126) and phosphoinositide 3-kinase (LY294002) prior to Epo treatment.
Analysis of protein expression
Proteins were extracted from endothelial cells and whole hearts, and concentration was determined using the BCA protein assay method (23227, Thermo Scientific, IL). After denaturation, protein was analyzed by gel electrophoresis (NuPAGE, Invitrogen Corp., Carlsbad CA) using reducing conditions, transferred to nitrocellulose membranes (Invitrogen) and probed using rabbit IgG specific for eNOS (Sigma, Saint Louis, MO), AKT, ERK44/42, p38 mitogen-activated protein kinase (MAPK), phospho-eNOS (Ser1177), phospho-AKT (S473), phospho-ERK44/42, phospho-p38MAPK and iNOS (Cell Signaling Technology, Inc) as the primary and horseradish peroxide-labeled anti–rabbit immunoglobulin G (IgG) (GE Healthcare Bio-Sciences Corp, Piscataway, NJ) as the secondary antibody, and visualized using ECL advance detection reagents (GE Healthcare Bio-Sciences Corp) and the FlourChemFC2 imager (Innotech, San Leandro, CA). Membranes treated with restore Western blot stripping buffer were re-probed using goat anti-β-actin polyclonal antibody and horseradish peroxide-labeled donkey anti-goat IgG (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). The Java image-processing program inspired by NIH image was used for quantitative analysis.
Gene expression analyses
Total RNA was extracted from cells and whole hearts using TRIzol (Invitrogen) and treated with Turbo DNase (Ambion, Austin, TX). RNA was reverse transcribed (MultiScribe Reverse Transcriptase, Applied Biosystems, Foster City, CA) and the resultant cDNA was used for Taqman RT-PCR reaction (7900HT Thermocycler, Applied Biosystems). The amount of target gene expressed was normalized to 16S levels. Primers used for mouse EpoR were forward: 5’-GCTCCGGGATGGACTTCA-3’, reverse: 5’-GAGCCTGGTGCAGGCTACAT-3’ and probe: 5’-ATACCAGCTCGAGGGTGAGTCACGAAAG-3’, and for human EpoR were forward: 5’-GCTCCCTTTGTCTCCTGCT-3’, reverse: 5’-CTCCCAGAAACACACCAAGTCCT-3’ and probe: 5’-AGCGGCCTTGCTGGCGG-3’
Epo and eNOS phosphorylation in mouse heart and coronary artery endothelial cells
WT-mice (n=16), ΔEpoR-mice (n=16) and eNOS-/--mice (n=16) (16 weeks old) were treated with Epo (Epoetin alpha, Amgen) (n=8) or saline (n=8), respectively. Fifteen minutes after intravenous injection (tail vein) of Epo (500 U/kg in 100 μl saline) or saline, mice were euthanized and hearts were excised from 5 mice and immediately snap frozen in liquid nitrogen for later western blot analysis. Hearts of 3 mice from each group were perfused with cold PBS buffer then cold 4% paraformaldehyde in PBS for preparation of paraffin sections.
Immuohistochemical and immunofluorescent staining
Paraffin sections were dewaxed, processed for antigen retrieval using citrate-based buffer (Vector Laboratories) and treated with hydrogen peroxidase to block endogenous peroxidase activity. Following blocking with normal goat serum, sections were incubated with primary antibodies specific for eNOS (Sigma), and phospho-eNOS (Ser1177) (Cell Signaling Technology), and then with horseradish peroxidase conjugated secondary antibody (Zymed). 3,3′-Diaminobenzidine tetrahydrochloride (DAB) Enhanced Liquid Substrate System (D6815, Sigma-Aldrich, Inc.) was used as peroxidase substrate for color development and methyl green was used as a nuclear counterstain. Cardiac endothelial cells cultured in 6-well plates were rinsed with cold PBS and fixed in 4% paraformaldehyde /PBS, pH 7.4, at 4°C for 30 min. After rinsing in cold PBS, the cells were incubated in 10% goat serum in PBS for 1hr at room temperature to block non-specific protein-protein interaction, then incubated with primary antibody, rat anti-mouse PECAM-1 (5μg/ml) (CBL1337, Cymbus Biotechnology) over night at 4°C. The secondary antibody was TRITC-conjugated goat anti-rat IgG used at 1/500 dilution for 1hr at room temperature.
Myocardial ischemic-reperfusion injury
Epo (10,000 U/ml; Amgen Inc., Thousand Oaks, CA) was diluted in normal (0.9%) saline to a dose of 4,000 U/kg in 200 μl. Normal saline or Epo was injected intraperitoneally 15 min prior to myocardial ischemia. Surgical ligation of the left coronary artery (LCA) on mice was performed following administration of ketamine/penotpbarbital anesthesia, intubation and connection to a rodent ventilator and median sternotomy as described previously [11]. After 45 minutes (30 minutes for eNOS-/-) of LCA ischemia, mice were reperfused for 24 hr. All mice analyzed survived this 24 hr ischemic-reperfusion injury procedure. Assessment of left ventricular area-at-risk (AAR) and infarct size (Inf) was performed as used previously [11].
Statistical analysis
Statistical analyses are expressed as mean ±STD or SEM values as indicated in the figure legends. Statistical significance was evaluated by Student’s t-test or one-way ANOVA with a Tukey’s Multiple Comparison Test.
Results
Epo induction of NO in coronary artery endothelial cells
The real time release of NO in primary human coronary artery endothelial cells (HCAEC) was monitored during treatment with saline as baseline using a sensitive chemiluminescence NOx analyzer (Figure 1A). Epo treatment resulted in an induction of NO release beginning at 15-20 minutes, reaching a peak at 15 minutes after the start of NO release and returning to baseline at about 30 minutes (Figure 1B). Epo induction of NO was lost with NG-nitro-L-arginine methyl ester (L-NAME) treatment (Figure 1C), suggesting that Epo-induced NO production is eNOS-dependent. Although inducible nitric oxide synthase (iNOS) has also been demonstrated to generate NO in endothelial cells. We did not detect iNOS expression in these cells.
Figure 1.
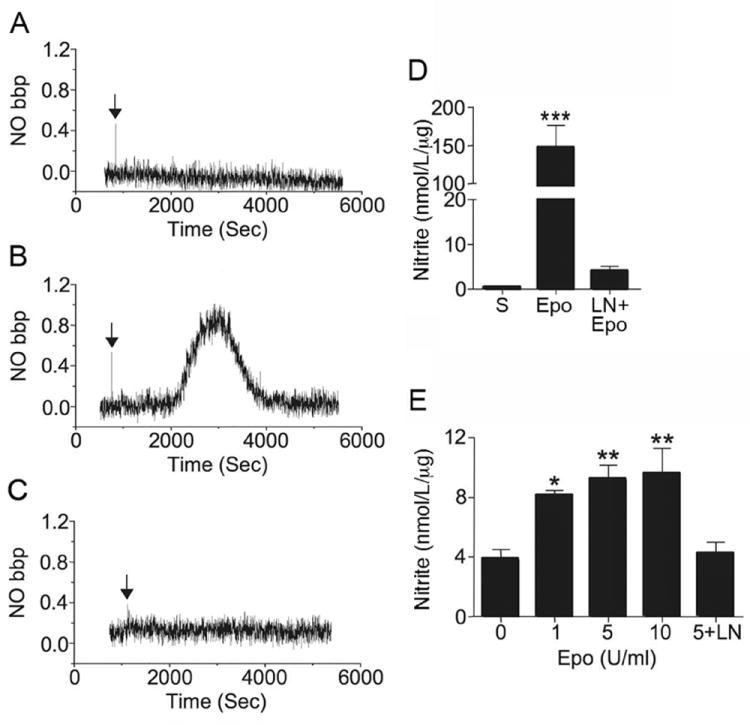
Epo effect on NO production in coronary artery endothelial cells (HCAEC). (A-C) Diffusion of NO release from the culture medium into the gas phase was directly monitored for primary coronary artery endothelial cells treated with A) saline (S), B) Epo (5 U/ml) and C) Epo plus L-NAME (pre-incubated at 1 mmol/L for 30 minutes) (LN+Epo). Arrow indicates the time of Epo or saline addition. (D) The amount of NO normalized to sample protein level is plotted. (E) Using a modified Griess reaction, the concentration of nitrite in culture medium of primary coronary artery endothelial cells treated with Epo (0, 1, 5 and 10 U/ml) under 2% O2 for 4 hours were determined. Cells cultured with Epo (5 U/ml) plus L-NAME (5+LN) were used as control. Values are ± STD. *** indicates p<0.001.
As an alternate measure of NO production, nitrite/nitrate levels determined by a modified Griess reaction were examined. NO production in coronary artery endothelial cells at 21% and 2% O2 was comparable. Epo-induced NO production at 2% O2 was statistically significant for Epo concentration at 1 to 10 U/ml with an increase of 2 fold or more at 4 hr (Figure 1E). Epo response appeared to be optimum at 5 U/ml with no further increase in NO production at 10 U/ml. L-NAME treatment abrogated the Epo induction of NO, confirming that NO production is mediated via activation of eNOS. These results are analogous to our previous observation of Epo response in human umbilical vein endothelial cells (HUVEC) that increased at low oxygen tension where EpoR expression was upregulated [3, 4].
Epo stimulation in coronary artery endothelial cells
Analysis of Epo/EpoR down stream signaling pathways in HCAEC by Western blotting revealed Epo stimulated phosphorylation of AKT, becoming significant at 15 minutes, while the level of AKT remained unchanged (Figures 2A-C). Epo-induced phosphorylation of AKT increased continuously even at 4 hours following Epo stimulation. Epo also activated ERK phosphorylation. Phosphorylation of ERK44 and ERK42 were increased at 5 minutes following Epo stimulation and were down regulated to a level close to baseline at 30 minutes while total ERK44 and ERK42 expression remained unchanged (Figures 2D-2H). In contrast to Epo stimulation of AKT and ERK phosphorylation, no differences in p38 mitogen-activated protein kinase (MAPK) or phosphorylated p38 MAPK (p-p38MAPK) levels were observed with Epo treatment (Figures 2I-2K).
Figure 2.
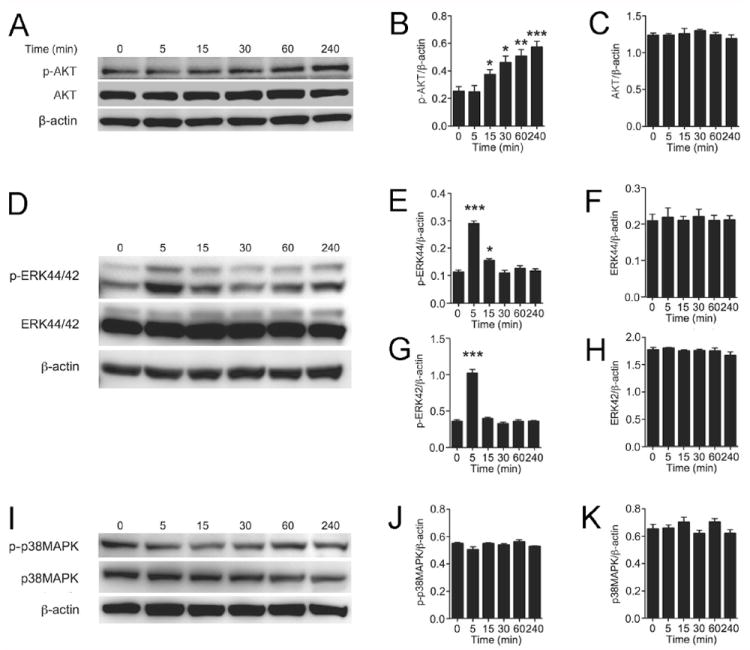
Epo activation of signaling pathways in coronary artery endothelial cells (HCAEC). (A-C) Representative Western blotting for phospho-AKT (p-AKT) and AKT (A) and quantification of p-AKT (B) and AKT (C) are shown. (D-H) Western blotting for phospho-ERK44/42 (p-ERK44/42) and ERK44/42 (D), and quantification of p-ERK44 (E), ERK44 (F), p-ERK42 (G) and ERK42 (H) are presented. (I-K) Western blotting for phospho-p38MAPK (p-p38MAPK) and p38MAPK (I), and quantification of p-p38MAPK (J) and p38MAPK (K) are shown. All quantitative analysis were normalized to β-actin. Mean values were determined from n=3 independent sets of measurements. Values are ± STD. * indicates p<0.05 and *** indicates P<0.001.
Epo activation of eNOS in coronary artery endothelial cells
To confirm that Epo induces NO production through stimulation of eNOS activity, eNOS expression and/or phosphorylation in Epo-treated HCAEC were examined by Western blotting. Epo treatment induced phosphorylation of eNOS at Ser1177, peaking at 15 minutes and returning to baseline level by 240 minutes (Figures 3A and 3B). Total eNOS expression was not affected (Figures 3A and 3C). The timing of Epo-stimulated increase in eNOS activity indicated by eNOS phosphorylation was consistent with the NO release detected in real-time measurement (Figure 1B). PI3K/protein kinase B (AKT) dependent phosphorylation of eNOS promotes NO production [15] and Epo stimulation of endothelial cells has been linked to PI3K/AKT signaling [39]. Treatment with PI3K inhibitor, LY294002, blocked Epo induction of NO (Figure 3D and 3F). In contrast, although Epo activated the MEK/ERK pathway and stimulated ERK phosphorylation, treatment with the MEK inhibitor, U0126, did not inhibit Epo stimulated NO production (Figures 3E-F). These results suggest that Epo-induced NO production in coronary artery endothelial cells is eNOS dependent and proceeds via the PI3K/AKT signaling pathway. Interestingly, Epo induced a transient phosphorylation of eNOS but had a more prolonged effect on AKT phosphorylation, suggesting that dephosphorylation of eNOS is initiated by a mechanism independent of AKT activity [18]. Alternatively, the possibility that activated AKT triggers dephosphorylation of eNOS (via activation of a phosphotase, for example) as a negative feedback mechanism cannot be excluded.
Figure 3.
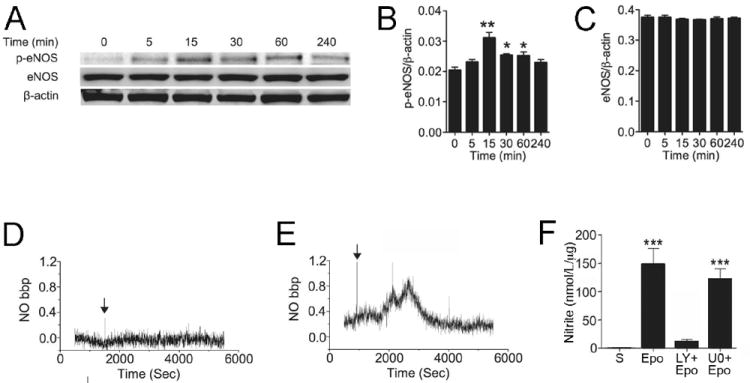
Epo activation of eNOS in coronary artery endothelial cells (HCAEC). (A-C) Representative Western blotting for phospho-eNOS (Ser1177) (p-eNOS) and total eNOS during Epo treatment up to 240 minutes (A) and quantification of p-eNOS (B) and total eNOS (C) are shown. All quantitative analysis were normalized to β-actin. (D-E) Diffusion of NO release from the culture medium into the gas phase was directly monitored for primary coronary artery endothelial cells treated with Epo (5U/ml) plus LY294002 (pre-incubated at 50 μmol/L for 1 hour) (LY+Epo) (D) and Epo plus U0126 (pre-incubated at 10 μmol/L for 30 minutes) (U0+Epo) (E). Arrow indicates the time of Epo. (F) The amount of NO normalized to sample protein level is plotted. S indicates saline control. Mean values were determined from n=3 independent sets of measurements. Values are ± STD. * indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001.
Epo induced eNOS phosphorylation in mouse coronary artery endothelial cells
To verify Epo induction of eNOS activity in heart in vivo, WT-mice were treated with Epo or saline. As observed in the culture studies, phosphorylated eNOS (phospho-eNOS) in heart from Epo-treated mice increased markedly compared with saline-treated mice at 15 minutes after Epo injection, although eNOS expression was not changed (Figures 4A-B). Immunohistochemical analysis of heart sections from Epo-treated mice localized the induction of eNOS phosphorylation in coronary artery endothelial cells but not in other tissues (Figure 4E). Epo treatment also promoted eNOS phosphorylation in heart of ΔEpoR-mice with no change in eNOS expression, similar to the levels observed in WT-mice (Figures 4A-B). Immunostaining for eNOS and phospho-eNOS in heart of ΔEpoR-mice was comparable to that of WT-mice with marked increase of phospho-eNOS staining in coronary artery endothelium after Epo treatment (Figure 4E). In contrast, Epo treatment in eNOS-/--mice showed no increase in either eNOS or phosphorylated eNOS (Figures 4A-B and E).
Figure 4.
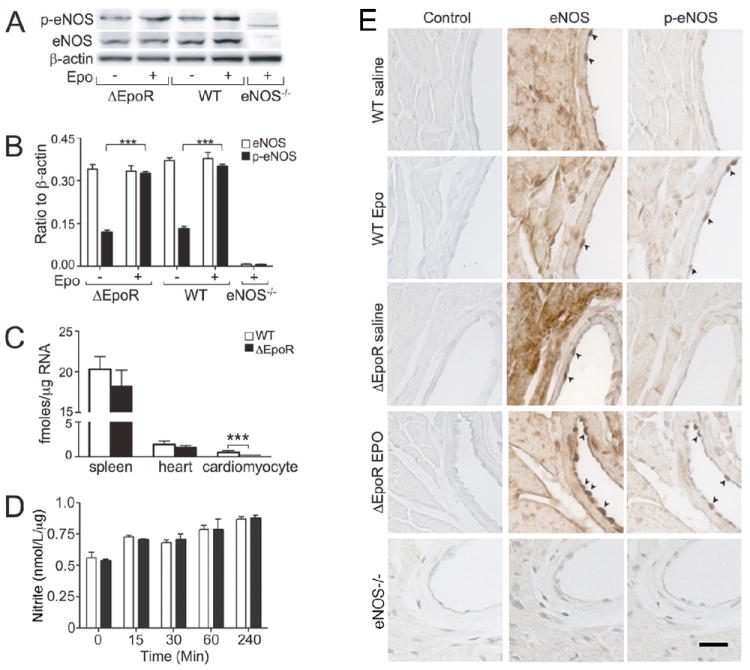
Epo induction of eNOS phosphorylation in heart. (A) Western blotting of phospho-eNOS (Ser1177) (p-eNOS) and total eNOS in whole heart tissue harvested from male ΔEpoR-, WT- and eNOS-/--mice at 15 minutes after intravenous administration of Epo or saline are shown. (B) Quantification of eNOS (open bar) and p-eNOS (filled bar) normalized to β-actin are presented. (C) Quantification of EpoR mRNA in spleen, heart and cardiomyocytes from adult WT-mice (open bar) and ΔEpoR-mice that lack EpoR expression in cardiomyocytes (filled bar) are presented. (D) The concentration of nitrite in culture medium of primary neonatal cardiomyocytes untreated (open bar) and treated (closed bar) with Epo (5 U/ml) was determined using a modified Griess reaction at time indicated. (E) Immunohistochemistry for eNOS and p-eNOS on heart sections prepared from WT-, ΔEpoR-mice treated with saline or Epo for 15 minutes and eNOS-/--mice is shown. Arrowhead indicates the staining specific for eNOS or p-eNOS in endothelial cells in the coronary artery. Scale bar represents 20 μm. Mean values were determined from n=3 independent sets of measurements. Values are ± STD. *** indicates p<0.001.
Quantification of EpoR mRNA shows EpoR expression in heart about 10 times lower than that in spleen, a hematopoietic tissue in mice. EpoR expression was even lower in isolated cardiomyocytes and cardiac fibroblasts from adult WT-mice (Figure 4C; Online resource Figure S2). In contrast to Epo stimulation of NO production in coronary artery endothelial cells (Figure 1), Epo treatment of primary neonatal cardiomyocytes (Figure 4D) and a cardiomyocyte cell line, HL-1 (data not shown), did not stimulate NO production as determined by a modified Griess reaction. In ΔEpoR-mice, EpoR expression in spleen and heart was similar to that in WT-mice (Figure 4C). However, lack of EpoR expression in isolated adult cardiomyocytes and cardiac fibroblasts from ΔEpoR-mice illustrates the restriction of EpoR expression to hematopoietic and endothelial tissue (Figure 4C; Online resource Figure S2). These data indicate that Epo induction of phospho-eNOS in heart of ΔEpoR-mice is also due primarily to an endothelial response rather than a direct effect of Epo on cardiomyocytes. The comparability of Epo effect in heart of WT- and ΔEpoR-mice suggests that coronary endothelium is the primary and/or initial target of Epo in inducing phosphorylation of eNOS.
Exogenous Epo in ischemia-reperfusion injury
Our analysis on Epo effect in coronary artery endothelial cells in both in vitro and in vivo conditions suggests an essential role of endothelium in Epo cardioprotection. To clarify this hypothesis, age and gender matched WT- and ΔEpoR-mice were divided into two groups each to assess ischemia-reperfusion injury with and without Epo treatment (Figures 5A and 5C). Plasma NO levels were determined by measuring nitrite/nitrate. No difference in plasma NO level among the groups was identified. Mice were treated with Epo 15 minutes prior to left coronary artery occlusion and evaluated after 24 hr of reperfusion, prior to the expected Epo induced increase in hematocrit. WT- and ΔEpoR-mice treated with Epo showed no change in hematocrit after 24 hr (Online resource Figure S3).
Figure 5.
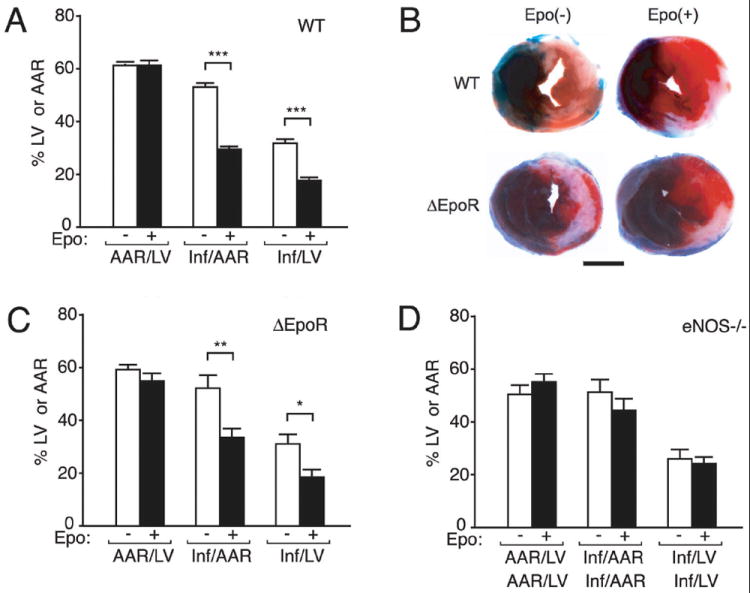
Epo protection of cardiac ischemia-reperfusion injury in mice. (A-D) WT (A), ΔEpoR (C) and eNOS-/- (D) male mice were subjected to cardiac ischemia-reperfusion injury. Area-at-risk (AAR) with respect to the left ventricle (LV) was similar between WT-mice without (open bar; n=15) or with (closed bar; n=12) Epo treatment (A) and ΔEpoR-mice without (open bar; n=7) or with (closed bar; n=5) Epo treatment (C). Epo treatment significantly attenuated myocardial infarct size (Inf) with respect to the AAR (Inf/AAR) in WT-mice (P<0.001) and in ΔEpoR-mice (P<0.008) and with respect to the LV (Inf/LV) in WT-mice (P<0.001) and in ΔEpoR-mice (P<0.018). (B) Representative mid-myocardial cross sections of TTC stained hearts for WT- and ΔEpoR-mice without or with Epo treatment are shown. Dark blue area (i.e., Evan’s blue-stained), nonischemic zone; remaining area, AAR; white area, infarcted tissue; red (i.e., TTC-positive), viable myocardium. (D) No Epo protective effect was observed in eNOS-/--mice. The ratios, (AAR/LV), (Inf/AAR) and (Inf/LV), were similar between eNOS-/--mice without (open bar; n=7) or with (closed bar; n=10) Epo treatment. Scale bar in C represents 2.0 mm. Values are ± SEM. * indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001.
The area-at-risk (AAR) relative to the entire left ventricle (LV) was similar in all of the groups (Figures 5A-C). In WT-mice, Epo decreased the infarct size (Inf) relative to the AAR (Inf/AAR) and infarct size relative to the entire LV (Inf/LV) by 44% when compared to vehicle treatment (Figures 5A-B). ΔEpoR-mice behaved analogous to age and sex matched WT-mice when subjected to heart ischemia-reperfusion injury (Figures 5B-C). Epo treatment in ΔEpoR-mice provided cardioprotection with reduction in infarct size at levels observed for WT-mice (Figures 5A-C). This suggests that preservation of endothelial response to Epo stimulation is sufficient for achieving Epo cardioprotection in this model of myocardial ischemia-reperfusion injury.
In contrast to WT- and ΔEpoR-mice, Epo cardioprotection was not observed in eNOS-/--mice (Figure 4D). In these animals, LV ischemia for 30 minutes and reperfusion for 24 hr resulted in similar extent of infarction relative to the AAR and the LV. Treatment with Epo did not significantly improve outcome when eNOS was no longer a contributing factor in the Epo effect. These data provide strong evidence that Epo response in endothelial cells is essential for cardioprotection of exogenous Epo that includes the up-regulation of eNOS activity and induction of NO production. Endogenous Epo signaling did not appear to provide additional protection to ischemia-reperfusion injury. Hematopoietic rescue of the EpoR-/- phenotype (TgEpoR) resulted in viable mice with no gross morphological defect of heart development [37]. At 5 months we observed no difference in myocardial ischemia-reperfusion injury between untreated WT and TgEpoR-mice (Online resource Figure S1). The damage in myocardial ischemia-reperfusion injury in TgEpoR-mice was not affected by loss of endogenous Epo signaling in non-hematopoietic tissue.
Discussion
Using a mouse model with EpoR expression restricted to hematopoietic and endothelial cells, we show specifically that the Epo effect in cardiac endothelial cells preserved the acute (24hr) Epo cardioprotection comparable to WT mice. This effect is unlikely due to hematopoietic response since we did not detect changes in hematocrit in the 24 hr period following Epo administration that was used for the ischemic-reperfusion injury model. In culture, we previously observed that Epo induction of eNOS activity in endothelial cells from select vascular beds. We demonstrate here for the first time the kinetic changes of NO production in coronary artery endothelial cells following Epo stimulation. The timing of Epo induced NO release beginning at 15 min. after stimulation appears concomitant with the change in eNOS activity and is dependent on the PI3K/AKT signaling pathway. These findings support the observation of PI3K dependent Epo protection in rat using the perfused isolated heart model and in vivo [6, 10]. In animal studies, we also demonstrate that change in eNOS phosphorylation in cardiac endothelial cells was evident 15 min. after Epo administration. The importance of eNOS in Epo activity is further suggested by the lack of Epo cardioprotection in eNOS-/--mice. The evidence from the current study demonstrates the Epo effect on coronary artery endothelial cells to stimulate eNOS activity and NO production and strongly suggests that endothelial response is sufficient to provide acute Epo cardioprotection.
NO is an important signaling molecule released from endothelial cells and contributes to a variety of fundamental cardiac activities. NO released from coronary artery endothelial cells can diffuse to coronary smooth muscle and initiate vasodilation of coronary arteries, which serves as a mediator of cardiac blood flow. In the capillaries, NO can diffuse to the cardiomyocytes, to affect timing of relaxation and myocardial oxygen consumption [33]. A number of studies have shown that enhancing NO availability by administration of NO donor reduces myocardial infarction size and improves the outcome of ischemia/reperfusion injury. This protective effect is attributed to contributions of exogenous NO to early preconditioning-like protection and induction of late phase preconditioning, involving both PI3K/Akt and ERK1/2, the so called reperfusion injury salvage kinase (RISK) pathways. Although endogenous NO is not necessary for early phase protection of ischemia-induced preconditioning, upregulation of NO production by eNOS is required for cardioprotective effects of late phase ischemic preconditioning [17, 20, 32, 44]. These NO associated activities suggest the therapeutic potential of mechanisms that increase NO levels in cardioprotection. Here, we observed a rapid Epo-activation of eNOS and the corresponding NO release in coronary artery endothelial cells. Epo treatment induced comparable cardioprotection against ischemia/reperfusion injury in both WT- and ΔEpoR-mice where EpoR expression in heart is restricted in endothelial cells, but is abrogated when eNOS is ablated (eNOS-/--mice). This indicates that Epo cardioprotection is eNOS dependent and requires endothelial stimulation by Epo, which may induce preconditioning-like effect through stimulating NO production. Unlike eNOS, iNOS is not constitutively expressed and its activity is regulated at the level of expression [22]. The lack of iNOS expression in our coronary artery endothelial cells after Epo treatment indicates that iNOS does not contribute to the immediate endothelial response observed here.
Long term Epo treatment increased constitutive eNOS expression and NO production in culture [3, 4] and transgenic mice overexpressing Epo exhibit elevated eNOS and NO [30]. However, the rapid response of coronary artery endothelial cells to Epo stimulation by increasing NO production in our current study is a result of increased activity of eNOS caused by phosphorylation without a change in overall eNOS expression. The data presented here support a key role for endothelial production of NO in Epo cardioprotection during the first 24 hr period in this model of ischemia-reperfusion injury. A sustained high level of Epo may also help recovery from ischemia-reperfusion injury though its pro-angiogenic activity, another major effect of Epo on endothelial cells [21]. Evidence that Epo treatment mobilizes endothelial progenitor cells from bone marrow and contributes to endothelial regeneration [39] may support a long-term recovery from heart infarction beyond the 24 hr period of ischemia-reperfusion injury model used here. In adult mice, EpoR deficiency in non-hematopoietic tissue caused impaired mobilization of endothelial progenitor cells and accelerated development of hypoxia-induced pulmonary hypertension[26, 31]. However, comparison between WT- and TgEpoR-mice suggests that endogenous Epo does not provide further protection in mice subjected to the 24 hr ischemia-reperfusion injury. In contrast to the report of greater susceptibility to cardiac ischemia/reperfusion injury in young TgEpoR-mice [38], the similarity in response to ischemia-reperfusion injury observed here among WT-, ΔEpoR- and TgEpoR-mice without Epo treatment (Online resource Figure S1), suggests a minimal or no cardioprotective effect of endogenous Epo, unlike that observed for exogenous Epo treatment.
Epo binding to its dimeric receptor on the cell surface results in activation of cytoplasmic associated JAK2 and phosphorylation of multiple tyrosines in the tail region of EpoR that triggers downstream signaling pathways such as PI3K/AKT and MEK/ERK. In endothelial cells, growth factors such as VEGF can stimulate AKT-mediated phosphorylation of eNOS [45]. Blocking this signaling pathway in isolated whole heart perfusion model, the protective effect of Epo was significantly attenuated [10]. We observed that Epo induced phosphorylation of eNOS (Ser1177) and NO production in coronary artery endothelial cells were blocked by PI3K inhibitor LY294002, providing evidence that the PI3K/AKT signaling pathway contributes specifically to Epo-induced endothelial response. Phosphorylation of eNOS via ERK44/42 has been suggested for adenosine induction of NO [43]. While we found Epo-stimulated ERK44/42 phosphorylation, this was not affected by blocking ERK activity with U0126. Estrogen activation of eNOS mediated by estrogen receptor alpha is p38 MAPK-dependent [2], but we found no change in phospho-p38MAPK.
A direct protective effect of Epo on cardiomyocytes and a potential cardioprotection by cardiac fibroblast cells stimulated by Epo have been suggested in previous studies, however, the lack EpoR expression in cardiomyocytes and cardiac fibroblast cells of ΔEpoR-mice suggests that the endothelial Epo response is the major contributor to Epo cardioprotection during the initial 24 hr of ischemia-reperfusion injury. While we previously observed high-level EpoR expression and Epo protection in primary neural progenitor cells and neurons [13, 46], we found low level of EpoR expression in primary cardiomyocytes. Lack of Epo stimulated eNOS response and Epo binding in cardiomyocytes was also observed during the initial 24 hr Epo protective response in a rat heterotropic heart transplantation model [23]. In contrast, direct Epo response by cardiomyocytes was previously reported for primary cardiomyocytes cultures treated with higher Epo concentrations (20 to 40 U/ml), but only after 24 hr of Epo treatment, and eNOS dependent Epo-protection from norepinephrine toxicity was observed at 48 hr [8]. This provides further evidence that Epo-stimulated cardioprotective eNOS response during the initial 24 hr is primarily mediated by endothelial cells while cardiomyocytes have been shown to contribute importantly to the late phase cardioprotection of Epo [40]. Although we show that endothelial Epo response is sufficient to provide acute protection in heart of ΔEpoR-mice, multiple factors may contribute to long term Epo cardioprotection in WT-mice and involve diverse cell types. Studies with other animal models of ischemia-reperfusion injury are warranted to fully understand the potential and limitations of EPO cardioprotection, including the role of RISK activation, for translation to a clinical setting [19, 35].
Epo stimulation of NO production in endothelial cells such as HUVEC and human umbilical artery and bone marrow microvascular endothelial cells was mediated by endothelial eNOS activation [4]. Epo induction of eNOS activity and endothelial NO production is likely an effect on multiple organ sites. This raises a possibility of systemically increased NO level following Epo treatment in vivo that may further contribute to Epo protection. With EpoR expression restricted to endothelial cells, EpoR expression in cardiomyocytes and direct cardiomyocyte response to Epo did not appear to be necessary for Epo cardioprotection in this mouse ischemia-reperfusion injury model. Whether EpoR expression in cardiomyocytes or other cell types without expression in endothelial cells is sufficient to provide a protective Epo response to cardiac injury requires further investigation.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, and grant support from NIH 2R01 HL-060849 to D.J.L. and NIH F32DK077380 to J.W.C.
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Anagnostou A, Lee ES, Kessimian N, Levinson R, Steiner M. Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proc Natl Acad Sci U S A. 1990;87:5978–5982. doi: 10.1073/pnas.87.15.5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anter E, Chen K, Shapira OM, Karas RH, Keaney JF., Jr p38 mitogen-activated protein kinase activates eNOS in endothelial cells by an estrogen receptor alpha-dependent pathway in response to black tea polyphenols. Circ Res. 2005;96:1072–1078. doi: 10.1161/01.RES.0000168807.63013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee D, Rodriguez M, Nag M, Adamson JW. Exposure of endothelial cells to recombinant human erythropoietin induces nitric oxide synthase activity. Kidney Int. 2000;57:1895–1904. doi: 10.1046/j.1523-1755.2000.00039.x. [DOI] [PubMed] [Google Scholar]
- 4.Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood. 2004;104:2073–2080. doi: 10.1182/blood-2004-02-0744. [DOI] [PubMed] [Google Scholar]
- 5.Brutsaert DL. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 6.Bullard AJ, Govewalla P, Yellon DM. Erythropoietin protects the myocardium against reperfusion injury in vitro and in vivo. Basic Res Cardiol. 2005;100:397–403. doi: 10.1007/s00395-005-0537-4. [DOI] [PubMed] [Google Scholar]
- 7.Bullard AJ, Yellon DM. Chronic erythropoietin treatment limits infarct-size in the myocardium in vitro. Cardiovasc Drugs Ther. 2005;19:333–336. doi: 10.1007/s10557-005-4595-5. [DOI] [PubMed] [Google Scholar]
- 8.Burger D, Lei M, Geoghegan-Morphet N, Lu X, Xenocostas A, Feng Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovasc Res. 2006;72:51–59. doi: 10.1016/j.cardiores.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- 10.Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109:2050–2053. doi: 10.1161/01.CIR.0000127954.98131.23. [DOI] [PubMed] [Google Scholar]
- 11.Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, Lefer DJ. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 12.Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, Cerami A, Brines M. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc Natl Acad Sci U S A. 2003;100:4802–4806. doi: 10.1073/pnas.0630444100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen ZY, Asavaritikrai P, Prchal JT, Noguchi CT. Endogenous erythropoietin signaling is required for normal neural progenitor cell proliferation. J Biol Chem. 2007;282:25875–25883. doi: 10.1074/jbc.M701988200. [DOI] [PubMed] [Google Scholar]
- 14.Contaldo C, Elsherbiny A, Lindenblatt N, Plock JA, Trentz O, Giovanoli P, Menger MD, Wanner GA. Erythropoietin enhances oxygenation in critically perfused tissue through modulation of nitric oxide synthase. Shock. 2009;31:599–606. doi: 10.1097/SHK.0b013e31818b9cc4. [DOI] [PubMed] [Google Scholar]
- 15.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 16.Gao E, Boucher M, Chuprun JK, Zhou RH, Eckhart AD, Koch WJ. Darbepoetin alfa, a long-acting erythropoietin analog, offers novel and delayed cardioprotection for the ischemic heart. Am J Physiol Heart Circ Physiol. 2007;293:H60–68. doi: 10.1152/ajpheart.00227.2007. [DOI] [PubMed] [Google Scholar]
- 17.Ghaboura N, Tamareille S, Ducluzeau PH, Grimaud L, Loufrani L, Croue A, Tourmen Y, Henrion D, Furber A, Prunier F. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3beta signaling. Basic Res Cardiol. 2011;106:147–162. doi: 10.1007/s00395-010-0130-3. [DOI] [PubMed] [Google Scholar]
- 18.Greif DM, Kou R, Michel T. Site-specific dephosphorylation of endothelial nitric oxide synthase by protein phosphatase 2A: evidence for crosstalk between phosphorylation sites. Biochemistry. 2002;41:15845–15853. doi: 10.1021/bi026732g. [DOI] [PubMed] [Google Scholar]
- 19.Hausenloy DJ, Baxter G, Bell R, Botker HE, Davidson SM, Downey J, Heusch G, Kitakaze M, Lecour S, Mentzer R, Mocanu MM, Ovize M, Schulz R, Shannon R, Walker M, Walkinshaw G, Yellon DM. Translating novel strategies for cardioprotection: the Hatter Workshop Recommendations. Basic Res Cardiol. 2010;105:677–686. doi: 10.1007/s00395-010-0121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heusch G, Boengler K, Schulz R. Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation. 2008;118:1915–1919. doi: 10.1161/CIRCULATIONAHA.108.805242. [DOI] [PubMed] [Google Scholar]
- 21.Kertesz N, Wu J, Chen TH, Sucov HM, Wu H. The role of erythropoietin in regulating angiogenesis. Dev Biol. 2004;276:101–110. doi: 10.1016/j.ydbio.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 22.Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase. Biol Chem. 2003;384:1343–1364. doi: 10.1515/BC.2003.152. [DOI] [PubMed] [Google Scholar]
- 23.Mihov D, Bogdanov N, Grenacher B, Gassmann M, Zund G, Bogdanova A, Tavakoli R. Erythropoietin protects from reperfusion-induced myocardial injury by enhancing coronary endothelial nitric oxide production. Eur J Cardiothorac Surg. 2009;35:839–846. doi: 10.1016/j.ejcts.2008.12.049. discussion 846. [DOI] [PubMed] [Google Scholar]
- 24.Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, Talan MI. Erythropoietin reduces myocardial infarction and left ventricular functional decline after coronary artery ligation in rats. Proc Natl Acad Sci U S A. 2003;100:11612–11617. doi: 10.1073/pnas.1930406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moon C, Krawczyk M, Paik D, Lakatta EG, Talan MI. Cardioprotection by recombinant human erythropoietin following acute experimental myocardial infarction: dose response and therapeutic window. Cardiovasc Drugs Ther. 2005;19:243–250. doi: 10.1007/s10557-005-3189-6. [DOI] [PubMed] [Google Scholar]
- 26.Nakano M, Satoh K, Fukumoto Y, Ito Y, Kagaya Y, Ishii N, Sugamura K, Shimokawa H. Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ Res. 2007;100:662–669. doi: 10.1161/01.RES.0000260179.43672.fe. [DOI] [PubMed] [Google Scholar]
- 27.Parsa CJ, Kim J, Riel RU, Pascal LS, Thompson RB, Petrofski JA, Matsumoto A, Stamler JS, Koch WJ. Cardioprotective effects of erythropoietin in the reperfused ischemic heart: a potential role for cardiac fibroblasts. J Biol Chem. 2004;279:20655–20662. doi: 10.1074/jbc.M314099200. [DOI] [PubMed] [Google Scholar]
- 28.Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB, Thompson RB, Petrofski JA, Annex BH, Stamler JS, Koch WJ. A novel protective effect of erythropoietin in the infarcted heart. J Clin Invest. 2003;112:999–1007. doi: 10.1172/JCI200318200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rui T, Feng Q, Lei M, Peng T, Zhang J, Xu M, Abel ED, Xenocostas A, Kvietys PR. Erythropoietin prevents the acute myocardial inflammatory response induced by ischemia/reperfusion via induction of AP-1. Cardiovasc Res. 2005;65:719–727. doi: 10.1016/j.cardiores.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 30.Ruschitzka FT, Wenger RH, Stallmach T, Quaschning T, de Wit C, Wagner K, Labugger R, Kelm M, Noll G, Rulicke T, Shaw S, Lindberg RL, Rodenwaldt B, Lutz H, Bauer C, Luscher TF, Gassmann M. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proc Natl Acad Sci U S A. 2000;97:11609–11613. doi: 10.1073/pnas.97.21.11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satoh K, Kagaya Y, Nakano M, Ito Y, Ohta J, Tada H, Karibe A, Minegishi N, Suzuki N, Yamamoto M, Ono M, Watanabe J, Shirato K, Ishii N, Sugamura K, Shimokawa H. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation. 2006;113:1442–1450. doi: 10.1161/CIRCULATIONAHA.105.583732. [DOI] [PubMed] [Google Scholar]
- 32.Schulz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:402–413. doi: 10.1016/j.cardiores.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 33.Seddon M, Shah AM, Casadei B. Cardiomyocytes as effectors of nitric oxide signalling. Cardiovasc Res. 2007;75:315–326. doi: 10.1016/j.cardiores.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 34.Shi Y, Rafiee P, Su J, Pritchard KA, Jr, Tweddell JS, Baker JE. Acute cardioprotective effects of erythropoietin in infant rabbits are mediated by activation of protein kinases and potassium channels. Basic Res Cardiol. 2004;99:173–182. doi: 10.1007/s00395-004-0455-x. [DOI] [PubMed] [Google Scholar]
- 35.Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, Schulz R, Heusch G. Ischemic postconditioning in pigs: no causal role for RISK activation. Circ Res. 2009;104:15–18. doi: 10.1161/CIRCRESAHA.108.186429. [DOI] [PubMed] [Google Scholar]
- 36.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki N, Ohneda O, Takahashi S, Higuchi M, Mukai HY, Nakahata T, Imagawa S, Yamamoto M. Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood. 2002;100:2279–2288. doi: 10.1182/blood-2002-01-0124. [DOI] [PubMed] [Google Scholar]
- 38.Tada H, Kagaya Y, Takeda M, Ohta J, Asaumi Y, Satoh K, Ito K, Karibe A, Shirato K, Minegishi N, Shimokawa H. Endogenous erythropoietin system in non-hematopoietic lineage cells plays a protective role in myocardial ischemia/reperfusion. Cardiovasc Res. 2006;71:466–477. doi: 10.1016/j.cardiores.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Urao N, Okigaki M, Yamada H, Aadachi Y, Matsuno K, Matsui A, Matsunaga S, Tateishi K, Nomura T, Takahashi T, Tatsumi T, Matsubara H. Erythropoietin-mobilized endothelial progenitors enhance reendothelialization via Akt-endothelial nitric oxide synthase activation and prevent neointimal hyperplasia. Circ Res. 2006;98:1405–1413. doi: 10.1161/01.RES.0000224117.59417.f3. [DOI] [PubMed] [Google Scholar]
- 40.Westenbrink BD, Ruifrok WP, Voors AA, Tilton RG, van Veldhuisen DJ, Schoemaker RG, van Gilst WH, de Boer RA. Vascular endothelial growth factor is crucial for erythropoietin-induced improvement of cardiac function in heart failure. Cardiovasc Res. 2010;87:30–39. doi: 10.1093/cvr/cvq041. [DOI] [PubMed] [Google Scholar]
- 41.Wright GL, Hanlon P, Amin K, Steenbergen C, Murphy E, Arcasoy MO. Erythropoietin receptor expression in adult rat cardiomyocytes is associated with an acute cardioprotective effect for recombinant erythropoietin during ischemia-reperfusion injury. Faseb J. 2004;18:1031–1033. doi: 10.1096/fj.03-1289fje. [DOI] [PubMed] [Google Scholar]
- 42.Wu H, Lee SH, Gao J, Liu X, Iruela-Arispe ML. Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development. 1999;126:3597–3605. doi: 10.1242/dev.126.16.3597. [DOI] [PubMed] [Google Scholar]
- 43.Wyatt AW, Steinert JR, Wheeler-Jones CP, Morgan AJ, Sugden D, Pearson JD, Sobrevia L, Mann GE. Early activation of the p42/p44MAPK pathway mediates adenosine-induced nitric oxide production in human endothelial cells: a novel calcium-insensitive mechanism. Faseb J. 2002;16:1584–1594. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- 44.Xuan YT, Guo Y, Zhu Y, Wang OL, Rokosh G, Bolli R. Endothelial nitric oxide synthase plays an obligatory role in the late phase of ischemic preconditioning by activating the protein kinase C epsilon p44/42 mitogen-activated protein kinase pSer-signal transducers and activators of transcription1/3 pathway. Circulation. 2007;116:535–544. doi: 10.1161/CIRCULATIONAHA.107.689471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Youn JY, Wang T, Cai H. An ezrin/calpain/PI3K/AMPK/eNOSs1179 signaling cascade mediating VEGF-dependent endothelial nitric oxide production. Circ Res. 2009;104:50–59. doi: 10.1161/CIRCRESAHA.108.178467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B, Lin CS, Nikodem VM, Hempstead B, Flanders KC, Costantini F, Noguchi CT. Erythropoietin receptor signalling is required for normal brain development. Development. 2002;129:505–516. doi: 10.1242/dev.129.2.505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.