

CUL4B up-regulates CDK2 by repressing miR-372 and miR-373, leading to increased phosphorylation and stabilization of CDC6, thus promoting replication licensing.
Abstract
Cullin-RING ubiquitin ligases (CRLs) participate in the regulation of diverse cellular processes including cell cycle progression. Mutations in the X-linked CUL4B, a member of the cullin family, cause mental retardation and other developmental abnormalities in humans. Cells that are deficient in CUL4B are severely selected against in vivo in heterozygotes. Here we report a role of CUL4B in the regulation of replication licensing. Strikingly, CDC6, the licensing factor in replication, was positively regulated by CUL4B and contributed to the loading of MCM2 to chromatin. The positive regulation of CDC6 by CUL4B depends on CDK2, which phosphorylates CDC6, protecting it from APCCDH1-mediated degradation. Thus, aside being required for cell cycle reentry from quiescence, CDK2 also contributes to pre-replication complex assembly in G1 phase of cycling cells. Interestingly, the up-regulation of CDK2 by CUL4B is achieved via the repression of miR-372 and miR-373, which target CDK2. Our findings thus establish a CUL4B–CDK2–CDC6 cascade in the regulation of DNA replication licensing.
Introduction
Complete and precise replication of DNA is one of the critical events in the cell cycle. In eukaryotic cells, the licensing of DNA replication is tightly regulated. During this process, pre-replication complexes (pre-RCs) assemble and bind to replication origins. In the late M phase of cycling cells, the six-subunit origin-recognition complexes (ORCs) bind to DNA to mark the positions of replication origins in genome. As a cell enters G1 phase, the licensing factor 6 (CDC6) will bind to ORC, which is followed by the recruitment of DNA replication factor 1 (CDT1) and the loading of the DNA replicative helicase minichromosome maintenance protein (MCM) complex to form the pre-RC (Bell and Dutta, 2002; Takeda and Dutta, 2005). In mammalian cells and Xenopus eggs, pre-RC is activated by CDK (cyclin-dependent kinase) and CDC7 (Dbf4-dependent kinase) at the onset of DNA replication (Arata et al., 2000; Walter, 2000; Tsuji et al., 2006). Loading of CDC45 to a preformed pre-RC leads to origin DNA unwinding and recruitment of the single-stranded DNA-binding protein (RPA), proliferating cell nuclear antigen (PCNA), and DNA polymerases onto the DNA to begin DNA synthesis (Takisawa et al., 2000). Therefore, gaining insight into how the formation of pre-RC is regulated is important for understanding DNA replication and cell cycling.
Cullins, which are evolutionarily conserved from yeast to mammals, function as “scaffolds” in cullin-RING–based E3 ubiquitin ligases (CRLs), the largest known class of E3 ubiquitin ligases that regulate diverse cellular processes, including cell cycle progression, transcription, signal transduction, and development (Petroski and Deshaies, 2005; Bosu and Kipreos, 2008; Sarikas et al., 2011). Through its C terminus, the cullin interacts with the RING domain protein, RBX1 or RBX2, which serves as a docking site for the ubiquitin-conjugating enzyme (E2); the N terminus of cullin binds to one of the adaptor proteins that position substrate receptors (SRs) and target proteins for ubiquitination (Petroski and Deshaies, 2005; Bosu and Kipreos, 2008).
Human genome encodes eight cullin members, CUL1, CUL2, CUL3, CUL4A, CUL4B, CUL5, CUL7, and PARC (Sarikas et al., 2011). CUL4A and CUL4B are derived from one ancestor, CUL4, and are 83% identical, with CUL4B having a unique N terminus of 149 amino acids in which the nuclear localization signal (NLS) is located (Zou et al., 2009). As both CUL4A and CUL4B can interact with the substrate adaptor DDB1, they may target the same substrates and function redundantly in some cellular functions, such as genome integrity maintenance (Jackson and Xiong, 2009; Chen et al., 2012). However, CUL4B has recently been shown to target substrates, such as WDR5 and peroxiredoxin III, that are not targeted by CUL4A (Ohtake et al., 2007; Li et al., 2011; Nakagawa and Xiong, 2011; Pfeiffer and Brooks, 2012). Mutations in human CUL4B cause X-linked mental retardation, short stature, and other developmental abnormalities (Tarpey et al., 2007; Zou et al., 2007). In addition, Cul4b knockout mice were embryonic lethal (Jiang et al., 2012; Liu et al., 2012b). Consistent with the importance of CUL4B function during development, heterozygous somatic cells in which the wild-type CUL4B/Cul4b allele is inactivated are severely selected against (Zou et al., 2007; Jiang et al., 2012; Ravn et al., 2012). Cul4a knockout mice, on the other hand, were not found to have remarkable abnormalities, except for failure in spermatogenesis (Liu et al., 2009; Kopanja et al., 2011; Yin et al., 2011). These results suggest that the two CUL4 genes are not entirely redundant in mammals.
We previously showed that CUL4B deficiency could lead to impairments in cell proliferation and S-phase progression in human cells (Zou et al., 2009). Here we investigated the role of CUL4B in DNA replication in mammalian cells and found that CUL4B is able to up-regulate CDC6 in promoting the DNA replication licensing. This positive regulation of CDC6 by CUL4B is achieved via the derepression of CDK2, which is responsible for phosphorylation and stabilization of CDC6. The expression level of CDK2 appeared to be suppressed by microRNAs, which themselves are subjected to the negative regulation at transcriptional level by CUL4B. Our findings thus revealed a novel function of CUL4B in maintaining the structural integrity of pre-RCs.
Results
Lack of CUL4B impedes the origin firing
We previously showed that constitutive knockdown of CUL4B inhibits cell proliferation by prolonging S-phase progression (Zou et al., 2009). To gain insight into the mechanism by which CUL4B regulates DNA replication, we first determined whether CUL4B was chromatin bound. As shown in Fig. 1 A, CUL4B protein was found to be mainly distributed in nuclei, and chromatin bound. This finding is consistent with the previous reports of ours (Zou et al., 2009) and others (Guerrero-Santoro et al., 2008). In addition, CUL4B is constitutively expressed and is localized in nuclei throughout all phases of the cell cycle (Fig. S1, A–E).
Figure 1.

Reduced CUL4B expression perturbs new origin firing. (A) Asynchronous cells were harvested and cytoplasmic (CF), nuclear-soluble (NF), and chromatin-bound (CBF) proteins were obtained and analyzed by Western blot. (B) Schematic double-labeled replication tracks and example of labeled replication tracks. Bar, 10 µm. (C) HeLa cells were transfected with CUL4B (siCUL4B) or negative control (siCon) siRNA. 72 h after transfection, levels of total cellular CUL4A and CUL4B were determined by Western blot analysis. (D) HeLa cells transfected with siCUL4B or siCon siRNA were synchronized in G1/S phase by double-thymidine block. 3 h after release into S phase, cells were labeled sequentially with CldU and IdU. At least 200 CldU/IdU-positive DNA fibers were visualized and quantified using a fluorescence microscope; results represent means ± SD from three independent experiments. (E) Indicated HeLa cells were synchronized with the double-thymidine block and released to progress through the cell cycle. Cells were harvested at the indicated times for analysis of cell cycle distribution by flow cytometry. The data shown are from a representative of three independent experiments.
We next examined the DNA replication dynamics in CUL4B-RNAi cells with a DNA fiber-labeling assay, using two modified nucleotide precursors, 5-chloro-2′-deoxyuridine (CldU) and 5-iodo-2′-deoxyuridine (IdU; Aten et al., 1992). The labeled chromatin was immunostained with anti-CldU and anti-IdU antibodies. Consecutive pulse labeling of the chromatin fibers that were actively synthesizing DNA with CldU and then IdU yields double-fluorescent–labeled tracks on the DNA (active replicons), which could be interpreted unambiguously as newly fired origins, ongoing forks, or terminations (Fig. 1 B).
RNAi of CUL4B in HeLa cells significantly reduced the expression of CUL4B (Fig. 1 C). Although replicons representing newly fired origins accounted for 49.8% of total active replicons 3 h after being released from double-thymidine block in the control siRNA-transfected cells, they only accounted for 31.4% of all labeled replicons in the CUL4B siRNA-transfected cells (Fig. 1 D). These data indicated that RNAi of CUL4B reduces the new origin firing.
We next determined whether CUL4B knockdown affects cell cycle progression from S to G2 phase. The miCUL4B HeLa cells that stably express CUL4B-RNAi vector and miNeg control cells (Zou et al., 2009) were released from G1/S boundary and cell cycle distribution was monitored by FACS analysis. As shown in Fig. 1 E, the transition from S to G2 was significantly delayed in the miCUL4B cells, suggesting that S phase progression is defective in CUL4B knockdown cells.
CUL4B is a positive regulator of nuclear CDC6
A six-subunit ORC binds DNA at origins of replication. As cells enter G1 phase, CDC6 binds to ORC, which is followed by the assembly of pre-RC from CDT1 and MCM complex (Bell and Dutta, 2002; Blow and Dutta, 2005; Takeda and Dutta, 2005). Importantly, CDT1 has been reported to be the substrate of CUL4–DDB1 ubiquitin ligases (Hu et al., 2004; Nishitani et al., 2006). We thus determined the levels of CDC6 and CDT1 in CUL4B knockdown HeLa cells. As shown in Fig. 2 A, though there was no significant difference in the level of nuclear CDT1 between miCUL4B and control cells, the level of nuclear CDC6 was significantly reduced in miCUL4B cells. Similar results were obtained with HEK293 and U2OS cells in which the expression of CUL4B was stably knocked down (Fig. 2 B). This was further confirmed through knocking down CUL4B expression in HeLa cells by synthetic 22-nucleotide small interfering RNA duplexes (unpublished data). We then transiently transfected 293T cells with plasmids expressing Flag-tagged CUL4B (Flag-CUL4B) to corroborate the positive regulation of CDC6 level by CUL4B. Consistent with the reduction of CDC6 when CUL4B is down-regulated, CUL4B overexpression resulted in an up-regulation of CDC6 (Fig. 2 C).
Figure 2.

The levels of CUL4B and nuclear CDC6 are positively correlated. (A) miCUL4B and miNeg HeLa cells were separated into nuclear and cytoplasmic fractions. The protein levels were determined with indicated antibodies. (B) Nuclear proteins prepared from stable CUL4B RNAi or negative control HEK293 and U2OS cells were immunoblotted with indicated antibodies. (C) Nuclear proteins prepared from 293T cells transiently transfected with indicated constructs were analyzed by Western blot. Band intensity given underneath gel images in A–C was measured using ImageJ software (NIH, Bethesda, MD), presented as fold change. (D) miCUL4B and miNeg HeLa cells were transfected with indicated plasmids. At 72 h after transfection, nuclear proteins were extracted and analyzed by Western blot. (E) miCUL4B and miNeg HeLa cells transfected with equal amounts of the indicated plasmids were separated into nuclear and cytoplasmic fractions and the protein expression was analyzed by Western blot.
To further confirm that the nuclear level of CDC6 was CUL4B dependent, we performed rescue experiment by transfecting RNAi-resistant expression vector (Flag-CUL4B (R)) into miCUL4B HeLa cells. As shown in Fig. 2 D, transient transfection of CUL4B expression vector could effectively restore the CDC6 level. Taken together, these results indicate that CUL4B has a positive role in maintaining the nuclear level of CDC6.
We previously showed that the N terminus of CUL4B is critical for CUL4B nuclear localization and functioning in cell proliferation (Zou et al., 2009). To determine whether the N terminus of CUL4B was required for its function in regulation of nuclear CDC6, we transiently transfected Flag-tagged full-length, N terminus–deleted (150–895) RNAi-resistant CUL4B expression vector or Flag empty vector into CUL4B RNAi or control HeLa cells, and measured CDC6 levels by Western blot. As CUL4A shares high homology with CUL4B, we also determined whether ectopic expression of CUL4A could offset the decrease of nuclear CDC6 protein caused by down-regulation of endogenous CUL4B. As shown in Fig. 2 E, only expression of full-length CUL4B efficiently restored nuclear CDC6 protein to normal level. Expression of N-terminal region–deleted CUL4B or CUL4A did not. These results suggest that the N terminus of CUL4B is required for its positive regulation of nuclear CDC6 and that CUL4A does not appear to play a redundant role in maintaining the level of nuclear CDC6.
CDC6 loading onto chromatin is dependent on CDC6 availability maintained by CUL4B
As binding of CDC6 protein to chromatin is critical for the subsequent loading of MCM proteins and therefore for DNA replication licensing in eukaryotic organisms (Cook et al., 2002), we next examined the levels of chromatin-bound CDC6 in CUL4B RNAi cells. CUL4B RNAi and control HeLa cells were synchronized at G1/S boundary, and cytoplasmic (CF), nuclear-soluble (NF), and chromatin-bound protein (CBF) extracts were separated and analyzed. As shown in Fig. 3 A, levels of chromatin-bound CDC6 were significantly decreased in miCUL4B HeLa cells. Moreover, the decrease of chromatin-bound CDC6 was accompanied by a reduced loading of MCM2, a subunit of MCM complex, onto chromatin (Figs. 3 A and S2). Similar results were obtained in asynchronous HeLa, HEK293, and U2OS cells (Fig. 3 B).
Figure 3.

CUL4B promotes CDC6 loading onto chromatin. (A) Indicated cells were synchronized at the G1/S boundary and cytoplasmic (CF), nuclear-soluble (NF), and chromatin-bound proteins (CBF) were prepared. Equal amounts of protein extracts were subjected to SDS-PAGE and detected with the indicated antibodies. (B) Western blot analysis of CUL4B, CDC6, and MCM2 protein levels in the chromatin-bound fraction prepared from asynchronous cells indicated. (C) miCUL4B and control HeLa cells were transfected with indicated plasmids. 72 h after transfection, chromatin-bound proteins were extracted and analyzed by Western blot. (D) Cells were transfected with indicated plasmids. At 72 h after transfection, chromatin-bound proteins were extracted and analyzed by Western blot. (E) HeLa cells cotransfected with indicated siRNAs and plasmids were synchronized in G1/S phase by double-thymidine block. 3 h after released into S phase, cells were labeled sequentially with CldU and IdU. DNA fibers were immunostained with anti-CldU and anti-IdU antibodies. More than 200 CldU/IdU-positive DNA fibers were visualized and quantified using a fluorescence microscope; results represent means ± SD from three independent experiments.
We further tested the dependence of chromatin loading of CDC6 and MCM2 on CUL4B by expressing RNAi-resistant Flag-tagged CUL4B. As shown in Fig. 3 C, expression of RNAi-resistant Flag-tagged CUL4B could completely restore the normal levels of chromatin-bound CDC6 and MCM2 proteins.
To determine whether the decrease in chromatin-bound MCM2 in CUL4B RNAi cells was a result of reduction in CDC6 protein, we expressed HA-tagged CDC6 in miCUL4B HeLa cells and determined the level of chromatin-bound MCM2. As expected, expression of exogenous wild-type CDC6 efficiently restored the level of chromatin-bound MCM2 in miCUL4B cells (Fig. 3 D). In addition, DNA fiber-labeling assay experiments also showed that expression of exogenous CDC6 could effectively restore the level of new origin firing in CUL4B RNAi cells (Fig. 3 E). Collectively, these results indicate that CUL4B is important for maintaining the level of CDC6 that is required for MCM2 loading to pre-RC.
CUL4B protects CDC6 from APCCDH1-mediated degradation
To determine the mechanism by which CUL4B maintains the level of nuclear CDC6, we first determined the CDC6 mRNA levels, but found that CDC6 mRNA level was not lower in miCUL4B cells than in control cells (Fig. 4 A), suggesting that CDC6 in CUL4B RNAi cells was down-regulated at posttranscriptional level.
Figure 4.

Proteolysis of CDC6 is accelerated in CUL4B knockdown cells. (A) The mRNA levels of CUL4A, CUL4B, and CDC6 in miCUL4B and miNeg HeLa cells were measured by real-time PCR. The normalized expression in miNeg cells was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (B) miCUL4B and miNeg HeLa cells were treated or untreated with 30 µM MG132 for 3 h or 10 µM LMB for 5 h and nuclear proteins were extracted and analyzed by Western blot. Band intensity given underneath gel image was measured using ImageJ software, presented as fold change compared with control cells. (C) miCUL4B and miNeg HeLa cells were transiently transfected with HA-tagged CDC6 construct. 72 h after transfection, cells were treated or untreated with 30 µM MG132 for 3 h and nuclear proteins were analyzed by Western blot. (D) miCUL4B and miNeg HeLa cells were treated with 50 µg/ml cycloheximide and harvested at the indicated time points. Equal amounts of nuclear proteins were analyzed by Western blot. The nuclear CDC6 proteins expression was quantified by densitometric analysis using ImageJ. Expression is represented as the percentage remaining relative to time zero; data are expressed as means ± SEM (n = 3). *, P < 0.05 vs. miNeg. (E) Western blot analyses of indicated protein levels in the chromatin-bound fraction prepared from miCUL4B and miNeg HeLa cells treated or untreated with 30 µM MG132 for 3 h. (F) miCUL4B and miNeg HeLa cells were transfected with indicated siRNAs. 48 h after transfection, cells were treated with 0.5 mM mimosine for 22 h and nuclear proteins were analyzed by Western blot.
CDC6 is translocated from the nucleus to the cytoplasm in a CRM1-dependent manner (Jiang et al., 1999; Petersen et al., 1999). To determine whether the reduction in nuclear CDC6 level is due to an accelerated nuclear export in CUL4B down-regulated cells, we treated miCUL4B HeLa cells and control cells with leptomycin B (LMB), which blocks CRM1-mediated protein export from the nucleus. As shown in Fig. 4 B, treatment with LMB could not reduce the difference in the level of nuclear CDC6 between miCUL4B and control cells, suggesting that the reduction of nuclear CDC6 in CUL4B knockdown cells is not due to increased nuclear export.
Because CDC6 is known to be targeted for ubiquitin-mediated proteolysis (Petersen et al., 2000), we next tested whether accelerated proteolysis is responsible for the reduced CDC6 level in CUL4B RNAi cells. As shown in Fig. 4 B, treatment with MG132 for 3 h completely blocked the reduction of nuclear CDC6 in CUL4B RNAi HeLa cells. Similar results were obtained in U2OS and HEK293 cells (Fig. S3, A and B). Treatment with MG132 also completely blocked the degradation of exogenous CDC6 in miCUL4B cells (Fig. 4 C). These data suggest that reduction of CDC6 in CUL4B RNAi cells is due to its accelerated degradation. Indeed, CUL4B down-regulation resulted in a significant decrease in the half-life of CDC6 compared with that in control cells (Fig. 4 D). As expected, MG132 treatment also significantly increased the chromatin-bound form of CDC6 (Fig. 4 E).
In mammalian cells, CDC6 is targeted for ubiquitin-mediated degradation by two ubiquitin E3 ligases, CDH1-associated form of the anaphase promoting complex, APC/C and HUWE1 enzyme (Petersen et al., 2000; Hall et al., 2007). Next, we transfected miCUL4B and miNeg HeLa cells with siRNA targeting CDH1 and HUWE1, respectively. To exclude possible bias due to changes in cell cycle distribution after RNAi of CDH1 and HUWE1, the cells were synchronized to G1 phase by treatment with mimosine for 22 h. The results showed that the transfection of siRNAs targeting CDH1, but not those targeting HUWE1, could efficiently restore the nuclear CDC6 in miCUL4B cells (Figs. 4 F and S3 C), suggesting that APCCDH1 contributes to the accelerated proteasomal degradation of CDC6 in CUL4B RNAi cells.
DDB1, the known adaptor protein of CUL4 CRL complexes, has also been reported to interact with CDH1 and modulates the activity of APCCDH1 (Lv et al., 2010). Therefore, we examined the level of nuclear DDB1. As shown in Fig. S3 D, there was no difference in nuclear DDB1 protein levels between CUL4B knockdown HeLa cells and control cells, though CUL4B was reported to assist the translocation of DDB1 into the nucleus in XP-E cells (Guerrero-Santoro et al., 2008). Moreover, the level of SKP2, which is a substrate of APCCDH1 and regulated by DDB1 (Lv et al., 2010), appeared to be unaffected by CUL4B RNAi (Fig. S3 D), indicating a specific role of CUL4B in protecting CDC6 from degradation through APCCDH1.
Protection of CDC6 by CUL4B is dependent on CDC6 phosphorylation by CDK2
Previous studies showed that CDC6 could become phosphorylated at S54, S74, and S106 by CDK2, and that such phosphorylation regulates subcellular localization and stabilization of CDC6 (Petersen et al., 1999; Mailand and Diffley, 2005). Therefore, we next determined the level of nuclear CDK2 protein in CUL4B knockdown cells. As seen in Fig. 5 A, constitutive RNAi of CUL4B could significantly decrease the levels of CDK2 protein in HeLa and HEK293 cells. Conversely, CUL4B overexpression resulted in higher levels of CDK2 (Fig. 5 B). We further determined the role of CDK2 in regulating the CDC6 level by transiently transfecting HeLa cells with HA-tagged CDK2 construct. As shown in Fig. 5 C, introduction of exogenous CDK2 remarkably elevated the level of CDC6 protein in miCUL4B cells. To confirm that the elevation of CDC6 by CUL4B is dependent on CDK kinase activity, we treated CUL4B-overexpressing 293T cells with a CDK kinase inhibitor, roscovitine. As shown in Fig. 5 D, roscovitine treatment decreased the basal level of CDC6, which is consistent with the previous report (Mailand and Diffley, 2005). Importantly, roscovitine efficiently attenuated the increase of CDC6 caused by CUL4B overexpression.
Figure 5.

Up-regulation of CDC6 by CUL4B is mediated by CDK2. (A) Nuclear proteins prepared from indicated cells were analyzed by Western blot. Band intensity presented as fold change compared with control cells was given underneath gel image. (B) 293T cells were transfected with indicated constructs and nuclear proteins were immunoblotted for the indicated proteins. (C) miCUL4B and miNeg HeLa cells were transfected with indicated constructs and nuclear proteins were analyzed by Western blot. (D) 293T cells were transfected with indicated constructs. 72 h after transfection, cells were treated with or without 25 µM roscovitine for 2 h and nuclear proteins were immunoblotted for the indicated proteins. (E) Nuclear proteins prepared from indicated HeLa cells treated or untreated with 30 µM MG132 for 3 h were analyzed by Western blot. Quantification of band intensity of p-CDC6 and total CDC6 is shown in the bar graph as the fold change compared with control cells. Data are expressed as means ± SEM (n = 3). (F) 293T cells were transiently transfected with indicated constructs and nuclear proteins were analyzed by Western blot. (G) HeLa cells were transfected with siCDK2 or control siRNA. 48 h after transfection, cells were treated with 0.5 mM mimosine for 22 h and total or chromatin-bound proteins were immunoblotted for the indicated proteins. (H) HeLa cells arrested in G1 phase by mimosine were incubated further for 2 h in the presence of 0.5 mM mimosine plus 25 µM roscovitine and nuclear or chromatin-bound proteins were analyzed by Western blot.
We next monitored the phosphorylation states of endogenous CDC6 at S54 and S106 in CUL4B RNAi cells. As seen in Fig. 5 E, the levels of S54- and S106-phosphorylated nuclear CDC6 proteins were greatly reduced in CUL4B RNAi cells when compared with control cells. In addition, after treatment with MG132, there was still a difference in the levels of S54- and S106-phosphorylated CDC6 between CUL4B RNAi cells and negative control cells. Correspondingly, when Flag-tagged CUL4B vector was transfected into 293T cells, the levels of nuclear S54- and S106-phosphorylated CDC6 exhibited a more pronounced increase than that of total nuclear CDC6 protein in the CUL4B overexpression cells (Fig. 5 F). These data suggest that CUL4B up-regulates CDC6 phosphorylation.
Phosphorylation of CDC6 by CDK2 was reported to be required for licensing in cells exiting quiescence (Mailand and Diffley, 2005). We next determined the role of CDK2 in promoting stabilization of CDC6 and pre-RC assembly during G1 phase in cycling cells. The results showed that knockdown of CDK2 dramatically reduced the levels of chromatin-bound CDC6 and MCM2 in cells synchronized in G1 phase (Figs. 5 G and S4 A). Similar results were obtained in roscovitine-treated cells (Figs. 5 H and S4 B). Thus, CDK2 appears to promote pre-RC assembly in G1 phase in cycling cells, as well. Collectively, our results indicate that the maintenance of a high level of nuclear CDC6 by CUL4B is mediated by CDK2.
CUL4B up-regulates CDK1/2 expression by repressing miR-372/373
Because CDK1 and CDK2 have the same cyclin-binding partners and CDK1 can compensate for the loss of CDK2 during S phase (Kaldis and Aleem, 2005; Satyanarayana and Kaldis, 2009), we also determined the level of nuclear CDK1 protein in CUL4B RNAi cells. Like CDK2, CDK1 was also down-regulated in CUL4B knockdown cells (Fig. 6 A).
Figure 6.

CUL4B up-regulates CDK2 expression by repressing miRNA. (A) Nuclear proteins prepared from miCUL4B and miNeg HeLa cells were analyzed by Western blot. (B) Luciferase reporter assay of miCUL4B and miNeg cells transiently transfected with pmir-GLO-CDK1-3′UTR, pmir-GLO-CDK2-3′UTR, and pmir-GLO empty vector. Values were corrected for the expression of Renilla luciferase and compared with that of pmir-GLO. Levels of luciferase activity were compared with those of miNeg-transfected cells, normalized to 1. The results are given as mean values from three independent experiments. Bars represent means ± SD. *, P < 0.05 vs. miNeg. (C) miR-372 expression is negatively regulated by CUL4B. The levels of indicated miRNAs in miCUL4B and miNeg HeLa cells were measured by real-time PCR. The normalized expression in miNeg cells was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (D) The levels of miR-372 in 293T cells transiently transfected with Flag-CUL4B or empty vector were analyzed by real-time PCR. The normalized expression in cells transfected with empty vector was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (E) The levels of indicated miRNAs in miCUL4B and miNeg HeLa cells were measured by real-time PCR. The normalized expression in miNeg cells was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (F) The levels of indicated miRNAs in 293T cells transiently transfected with Flag-CUL4B or empty vector were analyzed by real-time PCR. The normalized expression in cells transfected with empty vector was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (G) miR-372 or miR-373 target site in 3′-UTR of CDK2 predicted by TargetScan. (H) miCUL4B and miNeg HeLa cells were transfected with a control inhibitor (NC) or an inhibitor specific for miR-372 or miR-373. 48 h later, total proteins were extracted and analyzed by Western blot. (I) miCUL4B and miNeg HeLa cells were transiently transfected with wild-type (wt), miR-372/373 binding site mutant (372-mut) pmir-GLO-CDK2-3′UTR, and pmir-GLO empty reporter vector. 24 h later, luciferase assays were performed as above. Bars represent means ± SD. *, P < 0.05 vs. miNeg. (J) The levels of indicated mRNAs in miCUL4B and miNeg HeLa cells were measured by real-time PCR. The normalized expression in miNeg cells was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (K) The levels of indicated mRNAs in 293T cells transiently transfected with Flag-CUL4B or Flag empty vector were measured by real-time PCR. The normalized expression in cells transfected with Flag vector was set as 1. The assay was performed in triplicate, and relative means ± SE are shown. (L) The protein levels of β-catenin in indicated HeLa cells were analyzed by Western blot. (M) Indicated cells were transiently transfected with TOPFLASH or FOPFLASH along with pRL-TK vector. 24 h after transfection, cells were treated with or without 10 mM LiCl for 24 h, and the relative luciferase activity was determined. The activity of TOPFLASH in miNeg cells was set to 1. The assays were performed three times in triplicate. Each bar represents the value of means ± SD.
We next determined whether CDK1 and CDK2 were up-regulated by CUL4B at transcription level and found that the levels of CDK1/2 transcripts were not significantly reduced in miCUL4B cells (Fig. S5 A), suggesting that the regulation of CDK1/2 by CUL4B may occur at posttranscriptional level.
We then determined whether the decrease of CDK1 and CDK2 in nuclei was due to an accelerated nuclear export in miCUL4B cells. Treatment with LMB could not block the decrease of nuclear CDK1 or CDK2 in CUL4B RNAi cells (Fig. S5 B), suggesting that the reduction in nuclear CDK1/2 proteins in CUL4B RNAi cells was not due to deregulated nuclear transport.
We then determined nuclear CDK1/2 protein half-life by treatment with cycloheximide and found no difference in degradation rate between CUL4B RNAi cells and control cells (Fig. S5 C). In addition, treatment with MG132 could not block the reduction of nuclear CDK1/2 proteins in CUL4B RNAi cells (Fig. S5 D), indicating that the decrease in the amounts of CDK1/2 proteins in CUL4B RNAi cells did not result from accelerated protein degradation.
Altogether, these results suggest that the CDK1/2 down-regulation in CUL4B RNAi cells is due to translational inhibition, which prompted us to examine a possible regulatory role of the 3′ untranslated regions (UTRs) in CDK1/2. For this purpose, full-length CDK1 and CDK2 3′-UTRs were each subcloned into pmir-GLO dual-luciferase microRNA target expression vectors. These vectors and pmir-GLO empty vector were respectively used to transfect CUL4B RNAi and control cells, and the firefly activities of pmir-GLO-CDK1/2 3′-UTRs were determined. As shown in Fig. 6 B, CUL4B down-regulation significantly reduced the pmir-GLO-CDK1/2 3′-UTR reporter activity. These results suggest that the regulation of CDK1 and CDK2 expression by CUL4B may involve microRNAs (miRNAs).
Recently, several miRNAs have been reported to regulate the expression of CDKs by targeting their 3′-UTRs (Bueno and Malumbres, 2011). In particular, miR-410 targets CDK1 and miR-302d and miR-372 target CDK2 (Lin et al., 2010; Chien et al., 2011; Tian et al., 2011). miR-302d and miR-372 have the same seed sequence, and their responsive elements (REs) in the CDK2-3′-UTR are conserved across different species as shown by TargetScan 6.0 (Fig. S5 E). Real-time PCR showed that though there was no difference in the expression of miR-410 and miR-302d between CUL4B RNAi and control HeLa cells, the expression of miR-372 was elevated significantly in CUL4B RNAi cells (Fig. 6 C). Similar results were obtained in HEK293 and U2OS cells in which CUL4B was knocked down (Fig. S5 F). To corroborate the negative regulation of miR-372 by CUL4B, we analyzed the expression of miR-372 in 293T cells transiently transfected with plasmid expressing Flag-tagged CUL4B. Consistent with the increase in miR-372 expression in CUL4B down-regulated cells, CUL4B overexpression led to a decrease in level of miR-372 (Fig. 6 D). These results indicate that the expression of miR-372 is negatively regulated by CUL4B.
miR-372 belongs to miR-371–373 cluster, which is composed of miR-371-3p, miR-371-5p, miR-372, and miR-373. We next examined the effect of CUL4B on the expression of the three other genes of miR-371–373 cluster. Like that of miR-372, the levels miR-371-3p, miR-371-5p, and miR-373 were also increased in CUL4B knockdown cells (Fig. 6 E) and decreased in CUL4B-overexpressing cells (Fig. 6 F), suggesting that CUL4B can repress the miR-371–373 cluster.
miR-372 and miR-373 share the same seed sequence and are both predicted to target CDK2 by TargetScan (Fig. 6 G). We then tested whether the regulation of CDK2 by CUL4B is mediated by miR-372/373. As shown in Fig. 6 H, the down-regulation of CDK2 by CUL4B RNAi could be efficiently attenuated by inhibitors of hsa-miR-372 and hsa-miR-373. Furthermore, when mutation was introduced to the seed region of the miR-372/373 binding site, the luciferase activity of reporter construct containing full-length 3′-UTR of CDK2 remained unaltered in CUL4B RNAi cells (Fig. 6 I). Collectively, these data indicate that CUL4B up-regulates the expression of CDK2 by repressing miR-372/373.
We next examined the expression of the primary polycistronic miRNA transcript (pri-miRNA) of miR-371–373 cluster in CUL4B RNAi cells. As shown in Fig. 6 J, the level of pri-miR-371–373 was significantly increased in miCUL4B cells. Correspondingly, it was decreased in CUL4B-overexpressing cells (Fig. 6 K). These results suggest that CUL4B represses the initial step of miR-371–373 cluster biogenesis.
miR-371–373 cluster can be transactivated by β-catenin (Zhou et al., 2012). Moreover, the expression of β-catenin was reported to be affected by CUL4B in rat, mouse cells, and human brain (Tripathi et al., 2007; Liu et al., 2012a). We therefore examined the expression of β-catenin in CUL4B RNAi cells. As shown in Fig. 6 L, knockdown of CUL4B resulted in a slight decrease, but not an elevation, in the level of nuclear β-catenin, suggesting that the repression of miR-371–373 cluster by CUL4B may not be mediated by β-catenin. We further tested this by using the TOPFLASH/FOPFLASH system (a luciferase reporter with wild-type or mutated TCF response elements). miCUL4B and miNeg cells were transiently transfected with TOPFLASH or FOPFLASH reporter plasmid. As shown in Fig. 6 M, TOPFLASH activity was also slightly decreased in CUL4B RNAi cells. These results suggest that the transcriptional repression of miR-371–373 cluster by CUL4B is not mediated by β-catenin.
Cdk2 was down-regulated in Cul4b knockout mice
To determine whether the expression of CDK2 is positively regulated by CUL4B in vivo, we determined the Cdk2 expression in Cul4b knockout mouse. As loss of Cul4b results in early embryonic lethality, we analyzed the expression pattern of Cdk2 in E7.5 embryos by immunostaining. As shown in Fig. 7 A, the Cul4b-null embryos were devoid of staining by anti-Cul4b antibody. Correspondingly, the Cul4b-null embryos also showed significantly reduced Cdk2 expression when compared with wild-type embryos.
Figure 7.

Cdk2 expression is reduced in Cul4b knockout mice. (A) Immunohistochemistry of paraffin sections of wild-type and Cul4b-null embryos at E7.5 stained with anti-Cul4b antibody and anti-Cdk2 antibody. (B) Western blot analysis of indicated protein expression levels in nuclear lysates from liver of wild-type and Cul4b knockout (Cul4b flox/Y;Alb-cre) mice. Bar, 200 µm.
To further confirm this positive regulation of Cdk2 by CUL4B in vivo, we analyzed the expression of Cdc6 and Cdk1/2 in liver of mice with hepatocyte-specific deletion of Cul4b. As in human cells, the protein levels of mouse Cdc6, Cdk1, and Cdk2 were significantly decreased in Cul4b knockout hepatocyte cells (Fig. 7 B). Moreover, we found that the cyclin E protein was accumulated in Cul4b knockout hepatocytes, which was consistent with our previous report (Zou et al., 2009).
Discussion
CDC6 serves as an essential licensing factor for replication (Bell and Dutta, 2002; Takeda and Dutta, 2005; Lau et al., 2006). Together with CDT1, it promotes the loading of MCM2–7 onto pre-RCs at replication origins. In addition, the binding of CDC6 to origins may also stabilize the interaction of ORC with chromatin (Mizushima et al., 2000; Machida et al., 2005). Down-regulation of CDC6 was shown to block G1/S transition and perturb new origin firing (Lau et al., 2006; Luo et al., 2006). In quiescent cells, pre-RC assembly is prevented due to APC/C-dependent CDC6 proteolysis, and when cells exit quiescence, CDK2-dependent CDC6 stabilization allows pre-RC assembly (Mailand and Diffley, 2005). We showed here that CDK2-dependent stabilization of CDC6 is also important for pre-RC assembly in G1 phase of cycling cells (Fig. 5, G and H). Importantly, we identified CUL4B as a novel positive regulator of CDC6. We demonstrated that CUL4B can contribute to the maintenance of CDC6 level by repressing miR-372/373, inhibitors of CDK2 (Fig. 8). It should be noted that although CUL4A and CUL4B are closely related, down-regulation of CUL4A by RNAi led to no detectable change in the level of CDC6 (unpublished data). Interestingly, although ORC1 and CDT1, the other components of the pre-RC, were reported to be targeted for degradation by CUL1 and CUL4 containing E3 ubiquitin ligases (Méndez et al., 2002; Nishitani et al., 2006; Senga et al., 2006), the level of nuclear CDT1 was not affected by the down-regulation of CUL4B (Fig. 2 A). Thus, despite the high degree of homology between CUL4A and CUL4B in protein sequence, the two cullin members appear to regulate DNA replication licensing through different mechanisms. More studies are apparently needed to further distinguish between the functions of CUL4A and CUL4B in DNA replication licensing.
Figure 8.
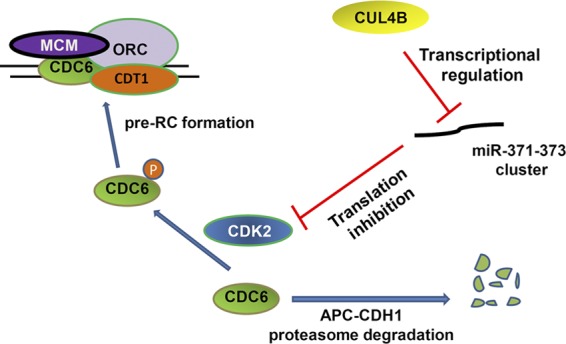
A model showing the CUL4B–miR-371/372–CDK2–CDC6 cascade in the regulation of DNA replication licensing. P, phosphate.
In human cells, CDC6 expression is strictly regulated at transcriptional and post-transcriptional levels (Hateboer et al., 1998; Yan et al., 1998; Jiang et al., 1999; Petersen et al., 1999, 2000; Hall et al., 2007; Jin and Fondell, 2009; Paolinelli et al., 2009), and its deregulated expression can lead to under- or overreplication, accumulation of DNA damage, and genetic instability (Blow and Gillespie, 2008; Borlado and Méndez, 2008). CDC6 can be targeted by two E3 ligases, APCCDH1 and HUWE1 in human cells (Petersen et al., 2000; Hall et al., 2007). Phosphorylation of CDC6 by CDK2 can block the association of CDC6 with the CDH1 protein, thus protecting CDC6 from APCCDH1-mediated proteolysis (Mailand and Diffley, 2005). Because CDK2 is down-regulated in CUL4B RNAi cells (Fig. 5 A), leading to reduced CDC6 phosphorylation, it is of no surprise that CUL4B knockdown only affects the CDC6 proteolysis mediated by APCCDH1, but not that by HUWE1 (Figs. 4 F and S3 C).
Progression through the cell cycle in mammalian cells is defined by sequential phosphorylation events in which CDK2 plays a central role. In the late G1 phase, CDK2 is activated by its association with cyclin E and phosphorylates retinoblastoma protein (Rb), resulting in the release of E2F transcription factor, inducing transcriptional activation of genes required for S-phase progression and DNA replication, leading to passage from G1/S phase to S phase (Berthet et al., 2006; Merrick et al., 2011). CDK2 also phosphorylates DNA replication factors to facilitate pre-RC assembly and origin firing (Sotillo et al., 2009). In S phase, CDK2 associates with cyclin A, which is required for the S-phase completion. Because of its critical function in cell cycle control, CDK2 kinase activity is tightly regulated by multiple mechanisms including cell cycle–dependent expression of cyclin A or cyclin E, CDK2 phosphorylation, and the expression of CDK inhibitors such as p21 and p27 (Lunn et al., 2010). In contrast, little is known about how CDK2 expression is regulated. Given the fact that CDK2 is shown to be overexpressed in a subset of human tumors (Kuźbicki et al., 2010), it is possible that control of CDK2 expression may also be involved in the regulation of cell proliferation. Our finding that CUL4B can up-regulate CDK2 expression (Fig. 5 B) illustrates a novel physiological role of CUL4B in cell cycle control.
It has recently been found that CDK2 expression is regulated by microRNAs, including miR-372, miR-885-5p, miR-302d, and miR-103 (Liao and Lönnerdal, 2010; Lin et al., 2010; Afanasyeva et al., 2011; Tian et al., 2011). In this study we demonstrate that CUL4B can up-regulate CDK2 expression through repressing miR-372 and miR-373 (Fig. 6, H and I). We observed that CUL4B RNAi led to an increased transcription of miR-371–373 cluster (Fig. 6 J) and an inhibition of CDK2 (Fig. 6 B). Furthermore, concomitant inhibition of miR-372 and miR-373 effectively attenuated the decrease in CDK2 level caused by CUL4B RNAi (Fig. 6 H). Moreover, mutation of miR-372/373 target site in 3′-UTR significantly offset the effect of CUL4B RNAi (Fig. 6 I). Our results presented here have established that CUL4B contributes to replication licensing by up-regulating the CDK2–CDC6 cascade, through the repression of miR-372/373.
miRNAs in the miR-371–373 cluster were shown to be up-regulated in some human tumors and overexpression of miR-372/373 can cause faster growth and increased invasiveness in tumor cells (Voorhoeve et al., 2006; Huang et al., 2008; Lee et al., 2009; Wu et al., 2011; Zhou et al., 2012); therefore, they are considered as oncogenes. However, miR-372 was recently reported to be down-regulated in hepatocellular and cervical cancer and up-regulation of miR-372 suppresses proliferation in HeLa cells (J. Wang et al., 2010; Tian et al., 2011). In addition, miR-373 was also reported to be down-regulated in some kinds of cancers and may function as a tumor suppressor (Chen et al., 2011; Tanaka et al., 2011; Keklikoglou et al., 2012). These findings suggest that miR-372/373 may function differently depending on cell types. Although miR-371–373 cluster was reported to be transactivated by β-catenin (Zhou et al., 2012), we were unable to detect any increased activity of β-catenin signaling in CUL4B RNAi cells (Fig. 6, L and M). Thus, CUL4B probably represses miR-371–373 cluster independent of β-catenin. Because CUL4 was shown to function in the regulation of histone methylation (Higa et al., 2006), which is involved in the regulation of miRNA expression in human cells (Agirre et al., 2012), CUL4B may also regulate the expression of miRNAs via histone code. The detailed mechanism governing CUL4B regulation of miR-371–373 cluster needs to be further investigated.
In summary, we demonstrated that CUL4B positively regulates the CDK2–CDC6 cascade in promoting DNA replication licensing. We also showed that the up-regulation of CDK2 by CUL4B is through the transcriptional repression of miR-372/373. To our knowledge, this is the first report that cullins may function in the regulation of miRNAs. Our findings provide a new insight into how CUL4B regulates cell cycle progression and DNA replication.
Materials and methods
Cell culture and manipulation
All cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere with 5% CO2. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. CUL4B stable knockdown and control cells were generated as described previously (Zou et al., 2009). In brief, 24 h after being transfected with CUL4B or control RNAi vectors, designated as miCUL4B and miNeg, respectively, HeLa, HEK293, and U2OS cells were incubated in the DMEM supplemented with 10% FBS containing 10 µg/ml blasticidin (Invitrogen). 3 wk after selection, cells resistant to blasticidin were isolated and analyzed for CUL4B expression by real-time PCR and Western blot. siRNAs targeting CDH1 and HUWE1 were purchased from Santa Cruz Biotechnology, Inc. The CUL4B, CDK2, and negative control siRNA duplexes were purchased from GenePharma, and the oligonucleotide sequences were as follows: CUL4B, 5′-GGUGAACACUUAACAGCAAdTdT-3′; CDK2, 5′-GGCAGCCCUGGCUCACCCUdTdT-3′; negative control, 5′-UUCUCCGAACGUGUCACGUdTdT-3′. The inhibitors of hsa-miR-372 and hsa-miR-373 were purchased from GenePharma.
To obtain G1/S phase synchronization, a double-thymidine block and release was performed. In brief, cells were treated with 2.5 mM thymidine for 16 h, and then released into fresh medium after being washed with phosphate-buffered saline (PBS) three times. After 8 h, the cells were retreated with thymidine for 16 h. Cells synchronized at late G1 phase were obtained by treatment with 0.5 mM mimosine for 22 h.
DNA fiber experiments
DNA fiber experiments were performed as described previously (Lau et al., 2006). In brief, cells were synchronized at the G1/S border by double-thymidine block and released for 3 h, and then the cells were sequentially pulsed with 10 mM CldU and then 15 mM IdU for 10 min each. After washing twice with PBS the cells were trypsinized, and mixed 1:8 with unlabeled cells to a final concentration of 625 cells/µl. Then 2.5 µl of cells were mixed with 7.5 µl spreading buffer (0.5% SDS in 200 mM Tris-HCl, pH 7.5, and 50 mM EDTA) on a glass slide. After being incubated for 9.5 min, the slides were tilted by 15°. The resulting DNA spreads were air-dried for 30 min, fixed in methanol/acetic acid (1:3), and incubated overnight at 4°C. IdU was detected with mouse anti-BrdU (1:200; BD) conjugated with FITC; CldU was detected with rat anti-BrdU (1:500; AbD Serotec) and goat anti–rat IgG conjugated with rhodamine (1:300; KDL).
Immunofluorescence
Immunofluorescence was performed as described previously (Zou et al., 2009). In brief, cells grown on a coverslip were washed with PBS and treated with 4% paraformaldehyde for 20 min. After washing with 2× PBS, the cells were permeabilized with 0.2% Triton X-100 for 20 min and blocked with 5% goat serum in PBS for 1 h at room temperature. Cells were then incubated with the primary antibodies at the appropriate dilution overnight at 4°C. The mouse anti-Flag antibody (1:1,000) and rabbit anti-CUL4B (1:500) were from Sigma-Aldrich. Cells were then washed with PBS and incubated for 60 min at 37°C with rhodamine-conjugated goat anti–mouse secondary antibody (1:500; Jackson ImmunoResearch Laboratories, Inc.). After washing with PBS three times, cells were further stained with DAPI for 10 min.
Preparation of protein lysates
Cytoplasmic, nuclear-soluble, and chromatin-bound proteins were fractionated using the Subcellular Protein Fractionation kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Preparation of cytoplasmic and nuclear protein extracts for Western blot analysis was performed as described previously (Zou et al., 2009). In brief, cells were lysed using cell lysis buffer containing 10 mM Hepes, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, 0.4% IGEPAL, and 1 mM phenylmethanesulfonyl fluoride. After centrifugation, supernatants (corresponding to cytoplasmic extracts) were collected. The pellets were further washed with cell lysis buffer and resuspended in nuclear extraction buffer (0.4 M NaCl, 20 mM Hepes, pH 7.9, 1 mM EDTA, 1 mM dithiothreitol, and 1 mM phenylmethanesulfonyl fluoride). After a vigorous shake for 30 min at 4°C, the nuclear extract was collected by centrifugation.
Western blot and antibodies
Western blot analysis was performed as described previously (Zou et al., 2009). In brief, equal amounts of extracts were separated by SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane (GE Healthcare), followed by incubation with specific primary antibodies overnight at 4°C. Then the membranes were incubated with HRP-conjugated secondary antibodies and detected using the ECL PLUS kit (GE Healthcare). The primary antibodies were mouse anti–α-tubulin (Santa Cruz Biotechnology, Inc.), anti–β-actin (Santa Cruz Biotechnology, Inc.), rabbit anti–cyclin E (Abcam), rabbit anti–β-catenin (Abcam), rabbit anti-CDK2 (Abcam), rabbit anti-CDT1 (Santa Cruz Biotechnology, Inc.), rabbit anti-DDB1 (Santa Cruz Biotechnology, Inc.), mouse anti-CDC6 (Santa Cruz Biotechnology, Inc.), mouse anti-CDK1 (Santa Cruz Biotechnology, Inc.), goat anti-MCM2 (Santa Cruz Biotechnology, Inc.), mouse anti-CDH1 (Santa Cruz Biotechnology, Inc.), mouse anti-Flag (Sigma-Aldrich), mouse anti-HA (Sigma-Aldrich), rabbit anti–p-54-CDC6 (Abcam), rabbit anti–p-106-CDC6 (Abcam), rabbit anti-H3 (Abcam), rabbit anti-SKP2 and anti-CUL4A (Abcam), rabbit anti-CUL4B (Sigma-Aldrich), and goat anti-ORC2 (Santa Cruz Biotechnology, Inc.).
Reverse-transcription PCR and real-time PCR assay
Extraction of total RNA from cultured dells, reverse-transcription PCR, and real-time quantitative PCR (qPCR) assay were performed as described previously (Zou et al., 2009). In brief, total RNA was isolated using TRIzol reagent (Invitrogen) and treated with RQ1 RNase-free DNase (Promega). After DNase treatment, RNA was transcribed to generate cDNA using RevertAid M-MuLV-RT (Thermo Fisher Scientific) with random hexamers as primers according to the manufacturer’s instructions. Real-time quantitative PCR was performed using the TaqMan 7500 instrument (Applied Biosystems).
The mRNA levels of genes were measured by a SYBR Green I assay using SYBR Green Universal PCR Master Mix (Applied Biosystems) according to the manufacturer’s instructions, and the human glyceraldehyde-3-phosphate dehydrogenase was used as a control housekeeping gene. Melting curve analysis was performed to confirm amplification of specific transcripts. Each reaction was run in triplicate and in parallel. All cDNA was measured in triplicate. The expression levels of transcripts were calculated by the relative quantification (ΔΔCt) study method by using 7500 System SDS software (Applied Biosystems). All primer sequences are listed in Table S1.
The levels of miRNAs were measured on a LightCycler 480 system (Roche) with miRNA qRT-PCR using an All-in-One miRNA Q-PCR detection kit (GeneCopoeia, Inc.) according to the manufacturer’s protocol. snRNA U6 was used as the endogenous control and the relative expression levels of the miRNAs were calculated using the 2−ΔΔCT method. Each reaction was run in triplicate and in parallel. All primers used for miRNA qPCR were from GeneCopoeia, Inc.
Plasmids
A CUL4A fragment, which was amplified using HeLa cDNA as template, was subcloned in-frame into pcDNA3.1/Myc-His B vector (Invitrogen) between the HindIII and BamHI sites to generate the pcDNA3.1/Myc-His B-CUL4A construct. pCMV-Tag 2B-CUL4A construct was generated by subcloning the PCR-amplified CUL4A fragment in-frame into the pCMV-Tag 2B vector (Agilent Technologies) between the BamHI and HindIII sites.
The pcDNA3.1/Myc-His A-CUL4B, pEGFP-C1-CUL4B, pEGFP-C1-CUL4B 150–895, pEGFP-C1-CUL4B RNAi-resistant vector, CUL4B-specific RNAi expression vector miCUL4B, and negative control vector miNeg were described previously (Zou et al., 2009). In brief, a PCR-amplified CUL4B fragment using HeLa cDNA as template was subcloned in-frame into the pcDNA3.1/Myc-His A vector (Invitrogen) between the BamHI and XhoI sites to generate the pcDNA3.1/Myc-His A-CUL4B. The KpnI and ApaI fragment from pcDNA3.1/Myc-His A-CUL4B was ligated into the pEGFP-C1 vector (Takara Bio Inc.) to generate pEGFP-C1-CUL4B. pEGFP-C1-CUL4B 150–895 was generated by ligating the PCR fragment in which the N-terminal fragment of CUL4B was removed by KpnI and ScaI digestion into the pEGFP-C1-CUL4B vector. Three silent point mutations, using overlap extension PCR (Ho et al., 1989), were introduced to the region of the CUL4B that is targeted by the RNAi to generate pEGFP-C1-CUL4B RNAi-resistant vector. The pCMV-Tag 2B-CUL4B was generated by subcloning the BamHI and XhoI fragment from pcDNA3.1/Myc-His A-CUL4B in-frame into the pCMV-Tag 2B vector. The pCMV-Tag 2B-CUL4B RNAi-resistant expression vector was constructed by ligating the BamHI and XhoI fragment from pEGFP-C1-CUL4B RNAi-resistant vector in-frame to the pCMV-Tag 2B vector. The pCMV-Tag 2B-CUL4B 150–895 RNAi-resistant vector was constructed by ligating the PCR fragment in which the N-terminal fragment of CUL4B was removed by digestion with BamHI and ScaI into the pCMV-Tag 2B-CUL4B RNAi-resistant vector. All primer sequences are listed in Table S2.
pCGN.CSH.FL42 plasmid encoding HA-tagged full-length human CDC6 were gifts from L. Drury (Clare Hall Laboratories, Cancer Research UK, London, England; Mailand and Diffley, 2005). pCMV-CDK2-HA plasmid encoding HA-tagged wild-type CDK2 was a gift from B.E. Clurman (University of Washington, Seattle, WA; Welcker et al., 2003). pCMV-HA plasmid was purchased from Beyotime.
Reporter constructs and luciferase assays
TOPFLASH and FOPFLASH constructs were from EMD Millipore. The pmir-GLO-CDK1/2 3′-UTR vectors were generated by subcloning PCR-amplified full-length 3′-UTR of CDK1 and CDK2 using HeLa cDNA as a template into SacI–XhoI sites of pmir-GLO vector (Promega) downstream of the firefly luciferase gene. The pmir-GLO-CDK1/2 3′-UTR vectors containing mutated miR-372 response elements were generated by site-directed mutagenesis using overlap extension PCR (Zou et al., 2009). Primer sequences used in construction of the vectors are listed in Table S3.
TOPFLASH/FOPFLASH assays were performed as described previously (H. Wang et al., 2010). In brief, cells were transiently transfected with TOPFLASH or FOPFLASH reporter plasmid using Lipofectamine 2000 (Invitrogen). 24 h after transfection, cells were treated with or without 10 mM LiCl for 24 h, and the relative luciferase activity was determined using the Dual-Luciferase Reporter Assay system (Promega) according to the manufacturer’s instructions, and measured by Victor 1420 Multilabel Counter (PerkinElmer). Each firefly luciferase activity was normalized to Renilla luciferase activity of the pRL-TK reporter (cotransfected internal control). All experiments were performed in triplicate and assayed in quadruplicates.
For 3′-UTR reporter luciferase assays, cells were plated in 96-well plates and transfected with pmir-GLO reporter plasmids using Lipofectamine 2000 (Invitrogen). Luciferase assays were performed 24 h after transfection using the Dual-Luciferase Reporter Assay system (Promega) with a multilabel counter (Victor 1420; PerkinElmer). Each firefly luciferase activity was normalized to Renilla luciferase activity of the pRL-TK reporter (cotransfected internal control). Transfections were performed in three independent experiments, and assayed in quadruplicates.
Mice
The Cul4b floxed mice were generated as reported previously (Jiang et al., 2012). In brief, Cul4b-floxed targeting vector in which the exons 3–5 of Cul4b were floxed by two loxP sites was electroporated into 129 embryonic stem (ES) cells (RW.4) for homologous recombination. Targeted ES cells were then injected into C57BL/6J blastocysts to establish chimeric mice for subsequent germline transmission to produce Cul4b-floxed mice. To generate constitutive Cul4b null mice, Cul4b+/flox females were first crossed to EIIa-Cre+/+ males (Model Animal Research Center of Nanjing University, Nanjing, China) to obtain mosaic females (Cul4b+/flox/Cul4b+/null; EIIa-Cre+/−) that were then crossed to wild-type males to generate Cul4b knockout male mice (Cul4bnull/Y). Mice with liver-specific deletion of Cul4b were generated by crossing the Cul4b-floxed mice with Alb-Cre transgenic mice (The Jackson Laboratory) that contain a Cre recombinase driven by albumin promoter (Postic et al., 1999). All animal experiments were performed in compliance with national regulations and by protocols approved by the institutional animal care and use committee.
Immunocytochemistry
Immunocytochemistry was performed as described previously (Jiang et al., 2012). In brief, specimens were fixed in 4% PFA at 4°C overnight, then dehydrated, embedded in paraffin, and sectioned. After deparaffinization and rehydration using xylene and decreasing grades of ethanol, the sections were boiled in sodium citrate buffer for 10 min for antigen recovery, and immersed in 3% H2O2 for 10 min to quench endogenous peroxidase. Sections were then blocked with 10% serum at 37°C for 1 h to reduce nonspecific staining and then incubated overnight at 4°C with primary antibodies. Afterward, the sections were rinsed and then coated with an HRP-conjugated second antibody (1:200; Jackson ImmunoResearch Laboratories, Inc.) and incubated at 37°C for 1 h. The DAB was used to visualize immunoreactions sites. Sections were counterstained with hematoxylin and mounted on glass slides. The rabbit anti-Cul4b antibody (1:500) was from Sigma-Aldrich; rabbit anti-CDK2 (1: 200) was from Abcam.
Image acquisition and processing
Images were acquired using a fluorescence microscope (model BX51; Olympus) equipped with a DP71 microscope digital camera (Olympus) and DP Controller version 3.1.1.267 software (Olympus). Immunofluorescence samples were viewed using a 10× NA 0.3 or 20× NA 0.5 air objective (Olympus). DNA fibers were viewed using a 100× NA 1.3 oil objective (Olympus). Histological stained embryo sections were viewed using a 20× NA 0.5 air or 100× NA 1.3 oil objective lens (Olympus). The mounting medium used for imaging was ProLong Antifade (Invitrogen). All digital microscopic images were acquired at room temperature. Images were processed using Photoshop CS5 (Adobe). If necessary, minor adjustments (brightness and contrast to the whole panel) were done for the entire image, the image was cropped, and individual color channels were extracted (when required) without color correction or gamma adjustments.
Statistical analysis
Statistical analysis was performed by a two-tailed unpaired t test to validate the data using SPSS13.0. Results were considered statistically significant where P < 0.05.
Online supplemental material
Fig. S1 shows expression pattern and subcellular localization of CUL4B during the cell cycle. Fig. S2 shows cell cycle distribution by FACS analysis of cells synchronized at the G1/S boundary by double-thymidine block. Fig. S3 shows impact of CUL4B RNAi on the proteasomal degradation of CDC6. Fig. S4 shows cell cycle distribution by FACS analysis of cells synchronized at the G1 phase by treating with mimosine. Fig. S5 shows that CUL4B regulates CDK2 expression through regulation of miR-372/373. Table S1 shows primers used for real-time PCR in this study. Table S2 shows primers used for CUL4A and CUL4B expression vectors construction in this study. Table S3 shows primers used for reporter construction in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201206065/DC1.
Supplementary Material
Acknowledgments
We would like to thank Dr. Lucy Drury for kindly providing the CDC6 expression vector and Dr. Bruce E. Clurman for kindly providing CDK2 expression vector.
This work was supported by National Basic Research Program of China grants (2011CB966200 to C. Shao and Y. Zou; and 2013CB910900 to Y. Gong), National Science Foundation Research grants (30830065 to Y. Zou, C. Shao, and Y. Gong; 30900804 to Y. Zou; 81021001 to Y. Gong and C. Shao; 81101522 to B. Jiang), and the Hong Kong Scholars Program (201104631 to Y. Zou).
The authors have declared that no competing interests exist.
Footnotes
Abbreviations used in this paper:
- CldU
- 5-chloro-2’-deoxyuridine
- CRL
- cullin-RING ubiquitin ligase
- IdU
- 5-iodo-2’-deoxyuridine
- LMB
- leptomycin B
- MCM
- minichromosome maintenance protein
- miRNA
- microRNA
- ORC
- origin-recognition complex
- pre-RC
- pre-replication complex
- UTR
- untranslated region
References
- Afanasyeva E.A., Mestdagh P., Kumps C., Vandesompele J., Ehemann V., Theissen J., Fischer M., Zapatka M., Brors B., Savelyeva L., et al. 2011. MicroRNA miR-885-5p targets CDK2 and MCM5, activates p53 and inhibits proliferation and survival. Cell Death Differ. 18:974–984 10.1038/cdd.2010.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agirre X., Martínez-Climent J.A., Odero M.D., Prósper F. 2012. Epigenetic regulation of miRNA genes in acute leukemia. Leukemia. 26:395–403 10.1038/leu.2011.344 [DOI] [PubMed] [Google Scholar]
- Arata Y., Fujita M., Ohtani K., Kijima S., Kato J.Y. 2000. Cdk2-dependent and -independent pathways in E2F-mediated S phase induction. J. Biol. Chem. 275:6337–6345 10.1074/jbc.275.9.6337 [DOI] [PubMed] [Google Scholar]
- Aten J.A., Bakker P.J., Stap J., Boschman G.A., Veenhof C.H. 1992. DNA double labelling with IdUrd and CldUrd for spatial and temporal analysis of cell proliferation and DNA replication. Histochem. J. 24:251–259 10.1007/BF01046839 [DOI] [PubMed] [Google Scholar]
- Bell S.P., Dutta A. 2002. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 71:333–374 10.1146/annurev.biochem.71.110601.135425 [DOI] [PubMed] [Google Scholar]
- Berthet C., Klarmann K.D., Hilton M.B., Suh H.C., Keller J.R., Kiyokawa H., Kaldis P. 2006. Combined loss of Cdk2 and Cdk4 results in embryonic lethality and Rb hypophosphorylation. Dev. Cell. 10:563–573 10.1016/j.devcel.2006.03.004 [DOI] [PubMed] [Google Scholar]
- Blow J.J., Dutta A. 2005. Preventing re-replication of chromosomal DNA. Nat. Rev. Mol. Cell Biol. 6:476–486 10.1038/nrm1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow J.J., Gillespie P.J. 2008. Replication licensing and cancer—a fatal entanglement? Nat. Rev. Cancer. 8:799–806 10.1038/nrc2500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlado L.R., Méndez J. 2008. CDC6: from DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis. 29:237–243 10.1093/carcin/bgm268 [DOI] [PubMed] [Google Scholar]
- Bosu D.R., Kipreos E.T. 2008. Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 3:7 10.1186/1747-1028-3-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno M.J., Malumbres M. 2011. MicroRNAs and the cell cycle. Biochim. Biophys. Acta. 1812:592–601 10.1016/j.bbadis.2011.02.002 [DOI] [PubMed] [Google Scholar]
- Chen Y., Luo J., Tian R., Sun H., Zou S. 2011. miR-373 negatively regulates methyl-CpG-binding domain protein 2 (MBD2) in hilar cholangiocarcinoma. Dig. Dis. Sci. 56:1693–1701 10.1007/s10620-010-1481-1 [DOI] [PubMed] [Google Scholar]
- Chen C.Y., Tsai M.S., Lin C.Y., Yu I.S., Chen Y.T., Lin S.R., Juan L.W., Chen Y.T., Hsu H.M., Lee L.J., Lin S.W. 2012. Rescue of the genetically engineered Cul4b mutant mouse as a potential model for human X-linked mental retardation. Hum. Mol. Genet. 21:4270–4285 10.1093/hmg/dds261 [DOI] [PubMed] [Google Scholar]
- Chien W.W., Domenech C., Catallo R., Kaddar T., Magaud J.P., Salles G., Ffrench M. 2011. Cyclin-dependent kinase 1 expression is inhibited by p16(INK4a) at the post-transcriptional level through the microRNA pathway. Oncogene. 30:1880–1891 10.1038/onc.2010.570 [DOI] [PubMed] [Google Scholar]
- Cook J.G., Park C.H., Burke T.W., Leone G., DeGregori J., Engel A., Nevins J.R. 2002. Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. Proc. Natl. Acad. Sci. USA. 99:1347–1352 10.1073/pnas.032677499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Santoro J., Kapetanaki M.G., Hsieh C.L., Gorbachinsky I., Levine A.S., Rapić-Otrin V. 2008. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res. 68:5014–5022 10.1158/0008-5472.CAN-07-6162 [DOI] [PubMed] [Google Scholar]
- Hall J.R., Kow E., Nevis K.R., Lu C.K., Luce K.S., Zhong Q., Cook J.G. 2007. Cdc6 stability is regulated by the Huwe1 ubiquitin ligase after DNA damage. Mol. Biol. Cell. 18:3340–3350 10.1091/mbc.E07-02-0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hateboer G., Wobst A., Petersen B.O., Le Cam L., Vigo E., Sardet C., Helin K. 1998. Cell cycle-regulated expression of mammalian CDC6 is dependent on E2F. Mol. Cell. Biol. 18:6679–6697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa L.A., Wu M., Ye T., Kobayashi R., Sun H., Zhang H. 2006. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 8:1277–1283 10.1038/ncb1490 [DOI] [PubMed] [Google Scholar]
- Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 77:51–59 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- Hu J., McCall C.M., Ohta T., Xiong Y. 2004. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6:1003–1009 10.1038/ncb1172 [DOI] [PubMed] [Google Scholar]
- Huang Q., Gumireddy K., Schrier M., le Sage C., Nagel R., Nair S., Egan D.A., Li A., Huang G., Klein-Szanto A.J., et al. 2008. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat. Cell Biol. 10:202–210 10.1038/ncb1681 [DOI] [PubMed] [Google Scholar]
- Jackson S., Xiong Y. 2009. CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 34:562–570 10.1016/j.tibs.2009.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B., Zhao W., Yuan J., Qian Y., Sun W., Zou Y., Guo C., Chen B., Shao C., Gong Y. 2012. Lack of Cul4b, an E3 ubiquitin ligase component, leads to embryonic lethality and abnormal placental development. PLoS ONE. 7:e37070 10.1371/journal.pone.0037070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Wells N.J., Hunter T. 1999. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc. Natl. Acad. Sci. USA. 96:6193–6198 10.1073/pnas.96.11.6193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F., Fondell J.D. 2009. A novel androgen receptor-binding element modulates Cdc6 transcription in prostate cancer cells during cell-cycle progression. Nucleic Acids Res. 37:4826–4838 10.1093/nar/gkp510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldis P., Aleem E. 2005. Cell cycle sibling rivalry: Cdc2 vs. Cdk2. Cell Cycle. 4:1491–1494 10.4161/cc.4.11.2124 [DOI] [PubMed] [Google Scholar]
- Keklikoglou I., Koerner C., Schmidt C., Zhang J.D., Heckmann D., Shavinskaya A., Allgayer H., Gückel B., Fehm T., Schneeweiss A., et al. 2012. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-κB and TGF-β signaling pathways. Oncogene. 31:4150–4163 10.1038/onc.2011.571 [DOI] [PubMed] [Google Scholar]
- Kopanja D., Roy N., Stoyanova T., Hess R.A., Bagchi S., Raychaudhuri P. 2011. Cul4A is essential for spermatogenesis and male fertility. Dev. Biol. 352:278–287 10.1016/j.ydbio.2011.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuźbicki L., Lange D., Stanek-Widera A., Chwirot B.W. 2010. Cyclin-dependent kinase 2 (CDK-2) expression in nonmelanocytic human cutaneous lesions. Appl. Immunohistochem. Mol. Morphol. 18:357–364 10.1097/PAI.0b013e3181d4069c [DOI] [PubMed] [Google Scholar]
- Lau E., Zhu C., Abraham R.T., Jiang W. 2006. The functional role of Cdc6 in S-G2/M in mammalian cells. EMBO Rep. 7:425–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.H., Goan Y.G., Hsiao M., Lee C.H., Jian S.H., Lin J.T., Chen Y.L., Lu P.J. 2009. MicroRNA-373 (miR-373) post-transcriptionally regulates large tumor suppressor, homolog 2 (LATS2) and stimulates proliferation in human esophageal cancer. Exp. Cell Res. 315:2529–2538 10.1016/j.yexcr.2009.06.001 [DOI] [PubMed] [Google Scholar]
- Li X., Lu D., He F., Zhou H., Liu Q., Wang Y., Shao C., Gong Y. 2011. Cullin 4B protein ubiquitin ligase targets peroxiredoxin III for degradation. J. Biol. Chem. 286:32344–32354 10.1074/jbc.M111.249003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Lönnerdal B. 2010. Global microRNA characterization reveals that miR-103 is involved in IGF-1 stimulated mouse intestinal cell proliferation. PLoS ONE. 5:e12976 10.1371/journal.pone.0012976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.L., Chang D.C., Ying S.Y., Leu D., Wu D.T. 2010. MicroRNA miR-302 inhibits the tumorigenecity of human pluripotent stem cells by coordinate suppression of the CDK2 and CDK4/6 cell cycle pathways. Cancer Res. 70:9473–9482 10.1158/0008-5472.CAN-10-2746 [DOI] [PubMed] [Google Scholar]
- Liu L., Lee S., Zhang J., Peters S.B., Hannah J., Zhang Y., Yin Y., Koff A., Ma L., Zhou P. 2009. CUL4A abrogation augments DNA damage response and protection against skin carcinogenesis. Mol. Cell. 34:451–460 10.1016/j.molcel.2009.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.C., Enikolopov G., Chen Y. 2012a. Cul4B regulates neural progenitor cell growth. BMC Neurosci. 13:112 10.1186/1471-2202-13-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Yin Y., Li Y., Prevedel L., Lacy E.H., Ma L., Zhou P. 2012b. Essential role of the CUL4B ubiquitin ligase in extra-embryonic tissue development during mouse embryogenesis. Cell Res. 22:1258–1269 10.1038/cr.2012.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn C.L., Chrivia J.C., Baldassare J.J. 2010. Activation of Cdk2/Cyclin E complexes is dependent on the origin of replication licensing factor Cdc6 in mammalian cells. Cell Cycle. 9:4533–4541 10.4161/cc.9.22.13789 [DOI] [PubMed] [Google Scholar]
- Luo F., Yee J.K., Huang S.H., Wu L.T., Jong A.Y. 2006. Downregulation of human Cdc6 protein using a lentivirus RNA interference expression vector. Methods Mol. Biol. 342:287–293 [DOI] [PubMed] [Google Scholar]
- Lv X.B., Xie F., Hu K., Wu Y., Cao L.L., Han X., Sang Y., Zeng Y.X., Kang T. 2010. Damaged DNA-binding protein 1 (DDB1) interacts with Cdh1 and modulates the function of APC/CCdh1. J. Biol. Chem. 285:18234–18240 10.1074/jbc.M109.094144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida Y.J., Hamlin J.L., Dutta A. 2005. Right place, right time, and only once: replication initiation in metazoans. Cell. 123:13–24 10.1016/j.cell.2005.09.019 [DOI] [PubMed] [Google Scholar]
- Mailand N., Diffley J.F. 2005. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 122:915–926 10.1016/j.cell.2005.08.013 [DOI] [PubMed] [Google Scholar]
- Méndez J., Zou-Yang X.H., Kim S.Y., Hidaka M., Tansey W.P., Stillman B. 2002. Human origin recognition complex large subunit is degraded by ubiquitin-mediated proteolysis after initiation of DNA replication. Mol. Cell. 9:481–491 10.1016/S1097-2765(02)00467-7 [DOI] [PubMed] [Google Scholar]
- Merrick K.A., Wohlbold L., Zhang C., Allen J.J., Horiuchi D., Huskey N.E., Goga A., Shokat K.M., Fisher R.P. 2011. Switching Cdk2 on or off with small molecules to reveal requirements in human cell proliferation. Mol. Cell. 42:624–636 10.1016/j.molcel.2011.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima T., Takahashi N., Stillman B. 2000. Cdc6p modulates the structure and DNA binding activity of the origin recognition complex in vitro. Genes Dev. 14:1631–1641 [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T., Xiong Y. 2011. X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyltransferase component WDR5 and regulates neuronal gene expression. Mol. Cell. 43:381–391 10.1016/j.molcel.2011.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H., Sugimoto N., Roukos V., Nakanishi Y., Saijo M., Obuse C., Tsurimoto T., Nakayama K.I., Nakayama K., Fujita M., et al. 2006. Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J. 25:1126–1136 10.1038/sj.emboj.7601002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F., Baba A., Takada I., Okada M., Iwasaki K., Miki H., Takahashi S., Kouzmenko A., Nohara K., Chiba T., et al. 2007. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 446:562–566 10.1038/nature05683 [DOI] [PubMed] [Google Scholar]
- Paolinelli R., Mendoza-Maldonado R., Cereseto A., Giacca M. 2009. Acetylation by GCN5 regulates CDC6 phosphorylation in the S phase of the cell cycle. Nat. Struct. Mol. Biol. 16:412–420 10.1038/nsmb.1583 [DOI] [PubMed] [Google Scholar]
- Petersen B.O., Lukas J., Sørensen C.S., Bartek J., Helin K. 1999. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 18:396–410 10.1093/emboj/18.2.396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen B.O., Wagener C., Marinoni F., Kramer E.R., Melixetian M., Lazzerini Denchi E., Gieffers C., Matteucci C., Peters J.M., Helin K. 2000. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev. 14:2330–2343 10.1101/gad.832500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski M.D., Deshaies R.J. 2005. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6:9–20 10.1038/nrm1547 [DOI] [PubMed] [Google Scholar]
- Pfeiffer J.R., Brooks S.A. 2012. Cullin 4B is recruited to tristetraprolin-containing messenger ribonucleoproteins and regulates TNF-α mRNA polysome loading. J. Immunol. 188:1828–1839 10.4049/jimmunol.1102837 [DOI] [PubMed] [Google Scholar]
- Postic C., Shiota M., Niswender K.D., Jetton T.L., Chen Y., Moates J.M., Shelton K.D., Lindner J., Cherrington A.D., Magnuson M.A. 1999. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274:305–315 10.1074/jbc.274.1.305 [DOI] [PubMed] [Google Scholar]
- Ravn K., Lindquist S., Nielsen K., Dahm T., Tumer Z. 2012. Deletion of CUL4B leads to concordant phenotype in a monozygotic twin pair. Clin. Genet. 82:292–294 10.1111/j.1399-0004.2011.01839.x [DOI] [PubMed] [Google Scholar]
- Sarikas A., Hartmann T., Pan Z.Q. 2011. The cullin protein family. Genome Biol. 12:220 10.1186/gb-2011-12-4-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyanarayana A., Kaldis P. 2009. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 28:2925–2939 10.1038/onc.2009.170 [DOI] [PubMed] [Google Scholar]
- Senga T., Sivaprasad U., Zhu W., Park J.H., Arias E.E., Walter J.C., Dutta A. 2006. PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281:6246–6252 10.1074/jbc.M512705200 [DOI] [PubMed] [Google Scholar]
- Sotillo E., Garriga J., Padgaonkar A., Kurimchak A., Cook J.G., Graña X. 2009. Coordinated activation of the origin licensing factor CDC6 and CDK2 in resting human fibroblasts expressing SV40 small T antigen and cyclin E. J. Biol. Chem. 284:14126–14135 10.1074/jbc.M900687200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda D.Y., Dutta A. 2005. DNA replication and progression through S phase. Oncogene. 24:2827–2843 10.1038/sj.onc.1208616 [DOI] [PubMed] [Google Scholar]
- Takisawa H., Mimura S., Kubota Y. 2000. Eukaryotic DNA replication: from pre-replication complex to initiation complex. Curr. Opin. Cell Biol. 12:690–696 10.1016/S0955-0674(00)00153-8 [DOI] [PubMed] [Google Scholar]
- Tanaka T., Arai M., Wu S., Kanda T., Miyauchi H., Imazeki F., Matsubara H., Yokosuka O. 2011. Epigenetic silencing of microRNA-373 plays an important role in regulating cell proliferation in colon cancer. Oncol. Rep. 26:1329–1335 [DOI] [PubMed] [Google Scholar]
- Tarpey P.S., Raymond F.L., O’Meara S., Edkins S., Teague J., Butler A., Dicks E., Stevens C., Tofts C., Avis T., et al. 2007. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 80:345–352 10.1086/511134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R.Q., Wang X.H., Hou L.J., Jia W.H., Yang Q., Li Y.X., Liu M., Li X., Tang H. 2011. MicroRNA-372 is down-regulated and targets cyclin-dependent kinase 2 (CDK2) and cyclin A1 in human cervical cancer, which may contribute to tumorigenesis. J. Biol. Chem. 286:25556–25563 10.1074/jbc.M111.221564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi R., Kota S.K., Srinivas U.K. 2007. Cullin4B/E3-ubiquitin ligase negatively regulates beta-catenin. J. Biosci. 32:1133–1138 10.1007/s12038-007-0114-0 [DOI] [PubMed] [Google Scholar]
- Tsuji T., Ficarro S.B., Jiang W. 2006. Essential role of phosphorylation of MCM2 by Cdc7/Dbf4 in the initiation of DNA replication in mammalian cells. Mol. Biol. Cell. 17:4459–4472 10.1091/mbc.E06-03-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voorhoeve P.M., le Sage C., Schrier M., Gillis A.J., Stoop H., Nagel R., Liu Y.P., van Duijse J., Drost J., Griekspoor A., et al. 2006. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 124:1169–1181 10.1016/j.cell.2006.02.037 [DOI] [PubMed] [Google Scholar]
- Walter J.C. 2000. Evidence for sequential action of cdc7 and cdk2 protein kinases during initiation of DNA replication in Xenopus egg extracts. J. Biol. Chem. 275:39773–39778 10.1074/jbc.M008107200 [DOI] [PubMed] [Google Scholar]
- Wang H., Zhou H., Zou Y., Liu Q., Guo C., Gao G., Shao C., Gong Y. 2010. Resveratrol modulates angiogenesis through the GSK3β/β-catenin/TCF-dependent pathway in human endothelial cells. Biochem. Pharmacol. 80:1386–1395 10.1016/j.bcp.2010.07.034 [DOI] [PubMed] [Google Scholar]
- Wang J., Liu X., Wu H., Ni P., Gu Z., Qiao Y., Chen N., Sun F., Fan Q. 2010. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 38:5366–5383 10.1093/nar/gkq285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M., Singer J., Loeb K.R., Grim J., Bloecher A., Gurien-West M., Clurman B.E., Roberts J.M. 2003. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell. 12:381–392 10.1016/S1097-2765(03)00287-9 [DOI] [PubMed] [Google Scholar]
- Wu N., Liu X., Xu X., Fan X., Liu M., Li X., Zhong Q., Tang H. 2011. MicroRNA-373, a new regulator of protein phosphatase 6, functions as an oncogene in hepatocellular carcinoma. FEBS J. 278:2044–2054 10.1111/j.1742-4658.2011.08120.x [DOI] [PubMed] [Google Scholar]
- Yan Z., DeGregori J., Shohet R., Leone G., Stillman B., Nevins J.R., Williams R.S. 1998. Cdc6 is regulated by E2F and is essential for DNA replication in mammalian cells. Proc. Natl. Acad. Sci. USA. 95:3603–3608 10.1073/pnas.95.7.3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y., Lin C., Kim S.T., Roig I., Chen H., Liu L., Veith G.M., Jin R.U., Keeney S., Jasin M., et al. 2011. The E3 ubiquitin ligase Cullin 4A regulates meiotic progression in mouse spermatogenesis. Dev. Biol. 356:51–62 10.1016/j.ydbio.2011.05.661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A.D., Diao L.T., Xu H., Xiao Z.D., Li J.H., Zhou H., Qu L.H. 2012. β-Catenin/LEF1 transactivates the microRNA-371-373 cluster that modulates the Wnt/β-catenin-signaling pathway. Oncogene. 31:2968–2978 10.1038/onc.2011.461 [DOI] [PubMed] [Google Scholar]
- Zou Y., Liu Q., Chen B., Zhang X., Guo C., Zhou H., Li J., Gao G., Guo Y., Yan C., et al. 2007. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am. J. Hum. Genet. 80:561–566 10.1086/512489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y., Mi J., Cui J., Lu D., Zhang X., Guo C., Gao G., Liu Q., Chen B., Shao C., Gong Y. 2009. Characterization of nuclear localization signal in the N terminus of CUL4B and its essential role in cyclin E degradation and cell cycle progression. J. Biol. Chem. 284:33320–33332 10.1074/jbc.M109.050427 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.