

Abstract
Objective:
A major motivation for seeking disease-associated genetic variation is to identify novel risk processes. Although rare copy number variants (CNVs) appear to contribute to attention deficit hyperactivity disorder (ADHD), common risk variants (single-nucleotide polymorphisms [SNPs]) have not yet been detected using genome-wide association studies (GWAS). This raises the concern as to whether future larger-scale, adequately powered GWAS will be worthwhile. The authors undertook a GWAS of ADHD and examined whether associated SNPs, including those below conventional levels of significance, influenced the same biological pathways affected by CNVs.
Method:
The authors analyzed genome-wide SNP frequencies in 727 children with ADHD and 5,081 comparison subjects. The gene sets that were enriched in a pathway analysis of the GWAS data (the top 5% of SNPs) were tested for an excess of genes spanned by large, rare CNVs in the children with ADHD.
Results:
No SNP achieved genome-wide significance levels. As previously reported in a subsample of the present study, large, rare CNVs were significantly more common in case subjects than comparison subjects. Thirteen biological pathways enriched for SNP association significantly overlapped with those enriched for rare CNVs. These included cholesterol-related and CNS development pathways. At the level of individual genes, CHRNA7, which encodes a nicotinic receptor subunit previously implicated in neuropsychiatric disorders, was affected by six large duplications in case subjects (none in comparison subjects), and SNPs in the gene had a gene-wide p value of 0.0002 for association in the GWAS.
Conclusions:
Both common and rare genetic variants appear to be relevant to ADHD and index-shared biological pathways.
Attention deficit hyperactivity disorder (ADHD) is a highly heritable disorder (heritability estimates range from 75% to 90% [1, 2]). Rare genetic variants, specifically large, rare copy number variants (CNVs), play an important role in ADHD (3–5), but so far, genome-wide searches have not identified common risk variants. Four published genome-wide association studies (GWAS) of ADHD (6–9) and a recent meta-analysis (10) of all available data have failed to yield genome-wide significant results for any single-nucleotide polymorphism (SNP).
There are several explanations as to why it has been difficult to identify common genetic risk variants for psychiatric disorders (11), including ADHD (12). One important factor is that the effect size of any individual SNP is likely to be small (13). This means that with currently available sample sizes, true common risk alleles are unlikely to achieve the stringent statistical thresholds required for genome-wide significance (14), although, as has repeatedly been demonstrated for other phenotypes, this can in part be overcome for at least a proportion of risk variants as larger samples become available for performing meta-analyses. For GWAS of childhood-onset psychiatric disorders, such as ADHD and autism, the types of sample sizes required, even with international collaboration, have yet to be achieved (15). Another possibility is that if ADHD is genetically heterogeneous (in the sense that there are multiple phenotypes with limited or no overlap at the level of common risk alleles), the effects of each allele might be diluted, resulting in lower apparent effect sizes. However, it is currently unclear how best to subdivide ADHD in a way that might overcome this problem or whether such subdivisions are possible.
An alternative explanation for the negative GWAS findings might be that ADHD risk is entirely explained by multiple low-frequency variants that are not well captured by the genotyping arrays. In reality, population genetics theory predicts that risk is most likely conferred by alleles that span the spectrum of frequencies (13). If it is the case that both common and rare variants contribute to ADHD risk, but genome-wide significant association cannot be a realistic goal with currently sized samples, we might expect to see a convergence of subthreshold signals from both types of variants influencing common biological risk pathways.
In the present study, we investigated whether specific biological pathways were enriched for associated SNPs and for CNVs, and whether these overlapped.
Method
Subjects and Clinical Measures
The ADHD patient sample consisted of 799 Caucasian children from Cardiff, Wales (N=559); St. Andrews, Scotland (N=44); and Dublin, Ireland (N=196). All children were recruited from community clinics and met DSM-IV or ICD-10 criteria for ADHD or hyperkinetic disorder. To be comparable with other GWAS, we excluded children with a major medical or neurological condition (including epilepsy), autism, bipolar disorder, or intellectual disability (IQ <70).
We obtained approval from North West England, Wales, NHS Tayside, and Eastern Regional Health Authority research ethics committees. Written informed consent from parents and assent from children were obtained.
Trained interviewers used the Child and Adolescent Psychiatric Assessment—Parent Version (16), a semistructured research diagnostic interview, to assess psychiatric diagnoses. Pervasiveness of ADHD symptoms (in school) was assessed using the Child Attention-Deficit Hyperactivity Disorder Teacher Telephone Interview (17) or the Conners Teacher Questionnaire (18). IQ was assessed using the WISC-IV (19).
The children were between 4 and 18 years old (mean=10 years 3 months [SD=3 years]). The sample consisted of 699 boys (87.4%) and 100 girls (12.6%). Table 1 summarizes ADHD subtypes and comorbidities.
TABLE 1.
ADHD Subtypes and Comorbid Disorder Rates in 799 Children With ADHDa
| Diagnosis | N | % |
|---|---|---|
| ADHD diagnoses (lifetime)a | ||
| DSM-IV ADHD, combined type | 498 | 64.8 |
| DSM-IV ADHD, predominantly inattentive type | 162 | 21.1 |
| DSM-IV ADHD, predominantly hyperactive-impulsive type | 62 | 8.1 |
| DSM-III-R ADHD | 46 | 6.0 |
| Other diagnoses (current) | ||
| DSM-IV conduct disorder | 107 | 13.7 |
| DSM-IV oppositional defiant disorder | 364 | 46.5 |
| DSM-IV anxiety disorder (generalized anxiety disorder, separation anxiety, or social phobia) | 37 | 4.7 |
| DSM-IV depressive disorder (any) | 22 | 2.8 |
a Because data on DSM subtypes for some case subjects are missing, the total is <799.
Genotype control data were obtained from the Wellcome Trust Case Control Consortium–Phase 2 (20). They comprised 3,000 individuals born in the United Kingdom during 1 week in 1958 (the 1958 British Birth Cohort) and 3,000 individuals from the U.K. Blood Services collection. It has previously been shown that it is valid to combine these two samples for use as comparison subjects in genetic association studies using U.K. case samples (20). The comparison subjects were not screened for psychiatric disorders. However, the potential loss of power that is attained by using unscreened comparison subjects is more than offset by the large numbers of comparison samples available (21).
SNP data for our 100 most strongly associated SNPs were requested from deCODE Genetics and the ADHD GWAS Consortium. The deCODE sample included 1,142 Icelandic individuals who met DSM-IV criteria for ADHD. Patients were recruited from outpatient psychiatric clinics in Iceland. Diagnoses were based on standardized diagnostic assessments and were reviewed by experienced clinicians as previously described (22). A total of 35,243 Icelandic individuals were available as comparison subjects (22). The second sample consisted of 2,064 parent-child trios, 896 case subjects, and 2,455 comparison subjects from the ADHD GWAS Consortium meta-analysis and has been described in detail elsewhere (10). This data set consists of four projects: the Children's Hospital of Philadelphia, phase I and phase II of the International Multisite ADHD Genetics Project, and a Pfizer-funded study from the University of California, Los Angeles, Washington University, and Massachusetts General Hospital.
Genotyping
DNA samples for our ADHD case subjects were genotyped on the Illumina (San Diego) Human660W-Quad BeadChip according to the manufacturer's instructions. Comparison subjects were genotyped by Wellcome Trust Case Control Consortium–Phase2 using the Illumina Human 1.2M BeadChip. BeadStudio (version 2.0) was used to call genotypes and inspect cluster plots. Analysis was based on 518,511 SNPs that were present on both chips.
Quality Control Assessment
Sample and SNP quality control assessments were performed using PLINK, version 1.07 (23). Sample quality control assessment was performed separately for case and comparison subjects. Full details are provided in the data supplement that accompanies the online edition of this article. In brief, case and comparison subjects were excluded if there was a call rate less than 0.99, low or high heterozygosity, evidence of relatedness, duplication, or non-European ancestry. Exclusions included one member of related pairs. Also, SNPs were excluded if they had a call rate less than 0.99, had a minor allele frequency less than 0.01, deviated from Hardy-Weinberg equilibrium at p<1×10–5, or had more than 1% discordant genotypes between the Illumina 550K and the Illumina Human 1.2M BeadChip arrays. After all the quality control, 502,702 SNPs were tested for association in 727 case subjects and 5,081 comparison subjects.
CNV Data
The ADHD sample is an extension of 366 cases previously examined for large, rare CNVs (5). All quality control and CNV detection protocols were identical to those previously described. BeadStudio was used to determine the log R ratio and B allele frequency at each SNP according to standard Illumina protocols. CNVs were defined by PennCNV (24) with loci spanning at least 15 consecutive informative SNPs, with those having copy number calls <2 and >2 being classed as deletions and duplications, respectively. Samples with a high standard deviation in their genome-wide log R ratio (>0.30) and carrying more than 30 apparent CNVs over 100 kb were also excluded. Large (classified as those >500 kb) and rare (<1% frequency) CNVs were used in this analysis because they are called with greater accuracy, have better concordance across different platforms, and show the most robust associations with neurodevelopmental disorders (5).
Statistical Analysis
GWAS.
SNPs were tested for association with ADHD using logistic regression in PLINK assuming an additive model. The EIGENSTRAT software package was used to calculate principle components by inferring continuous variation in allele frequencies reflecting ancestral differences in individuals (25). Two principle components were identified and used to control for population stratification since they had the maximum impact on the genomic control inflation factor λ. Genome-wide significance was considered to be achieved when the p value reached 5×10–8 (26).
Genotype data for the top 100 SNPs from the present GWAS were requested from the ADHD genetics consortium (10) and deCODE. In these samples, we tested for enrichment of association of our top 100 SNPs after linkage disequilibrium pruning (see the online data supplement). There were 204 samples from the present GWAS that overlapped with those included in the ADHD genetics consortium GWAS meta-analysis. Overlap was statistically accounted for in the analysis (see the online data supplement). There was no overlap between those two data sets and that of deCODE. Two methods were used to test for enrichment of association signal in the combined set of SNPs. The first of these was the Simes test (27), a more powerful and less conservative version of the Bonferroni method, which tests SNPs one at a time. The other was Fisher's method for combining p values, which aggregates the evidence for all SNPs simultaneously. Since odds ratios must be in the same direction as our GWAS to count as replication, one-sided p values were used in the analysis. Enrichment was tested in the ADHD genetics consortium and deCODE samples separately, and in both data sets combined (see the online data supplement). A meta-analysis of all three samples (Cardiff ADHD GWAS, ADHD genetics consortium, and deCODE) was also performed on each of the top 100 SNPs (without pruning) separately.
Pathway analysis of Cardiff GWAS data.
Gene sets used for pathway analyses of our GWAS data came from four sources (28): Gene Ontology (29), the Kyoto Encyclopedia of Genes and Genomes (ftp://ftp.genome.jp/pub/kegg/genes/organisms/hsa/hsa_pathway.list), the Mouse Genome Informatics database (30), and PANTHER (Protein Analysis Through Evolutionary Relationships [31]). Gene sets were required to contain between three and 1,000 genes to be included in the analysis, giving a total of 12,371 gene sets. Analysis was carried out using ALIGATOR (28), which converts a list of significant and nominally significant SNPs into a list of significant genes and tests this list for enrichment for genes within the gene sets, allowing for variable numbers of SNPs per gene. ALIGATOR generates p values for enrichment for each gene set and corrects these for testing multiple nonindependent gene sets. It also tests whether the number of significantly enriched gene sets is higher than expected given the observed set of SNP p values in the GWAS. Gene sets required at least two signals to be tested to remove the possibility of a small gene set being deemed significantly enriched based on one signal. An important modification to the original ALIGATOR method is that significant genes in the same gene set that are less than 1 Mb apart (and thus could be explained by the same association signal) were counted as a single signal. SNPs within the boundaries of a gene (genome build 36.3) were assigned to that gene: if SNPs mapped to more than one gene, they were assigned to all such genes. Using this method, 203,663 SNPs were assigned to 14,929 genes. As before (32), the significant genes included the top 5% of all genes represented by SNPs, which was a total of 746 genes with at least one SNP (p<0.0054).
Overlap of GWAS and CNV pathways.
Gene sets with nominally significant (p<0.05) enrichment in the pathway analysis of the GWAS data were tested for an excess of genes affected by large, rare CNVs in case subjects by fitting the following logistic model, which overcomes biases relating to gene and CNV size (33), to the combined set of CNVs:
logit (pr[case]) = CNV size + total number of annotated genes affected outside the gene set + number of genes affected in the gene set
and comparing the change in deviance between it and the model
logit (pr[case]) = CNV size + total number of annotated genes affected outside the gene set.
The comparison of case to control CNVs allows for the possibility of nonrandom CNV location unrelated to disease (i.e., CNVs tend to occur in specific locations of the genome, and this is unrelated to case status). A one-sided test for an excess of genes affected by case CNVs was performed. The inclusion of CNV size in the regression allows for case CNVs being of different size than typical CNVs (and thus likely to affect more genes, regardless of function). Inclusion of the total number of genes affected outside the gene set in the regression corrects for case CNVs affecting more genes overall (regardless of function) than control CNVs. Analysis was restricted to gene sets containing at least eight gene hits in total (case and control combined), since pathways with a large number of gene hits are more likely to be biologically meaningful. This criterion is different from that used for the ALIGATOR analysis of GWAS data (two significant genes) for two reasons. First, each gene is counted only once in ALIGATOR but can be counted multiple times in the CNV analysis (if hit by multiple CNVs). Second, two significant genes may be sufficient to flag a pathway of interest in a GWAS context if these gene associations are sufficiently significant. Correction for multiple testing was applied by randomly permuting case/control status of CNVs and repeating the analysis 5,000 times. This procedure gave a corrected p value for enrichment of gene hits in case CNVs for each gene set as well as a test of whether more gene sets than expected are significantly enriched. The latter gives a test of overlap in the pathways enriched for rare CNVs and common associated SNPs.
Results
GWAS
The quantile-quantile plot of the observed versus expected chi-square tests is presented in Figure 1. The genomic control inflation factor λ was 1.069. Standardized to a sample size of 1,000, λ1,000 was 1.054. No SNP achieved genome-wide significance. Table 2 lists the top 20 independent SNPs ordered by significance.
FIGURE 1.
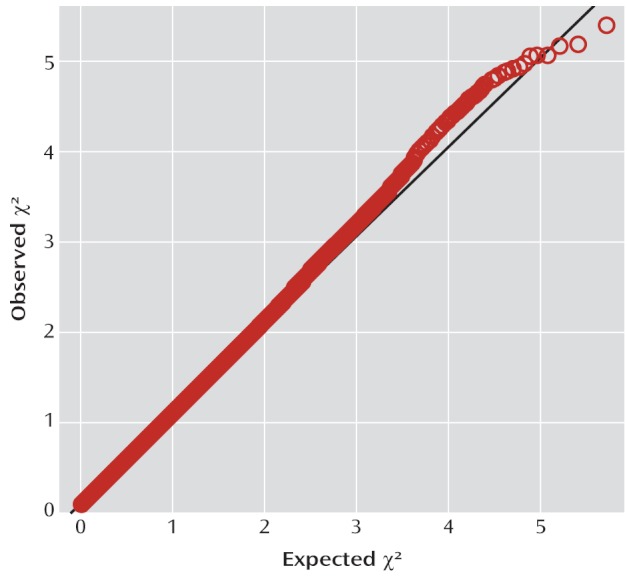
Quantile-Quantile Plot for 502,702 Single-Nucleotide Polymorphisms (SNPs) Genotyped in 727 Case Subjects and 5,081 Comparison Subjects With Genomic Control Inflation Factor λ=1.069 and λ1,000=1.054 in a Study of Common Genetic Variants and ADHD Risk
TABLE 2.
Top 20 Independent Single-Nucleotide Polymorphisms (SNPs) in an ADHD Genome-Wide Association Study (GWAS)
| Minor Allele |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chromosome | Position | Closest Gene | Location Relative to Gene | Minor Allele | Other Allele | Minor Allele Frequency | p | Odds Ratio | 95% CI |
| rs1744062 | 6 | 137350879 | IL20RA | Within noncoding gene | G | A | 0.43 | 4.16E-06 | 0.75 | 0.67–0.85 |
| rs11079828 | 17 | 43964102 | HOXB1 | Upstream | T | C | 0.47 | 6.54E-06 | 1.32 | 1.17–1.49 |
| rs42259 | 5 | 14439655 | TRIO | Intronic | T | C | 0.17 | 6.76E-06 | 1.41 | 1.22–1.64 |
| rs3779312 | 7 | 77549692 | MAGI2 | Intronic | T | C | 0.21 | 8.38E-06 | 1.37 | 1.19–1.57 |
| rs616668 | 12 | 110458663 | ATXN2 | Intronic | G | T | 0.20 | 8.62E-06 | 1.38 | 1.20–1.59 |
| rs11175219 | 12 | 62648986 | SRGAP1 | Intronic | T | C | 0.12 | 1.06E-05 | 1.46 | 1.23–1.73 |
| rs4238186 | 13 | 18588836 | LOC100128765 | Intergenic | A | G | 0.18 | 1.11E-05 | 1.39 | 1.20–1.61 |
| rs7746680 | 6 | 45885325 | - | Intergenic | A | G | 0.24 | 1.14E-05 | 1.35 | 1.18–1.54 |
| rs11686538 | 2 | 225526808 | DOCK10 | Intronic | G | A | 0.29 | 1.27E-05 | 0.74 | 0.64–0.84 |
| rs1304358 | 2 | 198677828 | PLCL1 | Intronic | C | T | 0.49 | 1.27E-05 | 1.30 | 1.16–1.47 |
| rs406742 | 10 | 8885947 | - | Intergenic | G | A | 0.26 | 1.49E-05 | 1.33 | 1.17–1.51 |
| rs790531 | 13 | 49623515 | DLEU2 | Within noncoding gene | G | A | 0.06 | 1.50E-05 | 1.62 | 1.30–2.02 |
| rs6815704 | 4 | 93693589 | GRID2 | Intronic | A | G | 0.15 | 2.09E-05 | 1.40 | 1.20–1.64 |
| rs9842394 | 3 | 181095930 | PEX5L | Intronic | T | C | 0.47 | 2.68E-05 | 0.77 | 0.69–0.87 |
| rs2636788 | 10 | 98866931 | SLIT1 | Intronic | G | A | 0.17 | 2.74E-05 | 0.70 | 0.59–0.83 |
| rs1490046 | 5 | 173888653 | - | Intergenic | A | G | 0.08 | 2.87E-05 | 1.56 | 1.27–1.92 |
| rs1050567 | 2 | 61559167 | XPO1 | 3′ untranslated region | T | C | 0.11 | 2.89E-05 | 1.44 | 1.22–1.72 |
| rs9384245 | 6 | 155201820 | TIAM2 | Intronic | T | C | 0.42 | 3.00E-05 | 0.77 | 0.68–0.87 |
| rs1370072 | 13 | 54739939 | - | Intergenic | T | C | 0.45 | 3.28E-05 | 1.29 | 1.14–1.45 |
| rs874836 | 22 | 15681843 | XKR3 | Intronic | A | G | 0.13 | 3.32E-05 | 1.41 | 1.20–1.67 |
We next sought replication for our top SNPs (see Table S1 in the online data supplement). To obtain 100 independent SNPs, we linkage disequilibrium pruned the GWAS data set using PLINK. For pairs of SNPs less than 1 Mb apart with r2>0.2, only the most significant SNP in the Cardiff GWAS in each pair was retained, leaving a total of 60 SNPs. No significant excess signal was observed among these SNPs in the ADHD Genetics Consortium meta-analysis data set (Simes p=0.176, Fisher's p=0.159) or in deCODE (Simes p=0.291, Fisher's p=0.621), or when both data sets (all published reports) were meta-analyzed (Simes p=0.135, Fisher's p=0.095). The individual p values for each of the 60 independent SNPs from these analyses are listed in Table S2 in the online data supplement. The individual p values for the top 100 Cardiff SNPs (see Table S1 in the online data supplement) and the p values from meta-analysis of the Cardiff ADHD GWAS, ADHD Genetics Consortium meta-analysis, and deCODE data sets are listed in Table S3 in the online data supplement. In the combined analysis of all data, no marker SNPs approached genome-wide significance (pmin=6.38×10–6 at rs11698703).
Pathway Analysis of Cardiff SNP Data
In the ALIGATOR analysis of our GWAS data set, 315 pathways were enriched at p<0.05 and 81 at p<0.01. More categories were enriched at the more stringent threshold (p=0.033) given the distribution of p values in the genes in the data set as a whole, but none was significant after correcting for multiple testing. Enrichment p values for the top 100 significant pathways are listed in Table S4 in the online data supplement.
Overlap of Enriched Pathways Between CNV and SNP Data
We included 727 ADHD case subjects and 1,047 comparison subjects in the CNV analysis. We observed a significantly (p=0.002) higher rate of large (>500 kb), rare CNVs in case subjects (N=85, 21 deletions, 64 duplications) than in comparison subjects (N=78, 13 deletions, 65 duplications). In the subset of the 727 ADHD case subjects (N=409) that had not been included in the previous report (5), the rate of large, rare CNVs was also significantly greater than in the comparison subjects (rate=0.112, compared with 0.075; p=0.02). More information is provided in Table S5 in the online data supplement.
More of the gene sets that were nominally significantly enriched in the ALIGATOR analysis of the SNP data were also significantly enriched for case CNVs (Figure 2). Thus, of the 315 pathways with enrichment at p<0.05 from the SNP data, in the CNV data 13 were enriched at p<0.05, eight at p<0.01, and seven at p<0.001. These numbers are significantly higher than expected by chance (p=0.0080, p=0.0022, p<0.0001, respectively). The 13 pathways significantly enriched (p<0.05) in both the SNP data and the CNV data are listed in Table 3. Although there was strong evidence of SNP and CNV signal convergence at the level of pathways, this was not evident at the individual gene level. Within the 13 significantly enriched pathways, 63 genes for which there were gene-wide (Simes) p values from the GWAS were affected by at least one CNV in a case subject or a comparison subject. Among these, there was some correlation (r=0.236) between genes showing evidence for association (-log GWAS Simes p and -log CNV enrichment p) at the level of SNPs and CNVs, but this did not quite achieve statistical significance (p=0.063).
FIGURE 2.
Significant Overlap of Biological Pathways, Including Cholesterol-Related and CNS Development, Enriched for Single-Nucleotide Polymorphism (SNP) Association, and Those Enriched for Rare Copy Number Variants (CNVs) in a Sample of 727 Children With ADHD and 5,081 Comparison Subjects
TABLE 3.
Pathways Showing Nominally Significant Enrichment (p<0.05) in Both the Single-Nucleotide Polymorphism Data and the Copy Number Variant (CNV) Data in a Genome-Wide Association Study of ADHD
| Pathway Numbera | Number of Genes | Gene Hits (Cases) | Gene Hits (Comparison) | p (CNV) | p (corr)b | p (GWAS) | Description |
|---|---|---|---|---|---|---|---|
| MGI:5278 | 188 | 14 | 0 | 1.47E-05 | 0.002 | 0.030 | Abnormal cholesterol homeostasis |
| MGI:3947 | 182 | 13 | 0 | 2.61E-05 | 0.004 | 0.023 | Abnormal cholesterol level |
| MGI:180 | 169 | 13 | 0 | 2.61E-05 | 0.004 | 0.026 | Abnormal circulating cholesterol level |
| GO:16746 | 214 | 14 | 0 | 1.42E-04 | 0.009 | 0.004 | Transferase activity, transferring acyl groups |
| GO:8415 | 203 | 13 | 0 | 1.43E-04 | 0.009 | 0.003 | Acyltransferase activity |
| GO:16747 | 205 | 13 | 0 | 1.43E-04 | 0.008 | 0.004 | Transferase activity, transferring acyl groups other than amino-acyl groups |
| GO:51298 | 11 | 8 | 0 | 6.95E-04 | 0.050 | 0.033 | Centrosome duplication |
| GO:32680 | 34 | 7 | 1 | 8.83E-03 | 0.341 | 0.014 | Regulation of tumor necrosis factor production |
| GO:5261 | 271 | 17 | 6 | 1.84E-02 | 0.547 | 0.042 | Cation channel activity |
| GO:7417 | 441 | 28 | 10 | 2.77E-02 | 0.683 | 0.002 | Central nervous system development |
| GO:16247 | 56 | 8 | 2 | 3.07E-02 | 0.719 | 0.026 | Channel regulator activity |
| GO:8233 | 572 | 25 | 8 | 4.89E-02 | 0.848 | 0.037 | Peptidase activity |
| GO:70011 | 553 | 25 | 8 | 4.89E-02 | 0.848 | 0.038 | Peptidase activity, acting on L-amino acid peptides |
a GO=Gene Ontology; MGI=Mouse Genome Informatics
b Represents the probability of obtaining by chance at least one pathway with a pathway-specific p value for enrichment of case CNV hits at least as significant as that observed in the actual data. Note that genes <1 Mb apart in the same pathway were collapsed into one signal.
The exception to this was CHRNA7, which is a member of the Gene Ontology (GO) categories “cation channel activity” (GO:5261; case CNV hits enrichment p=0.0184, GWAS enrichment p=0.042), “channel regulator activity” (GO:16247; p=0.0307, p=0.026), and “regulation of tumor necrosis factor production” (GO:32680; p=0.0088, p=0.014). CHRNA7 was affected by six duplications in case subjects but none in comparison subjects (p=9.08×10–4) and had a Simes-corrected gene-wide p value of 0.0002 from the GWAS.
Discussion
In a sample of 727 children with ADHD and 5,081 comparison subjects, there was no evidence of genome-wide significant association with any SNP. In keeping with previous results from a subsample of the present study, we found an increased burden of large and rare CNVs. Analysis of our top 100 SNPs in the ADHD genetics consortium meta-analysis and deCODE data sets yielded no significant evidence of association, after allowing for testing of individual SNPs, when the 100 SNPs were considered together and when the discovery GWAS data were combined with those from the other data sets. These results add to the four published GWAS studies of ADHD (6–10) that include meta-analyses in which no genome-wide significant findings had been found. The lack of significant GWAS findings could simply reflect sample sizes that are inadequate for the multiple testing burden, and it may be that when much larger samples are assembled for extended meta-analyses, common risk variants will be detected. That more sets of genes were significantly enriched for subthreshold association signals is consistent with this hypothesis, as it implies that the distribution of the association signals with respect to genes is not random.
One major motivation for undertaking genetic studies is to identify underlying biological risk mechanisms. In the present study, we sought evidence on whether the pathways enriched for SNP association converge with those enriched for rare CNVs. Our finding of significant evidence for such a convergence underscores our contention that it is premature to dismiss the contribution of SNP variation, but more importantly, it begins to provide evidence that genome-wide studies of ADHD, based on common or rare variants, are likely to inform processes of relevance to pathophysiology. At present, our study is not sufficiently powered to identify any of these categories unambiguously. Significant pathways included those related to cholesterol (four pathways) and CNS development. The latter has been previously implicated in ADHD (3), although different methods were used. The lack of a clear overlap at the level of individual genes may reflect true differences in the specific genes within pathways implicated by SNPs and CNVs, perhaps arising from the different mutational mechanisms responsible for generating large CNVs and SNPs, neither of which occur randomly with respect to the genomic sequence context. However, it is also likely that it reflects low power to identify specific risk genes. Although not supported at a genome-wide level of significance, the convergence of SNP and CNV association at CHRNA7, which encodes the cholinergic receptor nicotinic alpha 7, is intriguing. CHRNA7 is widely expressed in the brain, especially the hippocampus (34), and is involved in rapid synaptic transmission. CHRNA7 has been examined in relation to schizophrenia, associated cognitive deficits, and nicotine dependence (35, 36), although findings have not been entirely consistent. There has been little published work on ADHD, although incomplete evaluations of the gene in much smaller samples have not been supportive (37). Thus, to date this gene has yet to be comprehensively investigated in relation to ADHD.
Small duplications and deletions on 15q13.3 have been found to be associated with neuropsychiatric phenotypes that include ADHD. Recurrent deletions of chromosome 15q13.3 are associated with developmental delay and a variety of neuropsychiatric phenotypes. It has been suggested that haploinsufficiency of CHRNA7 may have a causal role (38). Duplications spanning CHRNA7 have also been found to be associated with a broad range of neuropsychiatric phenotypes that include ADHD (39, 40). Increased dosage of CHRNA7 in these microduplications has been considered to be responsible.
GWAS and CNV studies capture only a proportion of genetic variation and do not allow for the effects of unmeasured genetic and environmental risk factors. In the future, the next generation of sequencing studies will go some way toward addressing some of these gaps. The pathway analysis using ALIGATOR relies on Gene Ontology, the Kyoto Encyclopedia of Genes and Genomes pathways, Mouse Genome Informatics, and PANTHER-defined functional categories (28). The ability to detect enriched pathways will depend on how well and how accurately biological processes are defined, and again, this knowledge will evolve over time.
In summary, in keeping with similarly sized previous genome-wide association studies of ADHD, we failed to find significantly associated common variants. We previously found large, rare CNVs to be associated with ADHD, and the results remain similar in this newly extended sample. Contrary to what some might expect, we found a highly significant overlap of biological pathways hit by both CNVs and SNPS. This implies that both types of gene variants are relevant to ADHD risk. Finally, our results suggest that CHRNA7 is a promising candidate to examine further.
Acknowledgments
The authors thank the families who participated in this project and the clinicians who supported it; the field team members for sample collection (Charlotte Davies, Emma Evans, Elisabeth Felter, Kate Greening, Amy Hensey, Andrew Martin, Joanna Martin, Joanne Park, Rachel Roberts, Janet Robinson, Sarah Scott, Nina Smyth, Sharifah Syed, Lauren Whittington); Dr. Dobril Ivanov for setting up and maintaining the software and server; Dr. Kiran Mantripragada for assistance with genotyping; and Dr. Denise Harold for advice with analysis. The authors also acknowledge the support of the Health Research Board, Dublin (Drs. Gill and Hawi), the Dublin Molecular Medicine Centre, and the Hyperactive and Attention Disorder Group Ireland.
Footnotes
Received April 6, 2011; revision received June 9, 2011; accepted July 7, 2011.
This study was completed with the participation of the ADHD GWAS Consortium investigators: Stephen V. Faraone, Ph.D. (Chair, Departments of Psychiatry and of Neuroscience and Physiology, State University of New York Upstate Medical University, Syracuse, N.Y.); Benjamin M. Neale, Ph.D., Stephan Ripke, M.D., and Mark Daly, Ph.D. (Center for Human Genetic Research, Massachusetts General Hospital, Boston, and Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, Mass.); Sarah E. Medland, Ph.D., (Program in Medical and Population Genetics, Broad Institute of Harvard and MIT, Cambridge, and Genetic Epidemiology Unit, Queensland Institute of Medical Research, Brisbane, Australia); Philip Asherson, M.R.C.Psych., Ph.D. (MRC Social Genetic and Developmental Psychiatry, Kings' College London, U.K.); Barbara Franke, Ph.D. (Departments of Human Genetics and Psychiatry, Donders Institute for Brain, Cognition and Behavior, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands); Hans-Christoph Steinhausen, M.D., Ph.D., D.M.Sc. (Aalborg Psychiatric Hospital, Aarhus University Hospital, Denmark, and Clinical Psychology and Epidemiology, Institute of Psychology, University of Basel, Switzerland); Klaus-Peter Lesch, M.D. (ADHD Clinical Research Network, Unit of Molecular Psychiatry, Department of Psychiatry, Psychosomatics, and Psychotherapy, University of Wuerzburg, Wuerzburg, Germany); Christine Freitag, M.D., M.A. (Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University, Frankfurt am Main, Germany); Andreas Reif, M.D. (ADHD Clinical Research Network, Unit of Molecular Psychiatry, Department of Psychiatry, Psychosomatics, and Psychotherapy, University of Wuerzburg, Wuerzburg, Germany); Tobias J. Renner, M.D. (ADHD Clinical Research Network, Department of Child and Adolescent Psychiatry, Psychosomatics, and Psychotherapy, University of Wuerzburg, Wuerzburg, Germany); Thuy Trang Nguyen, Dipl. Math. oec. (Institute of Medical Biometry and Epidemiology, University of Marburg, Marburg, Germany); Marcel Romanos, M.D., and Jasmin Romanos, M.D. (Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, University of Munich, Munich, Germany); Susanne Walitza, M.D. (Department of Child and Adolescent Psychiatry, University of Zurich, Zurich, Switzerland); Helmut Schäfer, Ph.D. (Institute of Medical Biometry and Epidemiology, University of Marburg, Marburg, Germany); Andreas Warnke, M.D. (ADHD Clinical Research Network, Department of Child and Adolescent Psychiatry, Psychosomatics, and Psychotherapy, University of Wuerzburg, Wuerzburg, Germany); Jobst Meyer, Ph.D., and Haukur Palmason, Ph.D. (Institute of Psychobiology, Department of Neurobehavioral Genetics, University of Trier, Trier, Germany); Jan Buitelaar, M.D., Alejandro Arias Vasquez, Ph.D., and Nanda Lambregts-Rommelse, Ph.D. (Departments of Human Genetics and Psychiatry, Donders Institute for Brain, Cognition and Behavior, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands); Richard J. L. Anney, Ph.D. (Department of Psychiatry, Trinity Centre for Health Sciences, Dublin, Ireland); Joseph Sergeant, Ph.D. (Vrije Universiteit, De Boelelaan, Amsterdam); Herbert Roeyers, M.D., Ph.D. (Ghent University, Dunantlaan, Ghent, Belgium); Eric Mick, Sc.D. (Center for Human Genetic Research, Massachusetts General Hospital, Boston); Joseph Biederman, M.D. (Center for Human Genetic Research, Massachusetts General Hospital, Boston, and Clinical and Research Program in Pediatric Psychopharmacology and Adult ADHD at Massachusetts General Hospital, Boston); Alysa Doyle, Ph.D., Susan Smalley, Ph.D., and Sandra Loo, Ph.D. (UCLA Semel Institute for Neuroscience & Human Behavior, Los Angeles); Hakon Hakonarson, M.D., Ph.D., and Josephine Elia, M.D. (Center for Applied Genomics, The Children's Hospital of Philadelphia, Philadelphia); Alexandre Todorov, Ph.D. (Department of Psychiatry, Washington University School of Medicine, St. Louis); Ana Miranda, M.D. (Department of Developmental and Educational Psychology, University of Valencia, Spain); Fernando Mulas, M.D. (Department of Neuropaediatrics, La Fe University Hospital, Faculty of Medicine, Valencia, Spain); Richard Ebstein, Ph.D. (Singapore and Hebrew University, Jerusalem); Aribert Rothenberger, M.D., Ph.D. (Child and Adolescent Psychiatry, University of Goettingen, Germany); Tobias Banaschewski, M.D., Ph.D. (Department of Child and Adolescent Psychiatry, Psychosomatics and Psychotherapy, JW Goethe University, Frankfurt am Main, Germany); Robert Oades, Ph.D. (Clinic for Child and Adolescent Psychiatry, University of Duisburg-Essen, Essen, Germany); Edmund Sonuga-Barke, Ph.D. (MRC Social Genetic and Developmental Psychiatry, Kings' College London, and School of Psychology, Institute for Disorders of Impulse and Attention, University of Southampton, Highfield, Southampton, U.K.); James McGough, M.D. (UCLA Semel Institute for Neuroscience and Human Behavior, Los Angeles); Laura Nisenbaum, Ph.D. (Eli Lilly, Indianapolis); Frank Middleton, Ph.D. (Departments of Psychiatry and of Neuroscience and Physiology, State University of New York Upstate Medical University, Syracuse, N.Y.); Xiaolan Hu, Ph.D. (Pfizer, Molecular Medicine, Groton, Conn.); and Stan Nelson, M.D. (Department of Human Genetics and Neuroscience, University of California, Los Angeles).
All authors report no financial relationships with commercial interests.
Dr. Stergiakouli and Dr. Hamshere share first authorship.
Supported by Wellcome Trust grant 079711 and the Medical Research Council.
References
- 1.Faraone SV, Perlis RH, Doyle AE, Smoller JW, Goralnick JJ, Holmgren MA, Sklar P: Molecular genetics of attention-deficit/hyperactivity disorder. Biol Psychiatry 2005; 57:1313–1323 [DOI] [PubMed] [Google Scholar]
- 2.Stergiakouli E, Thapar A: Fitting the pieces together: current research on the genetic basis of attention-deficit/hyperactivity disorder (ADHD). Neuropsychiatr Dis Treat 2010; 6:551–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elia J, Gai X, Xie HM, Perin JC, Geiger E, Glessner JT, D'Arcy M, Deberardinis R, Frackelton E, Kim C, Lantieri F, Muganga BM, Wang L, Takeda T, Rappaport EF, Grant SF, Berrettini W, Devoto M, Shaikh TH, Hakonarson H, White PS: Rare structural variants found in attention-deficit hyperactivity disorder are preferentially associated with neurodevelopmental genes. Mol Psychiatry 2010; 15:637–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lesch KP, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T, Romanos M, Walitza S, Shoichet S, Dempfle A, Heine M, Boreatti-Hummer A, Romanos J, Gross-Lesch S, Zerlaut H, Wultsch T, Heinzel S, Fassnacht M, Fallgatter A, Allolio B, Schafer H, Warnke A, Reif A, Ropers HH, Ullmann R: Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Mol Psychiatry 2011; 16:491–503 [DOI] [PubMed] [Google Scholar]
- 5.Williams NM, Zaharieva I, Martin A, Langley K, Mantripragada K, Fossdal R, Stefansson H, Stefansson K, Magnusson P, Gudmundsson OO, Gustafsson O, Holmans P, Owen MJ, O'Donovan M, Thapar A: Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 2010; 376:1401–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lesch KP, Timmesfeld N, Renner TJ, Halperin R, Roser C, Nguyen TT, Craig DW, Romanos J, Heine M, Meyer J, Freitag C, Warnke A, Romanos M, Schafer H, Walitza S, Reif A, Stephan DA, Jacob C: Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J Neural Transm 2008; 115:1573–1585 [DOI] [PubMed] [Google Scholar]
- 7.Mick E, Todorov A, Smalley S, Hu X, Loo S, Todd RD, Biederman J, Byrne D, Dechairo B, Guiney A, McCracken J, McGough J, Nelson SF, Reiersen AM, Wilens TE, Wozniak J, Neale BM, Faraone SV: Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49:898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neale BM, Lasky-Su J, Anney R, Franke B, Zhou K, Maller JB, Vasquez AA, Asherson P, Chen W, Banaschewski T, Buitelaar J, Ebstein R, Gill M, Miranda A, Oades RD, Roeyers H, Rothenberger A, Sergeant J, Steinhausen HC, Sonuga-Barke E, Mulas F, Taylor E, Laird N, Lange C, Daly M, Faraone SV: Genome-wide association scan of attention deficit hyperactivity disorder. Am J Med Genet B Neuropsychiatr Genet 2008; 147B:1337–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neale BM, Medland S, Ripke S, Anney RJ, Asherson P, Buitelaar J, Franke B, Gill M, Kent L, Holmans P, Middleton F, Thapar A, Lesch KP, Faraone SV, Daly M, Nguyen TT, Schafer H, Steinhausen HC, Reif A, Renner TJ, Romanos M, Romanos J, Warnke A, Walitza S, Freitag C, Meyer J, Palmason H, Rothenberger A, Hawi Z, Sergeant J, Roeyers H, Mick E, Biederman J: Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49:906–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neale BM, Medland SE, Ripke S, Asherson P, Franke B, Lesch KP, Faraone SV, Nguyen TT, Schafer H, Holmans P, Daly M, Steinhausen HC, Freitag C, Reif A, Renner TJ, Romanos M, Romanos J, Walitza S, Warnke A, Meyer J, Palmason H, Buitelaar J, Vasquez AA, Lambregts-Rommelse N, Gill M, Anney RJ, Langely K, O'Donovan M, Williams N, Owen M, Thapar A, Kent L, Sergeant J, Roeyers H, Mick E, Biederman J, Doyle A, Smalley S, Loo S, Hakonarson H, Elia J, Todorov A, Miranda A, Mulas F, Ebstein RP, Rothenberger A, Banaschewski T, Oades RD, Sonuga-Barke E, McGough J, Nisenbaum L, Middleton F, Hu X, Nelson S: Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49:884–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craddock N, O'Donovan MC, Owen MJ: Genome-wide association studies in psychiatry: lessons from early studies of non-psychiatric and psychiatric phenotypes. Mol Psychiatry 2008; 13:649–653 [DOI] [PubMed] [Google Scholar]
- 12.Franke B, Neale BM, Faraone SV: Genome-wide association studies in ADHD. Hum Genet 2009; 126:13–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wray NR, Goddard ME, Visscher PM: Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res 2007; 17:1520–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang W, Barratt B, Clayton D, Todd J: Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 2005; 6:109–118 [DOI] [PubMed] [Google Scholar]
- 15.Psychiatric GWAS Consortium Coordinating Committee. Cichon S, Craddock N, Daly M, Faraone SV, Gejman PV, Kelsoe J, Lehner T, Levinson DF, Moran A, Sklar P, Sullivan PF: Genomewide association studies: history, rationale, and prospects for psychiatric disorders. Am J Psychiatry 2009; 166:540–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angold A, Costello EJ: The Child and Adolescent Psychiatric Assessment (CAPA). J Am Acad Child Adolesc Psychiatry 2000; 39:39–48 [DOI] [PubMed] [Google Scholar]
- 17.Holmes J, Lawson D, Langley K, Fitzpatrick H, Trumper A, Pay H, Harrington R, Thapar A: The Child Attention-Deficit Hyperactivity Disorder Teacher Telephone Interview (CHATTI): reliability and validity. Br J Psychiatry 2004; 184:74–78 [DOI] [PubMed] [Google Scholar]
- 18.Conners CK: Conners' Rating Scales—Revised. New York, Multi-Health Systems, 2001 [Google Scholar]
- 19.Wechsler D: WISC-IV Administration and Scoring Manual. San Antonio, Tex, Psychological Corporation, 2003 [Google Scholar]
- 20.The Wellcome Trust Case Control Consortium: Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007; 447:661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskvina V, Holmans P, Schmidt KM, Craddock N: Design of case-controls studies with unscreened controls. Ann Hum Genet 2005; 69(part 5):566–576 [DOI] [PubMed] [Google Scholar]
- 22.Ingason A, Rujescu D, Cichon S, Sigurdsson E, Sigmundsson T, Pietilainen OP, Buizer-Voskamp JE, Strengman E, Francks C, Muglia P, Gylfason A, Gustafsson O, Olason PI, Steinberg S, Hansen T, Jakobsen KD, Rasmussen HB, Giegling I, Moller HJ, Hartmann A, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Bramon E, Kiemeney LA, Franke B, Murray R, Vassos E, Toulopoulou T, Muhleisen TW, Tosato S, Ruggeri M, Djurovic S, Andreassen OA, Zhang Z, Werge T, Ophoff RA, Rietschel M, Nothen MM, Petursson H, Stefansson H, Peltonen L, Collier D, Stefansson K, Clair DM: Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry 2011; 16:17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M: PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007; 17:1665–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D: Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38:904–909 [DOI] [PubMed] [Google Scholar]
- 26.Pe'er I, Yelensky R, Altshuler D, Daly MJ: Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol 2008; 32:381–385 [DOI] [PubMed] [Google Scholar]
- 27.Simes RJ: An improved Bonferroni procedure for multiple tests of significance. Biometrika 1986; 73:751–754 [Google Scholar]
- 28.Holmans P, Green EK, Pahwa JS, Ferreira MA, Purcell SM, Sklar P, Owen MJ, O'Donovan MC, Craddock N: Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am J Hum Genet 2009; 85:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000; 25:25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bult CJ, Eppig JT, Kadin JA, Richardson JE, Blake JA: The Mouse Genome Database (MGD): mouse biology and model systems. Nucleic Acids Res 2008; 36:D724–D728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD: PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 2010; 38:D204–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones L, Holmans PA, Hamshere ML, Harold D, Moskvina V, Ivanov D, Pocklington A, Abraham R, Hollingworth P, Sims R, Gerrish A, Pahwa JS, Jones N, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Peters O, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Ruther E, Carrasquillo MM, Pankratz VS, Younkin SG, Hardy J, O'Donovan MC, Owen MJ, Williams J: Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One 2010; 5:e13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raychaudhuri S, Korn JM, McCarroll SA, Altshuler D, Sklar P, Purcell S, Daly MJ: Accurately assessing the risk of schizophrenia conferred by rare copy-number variation affecting genes with brain function. PLoS Genet 2010; 6:e1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burli T, Baer K, Ewers H, Sidler C, Fuhrer C, Fritschy JM: Single particle tracking of alpha7 nicotinic AChR in hippocampal neurons reveals regulated confinement at glutamatergic and GABAergic perisynaptic sites. PLoS One 2010; 5:e11507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiedler BJ, Debus OM, Neubauer BA, Kienle M, Kurlemann G: P50 sensory gating deficit in children with centrotemporal spikes and sharp waves in the EEG. Neurosci Lett 2006; 393:206–210 [DOI] [PubMed] [Google Scholar]
- 36.Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A, Ross RG, Adler LE, Freedman R: Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry 2002; 59:1085–1096 [DOI] [PubMed] [Google Scholar]
- 37.Kent L, Green E, Holmes J, Thapar A, Gill M, Hawi Z, Fitzgerald M, Asherson P, Curran S, Mills J, Payton A, Craddock N: No association between CHRNA7 microsatellite markers and attention-deficit hyperactivity disorder. Am J Med Genet 2001; 105:686–689 [DOI] [PubMed] [Google Scholar]
- 38.Shinawi M, Schaaf CP, Bhatt SS, Xia Z, Patel A, Cheung SW, Lanpher B, Nagl S, Herding HS, Nevinny-Stickel C, Immken LL, Patel GS, German JR, Beaudet AL, Stankiewicz P: A small recurrent deletion within 15q13.3 is associated with a range of neurodevelopmental phenotypes. Nat Genet 2009; 41:1269–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C, Cox GF, Dickinson H, Gentile J, Harris DJ, Hegde V, Hundley R, Khwaja O, Kothare S, Luedke C, Nasir R, Poduri A, Prasad K, Raffalli P, Reinhard A, Smith SE, Sobeih MM, Soul JS, Stoler J, Takeoka M, Tan WH, Thakuria J, Wolff R, Yusupov R, Gusella JF, Daly MJ, Wu BL: Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet 2009; 46:242–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Szafranski P, Schaaf CP, Person RE, Gibson IB, Xia Z, Mahadevan S, Wiszniewska J, Bacino CA, Lalani S, Potocki L, Kang SH, Patel A, Cheung SW, Probst FJ, Graham BH, Shinawi M, Beaudet AL, Stankiewicz P: Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat 2010; 31:840–850 [DOI] [PMC free article] [PubMed] [Google Scholar]