

Abstract
1-Deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) is a novel target for developing new antibacterial (including anti-tuberculosis) and antimalaria drugs. 41 lipophilic phosphonates, representing a new class of DXR inhibitors, were synthesized, among which 5-phenylpyridin-2-ylmethylphosphonic acid possesses the most activity against E. coli DXR (EcDXR) with a Ki of 420 nM. Structure activity relationships (SAR) are discussed, which can be rationalized using our EcDXR:inhibitor structures, and a predictive quantitative SAR (QSAR) model is also developed. Since inhibition studies of DXR from Mycobacterium tuberculosis (MtDXR) have not been well performed, 48 EcDXR inhibitors with a broad chemical diversity were found, however, to generally exhibit considerably reduced activity against MtDXR. The crystal structure of a MtDXR:inhibitor complex reveals the flexible loop containing the residues 198–208 has no strong interactions with the 3,4-dichlorophenyl group of the inhibitor, representing a structural basis for the reduced activity. Overall, these results provide implications in the future design and development of potent DXR inhibitors.
Introduction
Isoprenoid biosynthesis, producing very important substances such as isoprenyl (e.g., farnesyl and undecaprenyl) diphosphates, vitamins, steroids and terpenoids, is essential to the growth of all organisms. Isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP) are two common precursors for isoprenoid biosynthesis. In humans and animals, the mevalonate pathway is used to synthesize IPP and DMAPP (Figure 1). On the other hand, the vast majority of bacteria and apicomplexan parasites such as Plasmodium falciparum, the causative agent of malaria, use the non-mevalonate (or methylerythritol phosphate, MEP) isoprene biosynthesis pathway discovered in 1990s (Fig. 1).1 Since humans do not have the 7 enzymes in the pathway, the non-mevalonate pathway, especially the second enzyme 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR), has attracted much interest for its potential to be a new anti-infective drug target.2
Figure 1.

The non-mevalonate and mevalonate isoprene biosynthesis pathways.
DXR catalyzes the isomerization and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol-4-phosphate (MEP) with a Mg2+ and NADPH being the enzyme cofactors (Fig. 1). Fosmidomycin (1, Chart 1) (as well as its analog FR900098 (2)) is a very potent DXR inhibitor3 and has strong antibacterial activity against many Gram-negative bacteria such as Escherichia coli and Pseudomonas aeruginosa.4 Particularly remarkable is its profound antimalaria activity in preclinical studies and recent clinical trials.5 However, since 1 is a highly polar molecule with great water solubility, it has a short half-life (~1 h) in plasma as well as poor oral availability. In addition, many pathogens, such as Mycobacterium tuberculosis, the causative agent of tuberculosis that kills ~1.6 million people each year, are completely resistant to 1 and sensitive bacteria can also easily develop drug resistance. This is because 1 is actively transported into cells via glycerol-3-phosphate transporter GlpT.6 Pathogens without GlpT or with a mutant GlpT are therefore not affected by 1 due to limited cellular uptake.2d,7 Great interest has therefore been generated to develop potent inhibitors of DXR.8–10
Chart 1.

Structures of known DXR inhibitors 1 – 10.
A potential solution is to develop lipophilic DXR inhibitors that are able to passively permeate into bacterial cells. Medicinal chemistry studies based on the structures of 1 and 2 have been carried out for the past decade.8 Although most modifications resulted in compounds with largely reduced activity, derivatives of 1 and 2 having an α-substituent still possess the majority of the inhibitory activity.8j The best in this series are compounds 3 and 4 with a 3,4-dichlorophenyl group, which have similar enzyme activity as 1 but showed greatly increased activity against P. falciparum (EC50 of 90 nM for 3 and 250 nM for 4 vs. 1100 nM for 1),8j presumably due to enhanced cellular uptake resulted from their lipophilicity. In addition, only a few DXR inhibitors have been identified that are structurally distinct from 1 and 2.9 Two bisphosphonates (5 and 6, Chart 1) bearing a hydrophobic pyridine and isoquinoline sidechain are relatively weak inhibitors (IC50: 4 and 7 μM).9a We recently used a coordination chemistry based design to find a lipophilic, drug-like DXR inhibitor 7 (IC50: 1.4 μM) without a phosphonate group, which exhibited good activity against a broad spectrum of pathogenic bacteria.9b Moreover, in order to probe the hydrophobic nature of the DXR active site, we identified three new lipophilic phosphonate inhibitors 8 – 10 having a pyridine or quinoline group with IC50 values as low as 0.84 μM.9c
Our x-ray crystallographic studies of E. coli DXR (EcDXR) in complex with 8 – 10 reveal a new inhibitor binding mode as well as an important role of Trp211 in inhibitor recognition.9c The great plasticity of a flexible loop (residues 205 – 215) of EcDXR allows the indole ring of Trp211 to form π-π stacking and charge transfer interactions with the electron-deficient pyridine/quinoline group of these inhibitors. Previously reported crystal structures of EcDXR in complex with 1,11 5 and 69a also show favorable hydrophobic interactions between Trp211 and the inhibitors. In addition, our structures show that the conformational change of the flexible loop opens up a mainly hydrophobic pocket, where the pyridine/quinoline ring of 8 – 10 is located. These structures have been used to model the binding modes of other lipophilic DXR inhibitors, such as compounds 3 and 4.
Here we report synthesis, inhibitory activity, structure activity relationships (SAR) and quantitative SAR (QSAR) of 41 lipophilic phosphonate and related compounds against E. coli DXR. In addition, since M. tuberculosis is a Gram-positive bacterium (which behaves very differently from Gram-negative bacteria, e.g., E. coli) and causes enormous human mortalities, we also report the activity of 48 EcDXR inhibitors with a broad range of chemical diversity against M. tuberculosis DXR (MtDXR). The results show that these inhibitors generally have considerably lowered affinity to MtDXR. In addition, to investigate the possible structural basis for the reduced activity, a crystal structure of MtDXR:inhibitor complex is disclosed, showing the flexible loop of MtDXR, especially the residue Trp203 (the aligned counterpart of EcDXR Trp211) does not have strong interactions with the hydrophobic group of the inhibitor.
Results and Discussion
SARs of lipophilic phosphonates in EcDXR inhibition
In our previous study,9c pyridine/quinoline containing phosphonate compounds 8 – 10 were identified to be a new class of EcDXR inhibitors. Based on the structure of 8, we synthesized a total of 41 compounds to analyze the structure activity relationships (SAR) of this class of compounds. These compounds were tested to inhibit recombinant EcDXR and their inhibition constants (Ki) are shown in Table 1.
Table 1.
Structures of compounds 8 – 48 and their Ki values against EcDXR and MtDXR.
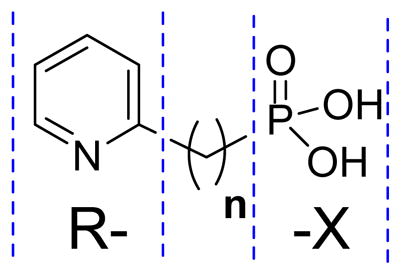
| |||||
|---|---|---|---|---|---|
| Compound ID | R- | n | -X | EcDXR Ki (μM) | MtDXR Ki (μM) |
| 11 | Pyridin-2-yl | 0 | -P(O)(OH)2 | NI a | NI |
| 8 | Pyridin-2-yl | 1 | -P(O)(OH)2 | 2.3 | 1.6 |
| 12 | Pyridin-2-yl | 2 | -P(O)(OH)2 | 18.6 | NI |
| 13 | Pyridin-2-yl | 3 | -P(O)(OH)2 | 9.5 | NI |
| 14 | Pyridin-3-yl | 1 | -P(O)(OH)2 | 5.0 | NI |
| 15 | Pyridin-4-yl | 1 | -P(O)(OH)2 | 3.5 | 6.8 |
| 10 | Quinolin-2-yl | 1 | -P(O)(OH)2 | 7.9 | 6.9 |
| 16 |
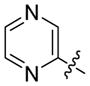
|
1 | -P(O)(OH)2 | 11.2 | NI |
| 17 |
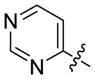
|
1 | -P(O)(OH)2 | 3.5 | 12.4 |
| 18 | Phenyl- | 0 | -P(O)(OH)2 | NI | NI |
| 19 | Phenyl- | 1 | -P(O)(OH)2 | NI | NI |
| 20 | Phenyl- | 2 | -P(O)(OH)2 | NI | NI |
| 21 | 3,4-Dichlorophenyl- | 1 | -P(O)(OH)2 | NI | NI |
| 22 | 2-Hydroxyphenyl- | 2 | -P(O)(OH)2 | NI | NI |
| 23 | 2,3-Dihydroxyphenyl- | 1 | -P(O)(OH)2 | NI | NI |
| 24 |
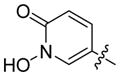
|
1 | -P(O)(OH)2 | NI | NI |
| 25 |
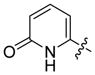
|
2 | -P(O)(OH)2 | NI | NI |
| 26 | 4-Bromopyridin-2-yl | 1 | -P(O)(OH)2 | 3.5 | 32.7 |
| 27 | 4-Phenylpyridin-2-yl | 1 | -P(O)(OH)2 | 1.1 | 6.9 |
| 28 | 4-(4-Fluorophenyl)pyridin-2-yl | 1 | -P(O)(OH)2 | 3.5 | NI |
| 29 | 5-Bromopyridin-2-yl | 1 | -P(O)(OH)2 | 6.7 | 45.2 |
| 9 | 5-Phenylpyridin-2-yl | 1 | -P(O)(OH)2 | 0.42 | 3.2 |
| 30 | 5-Hydroxypyridin-2-yl | 1 | -P(O)(OH)2 | 0.7 | 17.3 |
| 31 | 5-Benzyloxypyridin-2-yl | 1 | -P(O)(OH)2 | NI | NI |
| 32 | 6-Bromopyridin-2-yl | 1 | -P(O)(OH)2 | 3.6 | NI |
| 33 | 6-Phenylpyridin-2-yl | 1 | -P(O)(OH)2 | 3.5 | NI |
| 34 | 5-(4-Fluorophenyl)pyridin-2-yl | 1 | -P(O)(OH)2 | 1.1 | 50.7 |
| 35 | 5-(4-Chlorophenyl)pyridin-2-yl | 1 | -P(O)(OH)2 | 1.6 | NI |
| 36 | 5-(4-Methylphenyl)pyridin-2-yl | 1 | -P(O)(OH)2 | 3.9 | NI |
| 37 | 5-(3-Methoxyphenyl)pyridin-2-yl | 1 | -P(O)(OH)2 | 3.6 | NI |
| 38 | 5-(2-Naphthyl)pyridin-2-yl | 1 | -P(O)(OH)2 | 13.9 | NI |
| 39 | Pyridin-2-yl | 0 | -COOH | NI | NI |
| 40 | Pyridin-2-yl | 1 | -COOH | NI | NI |
| 41 | Pyridin-3-yl | 1 | -COOH | NI | NI |
| 42 | Pyridin-4-yl | 1 | -COOH | NI | NI |
| 43 | Pyridin-2-yl | 0 | -CONHOH | NI | NI |
| 44 | Pyridin-2-yl | 1 | -CONHOH | NI | NI |
| 45 | 5-Phenylpyridin-2-yl | 0 | -COOH | NI | NI |
| 46 | 5-Phenylpyridin-2-yl | 1 | -COOH | NI | NI |
| 47 | 5-Phenylpyridin-2-yl | 1 | -CONHOH | NI | NI |
| 48 | 5-Phenylpyridin-2-yl | 1 | -SO3H | NI | NI |
No inhibition observed at 100 μM.
First, compounds 11 – 13 (Table 1) were synthesized to find the optimal linker length between the pyridine ring and the phosphonate group. Compound 11 without a linker has essentially no activity on EcDXR at 100 μM, while 12 and 13 having two and three methylenes are active with Ki values of 18.6 and 9.5 μM, respectively. However, compared to 8 with a Ki of 2.3 μM, they are ~8× and 4× less potent, suggesting one methylene linker is best for DXR inhibition.
Second, we optimized the aromatic ring of 8 in order to find more potent DXR inhibitors. Compounds 14 and 15 with a pyridin-3-yl and pyridin-4-yl group were found to be active (Ki = 5.0 and 3.5 μM). In addition, other N-containing aromatic heterocyclic compounds 10 (with quinoline), 16 (with pyrazine) and 17 (with pyrimidine) also exhibit inhibitory activity against EcDXR, with Ki values ranging from 3.5 to 11.2 μM. However, phenyl ring containing compounds 18 – 20 with a variety of linker length (n = 0 – 2) are inactive. Since 3,4-dichlorophenyl group is present in potent EcDXR inhibitors 3 and 4 (Chart 1) with antimalaria activity,8j compound 21 having a 3,4-dichlorophenyl group was prepared but it was found to have no activity. Compounds 22 and 23 containing electron-donating hydroxyl groups also possess no inhibitory activity. To further probe the electronic effects of the aromatic ring, compounds 24 and 25 were synthesized, which have hydroxyl (or tautomeric carbonyl) substituted pyridine rings that are more electron-rich. Compared to unsubstituted parent compounds 14 and 12, these two compounds are essentially inactive. These results indicate that a highly electron-deficient aromatic ring, such as pyridine, quinoline and pyrimidine, is needed for DXR inhibition.
Third, compounds 26 – 38 were synthesized to see which substituents on the pyridine ring are favored. Compared to the parent compound 8, compound 26 with a 4-bromo substituent has a slightly weakened inhibitory activity with a Ki of 3.5 μM. 27 with a 4-phenyl group exhibits better activity (Ki = 1.1 μM), while compound 28 bearing a 4-(4-fluorophenyl) group possesses a reduced activity (Ki = 3.5 μM). For compounds 9, 29 – 31 that have a 5-substituent, compound 9 with a 5-phenyl group shows a Ki of 0.42 μM against EcDXR, which is 5× as potent as 8, while compound 29 with a 5-bromo group exhibits a weaker inhibition (Ki = 6.7 μM). Compound 30 with a 5-hydroxyl group has a strong activity with a Ki of 0.7 μM, being ~3× as potent as 8. This is rather unexpected given that 5-hydroxylpyridine is more electron-rich and, according to the SAR found above, should lead to a reduced activity. However, the high activity of 30 could be rationalized by molecular modeling using the crystal structure of EcDXR:8 complex, as discussed below. Compound 31 is inactive likely due to its electron-donating 5-benzyloxy group, which is also sterically more demanding. Compounds 32 and 33 bearing a 6-bromo and -phenyl group, respectively, show slightly weaker inhibitory activity (Ki = 3.6 and 3.5 μM) against EcDXR than does 8. Since compound 9 possesses a greatly improved potency, five derivatives 34 – 38 were made in an effort to find more active inhibitors. However, neither electron-withdrawing -F/-Cl (for 34 and 35, Ki = 1.1 and 1.6 μM) nor electron-donating -Me/-OMe (for 36 and 37, Ki = 3.9 and 3.6 μM) on the 5-phenyl ring provide enhanced potency. 38 with a bulkier naphthyl substituent is also a weak inhibitor with a Ki of 13.9 μM. These results show that introducing a 5-phenyl group leads to the strongest EcDXR inhibitor 9 among this class of compounds.
Finally, the phosphonate group in compounds 8 and 9 was replaced by an isosteric carboxyl, hydroxamate or sulfonate group with a rationale that the replacement with a less polar, negatively charged group could result in a higher lipophilicity, thereby improving cellular uptake. However, compounds 39 – 44 that are analogs of 8 with either a carboxyl or a hydroxamate group exhibit no inhibitory activity against EcDXR at 100 μM. Similarly, compounds 45 – 48, which are analogs of 9 containing a carboxyl, hydroxamate or sulfonate group, are also inactive. These results show the phosphonate group is essential to the tight binding of this class of inhibitors.
SAR rationalization
With the availability of the crystal structures of EcDXR:8 – 10 complexes, it is possible to rationalize the structure activity relationships observed above. Our crystallographic studies show that there are two common interactions between EcDXR and these inhibitors.9c The first one is the π-π stacking and charge transfer interactions between the electron-deficient pyridine/quinoline group of 8 – 10 and the indole ring of the residue Trp211, with the two parallel rings having a distance of ~3.5 Å. To see if compounds 11 – 13 are potentially able to form such interactions, we docked these compounds into the crystal structure of DXR:8 complex, using the program Glide12 in Schrödinger,13 and the results are shown in Figures 2a–c. The phosphonate group of 11 is predicted to be located very close to the phosphonate binding site of 8. However, its pyridine ring cannot form a π-π interaction with the indole ring of Trp211, which might be responsible for its non-activity. The docking results of compounds 12 and 13 indicate their pyridine rings can be parallel to the indole, thanks to their longer, flexible linkers. The modeling studies are in line with the experimental data showing 12 and 13 are active against EcDXR (Table 1). In addition, importance of Trp211 in inhibitor recognition also determines that an electron-deficient aromatic ring is needed for EcDXR inhibition, because of its stronger electrostatic interaction with the electron-rich indole ring. Moreover, the crystal structure of DXR:9 complex reveals that the conformation of Trp211 is different from that of DXR:8, with the indole ring flipping ~180° in order to have more hydrophobic interactions with the 5-phenyl group of 9.9c This might account for the improved activity of compound 9.
Figure 2.

The lowest energy docking structures of compounds (A) 11, (B) 12, (C) 13, and (D) 30 in the EcDXR:8 structure, together with superimposed structure of 8 (in yellow).
The second key interaction is the tight binding of the phosphonate group in 8 – 10, with all of the three negatively charged O atoms forming a network of electrostatic and H-bond interactions with the residues Lys227, Asn226, Ser221 and Ser185. However, a carboxylate, hydroxamate, or sulfonate group possesses considerably reduced polarity as well as negative charges at the physiological pH. Lack of activity of compounds 39 – 48 against EcDXR can therefore be rationalized.
Moreover, molecular modeling could be used to explain why compound 30 with the electron-donating 5-hydroxy group possesses unexpected, strong inhibition (Ki = 0.7 μM). As shown in Figure 2d, the docking result of 30 in the DXR:8 structure indicates that in addition to the phosphonate binding and the π-π stacking with Trp211, the 5-hydroxy group of 30 forms a hydrogen bond with the carbonyl of Asn210. This extra H-bond could compensate the weakened π-π/charge transfer interactions between 30 and the protein.
QSAR study in EcDXR inhibition
The above qualitative SAR analysis and rationalization demonstrate that a complex combination of electronic, steric, hydrophobic and H-bonding features of the substituents affect the activity of these compounds. In order to understand these observations in a quantitative and predictive manner, we next carried out a 3-D quantitative structure activity relationship (QSAR) study, using the program Phase14 in Schrödinger. Since compounds 1 – 4 (Chart 1) are more potent EcDXR inhibitors (Table 2, Ki: 19 – 58 nM) than 8 – 10 and known to occupy the same binding site, they were also included in an effort to generate a broader and more predictive QSAR model. In addition, for those inactive compounds in Table 1 against EcDXR, their Ki values were arbitrarily assigned to be 300 μM for QSAR calculation. The 45 compounds (1 – 4 and 8 – 48) were constructed, minimized, and aligned with compound 8 as a template, using the “flexible ligand alignment” module in Maestro15 in Schrödinger, which recognizes common features within the molecules (e.g., similar partial charge, hydrophobicity, aromaticity and H-bond donor/acceptor). The aligned compounds were imported into Phase and a partial least-squares (PLS) method was used to correlate the EcDXR activities of these compounds with the Phase field data calculated from their 3-D structures. The QSAR model yielded r2 = 0.95, q2 (no. of factors) = 0.78 (5), F-test = 158.5, and a pKi error of 0.30, as shown in Table 3 and Figure 3a. Five leave-5-out tranining/test sets were applied to further validate the model, obtaining a good predictivity with r2 ≥0.94, q2 ≥0.62, F-test ≥123.5, as shown in detail in Table 3. The compound alignment and the 3-D QSAR virtualization showing negative ionic, electron-withdrawing, hydrophobic, H-bond donor Phase fields are shown in Figures 3b–f. The results are in good agreement with the qualitative SAR observations. For example, the negatively charged phosphonate group (Fig. 3c) and the electron-deficient aromatic ring (Fig. 3d) are important for the activity. A hydrophobic group (e.g., phenyl) at the 5-position of the pyridine ring of compound 9 is favored (Fig. 3e). In addition, the negative charge (due to chelating Mg2+) and the electron-withdrawing feature of the hydroxamate group as well as the hydrophobic nature of the carbon skeleton of compounds 1 – 4 contribute together to their superior EcDXR inhibitory activities (Fig. 3c–e).
Table 2.
Ki values of 1 – 7 against EcDXR and MtDXR.
| Compound ID | EcDXR Ki (μM) | MtDXR Ki (μM) |
|---|---|---|
| 1 | 0.027 | 0.14 |
| 2 | 0.019 | 0.13 |
| 3 | 0.058 | 1.2 |
| 4 | 0.036 | 1.5 |
| 5 | 6.3 | 32.7 |
| 6 | 15.3 | NI a |
| 7 | 0.70 | 21.8 |
No inhibition observed at 100 μM.
Table 3.
QSAR results for EcDXR inhibition using Phase.
| Phase pKi predictions | |||||||
|---|---|---|---|---|---|---|---|
| compound | pKi (M)a | training set | test set 1b | test set 2 | test set 3 | test set 4 | test set 5 |
| 1 | 7.57 | 7.67 | 7.58 | 7.82 | 7.42 | 7.78 | 7.62 |
| 2 | 7.72 | 7.95 | 7.84 | 7.48 | 7.99 | 8.07 | 7.93 |
| 3 | 7.24 | 7.13 | 6.94 | 7.06 | 7.14 | 7.15 | 7.16 |
| 4 | 7.44 | 7.13 | 7.10 | 6.93 | 7.20 | 6.97 | 7.17 |
| 8 | 5.64 | 5.57 | 5.51 | 5.66 | 5.59 | 5.62 | 5.64 |
| 9 | 6.38 | 5.85 | 5.81 | 5.77 | 5.86 | 5.91 | 5.94 |
| 10 | 5.10 | 4.76 | 4.75 | 4.77 | 4.80 | 4.65 | 4.82 |
| 11 | 3.52 | 3.72 | 3.65 | 3.79 | 3.80 | 3.62 | 4.06 |
| 12 | 4.73 | 4.54 | 4.57 | 4.59 | 4.48 | 4.54 | 4.62 |
| 13 | 5.02 | 5.18 | 5.22 | 5.24 | 5.15 | 5.18 | 4.74 |
| 14 | 5.30 | 4.88 | 4.88 | 4.93 | 4.92 | 4.83 | 4.89 |
| 15 | 5.46 | 5.03 | 4.97 | 5.10 | 5.10 | 4.93 | 5.06 |
| 16 | 4.95 | 5.40 | 5.34 | 5.69 | 5.40 | 5.46 | 5.43 |
| 17 | 5.46 | 6.09 | 6.07 | 6.15 | 6.19 | 6.05 | 6.15 |
| 18 | 3.52 | 3.29 | 3.50 | 3.36 | 3.33 | 3.27 | 3.32 |
| 19 | 3.52 | 4.20 | 4.12 | 4.36 | 4.13 | 4.18 | 4.19 |
| 20 | 3.52 | 3.72 | 3.74 | 3.61 | 3.46 | 3.62 | 3.71 |
| 21 | 3.52 | 3.78 | 3.72 | 3.91 | 3.76 | 3.68 | 3.76 |
| 22 | 3.52 | 3.51 | 3.58 | 3.34 | 3.55 | 3.47 | 3.58 |
| 23 | 3.52 | 3.68 | 3.59 | 3.84 | 3.75 | 3.64 | 3.65 |
| 24 | 3.52 | 3.48 | 3.45 | 3.57 | 4.35 | 3.46 | 3.48 |
| 25 | 3.52 | 3.53 | 3.55 | 3.56 | 3.53 | 3.50 | 3.56 |
| 26 | 5.46 | 5.61 | 5.66 | 5.75 | 5.61 | 5.64 | 5.65 |
| 27 | 5.96 | 5.60 | 6.09 | 5.84 | 5.53 | 5.57 | 5.57 |
| 28 | 5.46 | 5.53 | 6.06 | 5.80 | 5.46 | 5.49 | 5.49 |
| 29 | 5.17 | 5.16 | 5.10 | 5.22 | 5.16 | 5.18 | 5.20 |
| 30 | 6.15 | 5.50 | 5.47 | 5.58 | 5.57 | 5.52 | 5.56 |
| 31 | 3.52 | 3.29 | 3.22 | 3.38 | 3.43 | 3.29 | 3.28 |
| 32 | 5.46 | 5.29 | 5.25 | 5.42 | 5.28 | 5.27 | 5.35 |
| 33 | 5.46 | 5.60 | 5.56 | 5.74 | 5.58 | 5.61 | 5.57 |
| 34 | 5.96 | 5.88 | 5.86 | 5.77 | 5.85 | 5.90 | 5.97 |
| 35 | 5.80 | 5.82 | 5.82 | 5.69 | 5.86 | 5.76 | 5.91 |
| 36 | 5.41 | 5.80 | 5.79 | 5.67 | 5.84 | 5.78 | 5.88 |
| 37 | 5.46 | 5.75 | 5.71 | 5.66 | 5.70 | 5.88 | 5.82 |
| 38 | 4.86 | 4.67 | 5.09 | 4.58 | 4.79 | 4.66 | 4.72 |
| 39 | 3.52 | 3.36 | 3.29 | 3.33 | 3.41 | 3.30 | 3.41 |
| 40 | 3.52 | 3.53 | 3.52 | 3.81 | 3.53 | 4.05 | 3.48 |
| 41 | 3.52 | 3.59 | 3.59 | 3.50 | 3.58 | 3.63 | 3.57 |
| 42 | 3.52 | 3.57 | 3.59 | 3.50 | 3.54 | 3.57 | 3.53 |
| 43 | 3.52 | 3.38 | 3.33 | 3.32 | 3.42 | 3.36 | 3.33 |
| 44 | 3.52 | 3.31 | 3.33 | 3.36 | 3.24 | 3.50 | 3.41 |
| 45 | 3.52 | 3.68 | 3.70 | 3.32 | 3.62 | 3.83 | 3.68 |
| 46 | 3.52 | 3.50 | 3.48 | 4.02 | 3.52 | 3.80 | 3.44 |
| 47 | 3.52 | 3.59 | 3.58 | 3.35 | 3.55 | 4.19 | 3.59 |
| 48 | 3.52 | 3.89 | 3.80 | 3.87 | 3.86 | 3.95 | 3.86 |
| r2 | 0.95 | 0.95 | 0.94 | 0.95 | 0.95 | 0.96 | |
| q2 | 0.78 | 0.68 | 0.65 | 0.62 | 0.76 | 0.76 | |
| F-test | 158.5 | 131.5 | 128.3 | 123.5 | 165.8 | 151.7 | |
| N | 5 | 5 | 4 | 5 | 4 | 5 | |
| n | 45 | 40 | 40 | 40 | 40 | 40 | |
An arbitrary pKi value of 3.52 (Ki = 300 μM) is given to all inactive compounds.
Bold values represent predicted activities of compounds that were not included in the training set.
Figure 3.

(A) Correlation between the experimental EcDXR inhibitory activity and predicted activity by the Phase QSAR model, showing r2 = 0.95; (B) Flexible alignment of compounds (only heavy atoms and polar hydrogens displayed for clarity), with compound 9 shown in a ball & stick model and 1 in a tube model; (C) Phase negative ionic fields, pink favorable, yellow disfavored; (D) electron-withdrawing fields, red favorable; (E) hydrophobic fields, cyan favorable, purple disfavored; (F) H-bond donor fields, orange disfavored.
Inhibition of MtDXR
Since the discovery of the non-mevalonate isoprene biosynthesis pathway, DXRs from many species, including bacteria, plants and parasites, have been cloned and characterized.3, 5a, 7a, 16 However, inhibition studies of DXRs other than E. coli DXR have not been well performed, except that 1 has been tested and shown broad activity on these enzymes. To this end, we are particularly interested in DXR from M. tuberculosis (MtDXR). This bacterium is a major human pathogen, causing deaths of ~1.6 million people a year worldwide. In addition, it has become more and more resistant to first line treatments such as isoniazid and rifampicin during the past decade.17 Previous studies demonstrated that M. tuberculosis exclusively uses the non-mevalonate pathway and DXR is essential for its survival,2d,7 suggesting DXR is a target for developing new anti-tuberculosis drugs. But the bacterium is completely resistant to the treatment of 1 due to lack of cellular uptake. Without GlpT (glycerol-3-phosphate transporter), 1 cannot penetrate M. tuberculosis cell membrane and reach the target in cytosol. Nevertheless, 1 was found to be a strong inhibitor of MtDXR. However, no other EcDXR inhibitors have been tested against MtDXR.
We examined the activity of 48 EcDXR inhibitors (compounds 1 – 48) on recombinant DXR from M. tuberculosis. It is also noteworthy that these inhibitors represent a wide range of chemical diversity. Compounds 1 and 2 are highly polar molecules containing a hydroxamate and a negatively charged phosphonate group, while 3 and 4 are their derivatives with a hydrophobic 3,4-dichlorophenyl sidechain.8j Compounds 5 and 6 are isoquinoline/pyridine containing bisphosphonates,9a which are extremely polar with 4 negative charges at the physiological pH. Distinct from the above compounds, 7 is a lipophilic, drug-like DXR inhibitor without a phosphonate.9b Moreover, all of compounds 8 – 48 consist of a lipophilic sidechain and a polar headgroup (phosphonate, carboxylate, hydroxamate, or sulfonate).
Activities of compounds 1 – 7 against MtDXR as well as EcDXR are shown in Table 2. In our study, the reference compound 1 exhibited a Ki value of 140 nM against MtDXR, which is very close to that of 150 nM found in a previous study2d (calculated from the IC50 of 310 nM). It is noted that in both assays physiologically relevant Mg2+ (4 mM) was used as the cofactor. 1 was also reported to have a lower IC50 value of 80 nM using 2.1 mM of Mn2+.18 However, such a high concentration of Mn2+ is not present in any organisms and the Km value for the substrate DXP was not measured in the experiment. This result therefore cannot be used to compare to our data. In addition, we determined the Ki value of 1 for EcDXR to be 27 nM, which is also similar to a literature value of 21 nM.19 Therefore, our study shows 1 binds to MtDXR ~5× weaker than EcDXR. Likewise, compound 2, another extremely potent EcDXR inhibitor,8j was found to possess a Ki of 19 nM against EcDXR in our experiment, while the Ki of 2 for MtDXR is 130 nM, indicating again the inhibitor has ~6× less affinity to the M. tuberculosis enzyme. Even more drastic are the differences between the Ki values of compounds 3 and 4 for these two DXR proteins. The two compounds bearing a bulky α-(3,4-dichloro)phenyl group are potent inhibitors of EcDXR with Ki values of 58 and 36 nM, approaching the activities of their parent compounds 1 and 2. However, their inhibition constants against MtDXR are 1.2 and 1.5 μM, being ~10× less active than 1 and 2 (KiMtDXR = 0.14 and 0.13 μM). In addition, compared to EcDXR, the binding of compounds 3 and 4 to MtDXR exhibits enormous 21- and 42-fold affinity losses. These results show that introducing an α-(3,4-dichloro)phenyl group is highly disfavored with respect to targeting MtDXR. Similarly, bisphosphonates (5 and 6) and compound 7 (Table 2) were also found to be >5× and 31× less active against the M. tuberculosis enzyme, respectively.
The Ki values of compounds 8 – 48 against MtDXR are shown in Table 1. Except for compounds 8 and 10, these compounds possess considerably reduced binding affinities to MtDXR. The best EcDXR inhibitor 9 (KiEcDXR = 0.42 μM) in this series has a 7-fold affinity loss to MtDXR (KiMtDXR = 3.2 μM). In addition, it appears that the M. tuberculosis enzyme is very sensitive to any additional substituents on 9. For example, compound 34 with only a small 4′-fluoro substituent, which is still a strong EcDXR inhibitor (KiEcDXR = 1.1 μM), is much less active against MtDXR with a KiMtDXR of 50.7 μM. Other derivatives of 9, compounds 35 – 37 with a 4′-chloro, 4′-methyl and 3′-methoxy substituent, are essentially inactive against MtDXR. On the other hand, compounds 8 and 10 possess slightly improved Ki values (1.6 and 6.9 μM, respectively) for MtDXR, compared to their KiEcDXR values of 2.3 and 7.9 μM.
The inhibition study of M. tuberculosis DXR is of interest, since it shows most inhibitors, including the reference compounds 1 and 2, have significantly reduced binding affinities to the enzyme (KiEcDXR ≫ KiMtDXR). More problematic is that the substrate DXP, however, binds to MtDXR 2× as tightly as EcDXR (KmMtDXR = 47.1 μM2d vs. KmEcDXR = 99 μM20). These two factors render the inhibition of M. tuberculosis DXR in the presence of DXP even more difficult and make it a great challenge to design potent MtDXR inhibitors.
Crystallographic studies of MtDXR
Due to the limited number of active MtDXR inhibitors, our attempts for QSAR studies in MtDXR inhibition did not yield a predictive model. X-ray crystallography was used to investigate the possible structural basis for the generally reduced inhibitor affinity to the protein. DXR is highly conserved across the species using the non-mevalonate pathway.9c MtDXR and EcDXR share an overall ~40% sequence identity and 69% similarity (Supporting Information Figure S1), with the active site showing even higher homology (~50% identity). In fact, despite the 5× affinity difference, 1 binds to the two DXRs in a very similar fashion, as shown in the aligned MtDXR:1 (PDB code: 2JCV)18 and EcDXR:1 (PDB code: 2EGH)11d structures (Figure S2). Three common interactions are 1) the electrostatic/H-bonds between the phosphonate and the protein; 2) metal ion (Mg2+ or Mn2+) chelation by the hydroxamate group; and 3) the hydrophobic interaction of the inhibitor backbone with a conserved Trp residue (Trp211 for EcDXR and Trp203 for MtDXR), which also shields the active site from the solvent. From the two DXR:1 structures, it is still unclear what factor determines the affinity difference.
We turned to compound 4 for its larger affinity difference (KiEcDXR = 36 nM vs. KiMtDXR = 1500 nM). Besides, 4 is also 10× less active than 1 against MtDXR, while these two compounds have similar activity on EcDXR. We determined the x-ray crystal structure of MtDXR in complex with 4, Mn2+, and NADPH, at 2.5 Å. Details of data processing and refinement are shown in Table 4 and the overall structure of the MtDXR:4 complex illustrated in Figure S3a. Although the protein crystallizes as a homodimer, only one monomer (monomer A) contains inhibitor 4 as well as Mn2+ and NADPH in the active site. In addition, a flexible loop consisting of the residues 198 – 208, which acts as a “cap” opening and closing when the substrate or an inhibitor enters the active site, adopts a closed conformation (Fig. S3b). The other monomer (monomer B) includes a Mn2+, SO42− and NADPH in the active site, with the flexible loop in a widely open conformation due to crystal packing interactions (Fig. S3c).18 No inhibitor is found in the active site of monomer B, while a SO42− ion is located in the phosphonate binding site of 1. The above phenomena can also be observed in the MtDXR:1 structure.18 There is one molecule of 4 found on the surface of monomer B, likely due to non-specific binding. Each of the two Mn2+ ions is anchored to the protein via coordination to three carboxylate groups of the residues Asp151, Glu153 and Glu222. Moreover, the binding of both NADPH molecules in the monomers A and B closely resemble those found in the MtDXR:1 structure. Since monomer A mimics the solution state of the MtDXR:4 complex better, the discussion below is focused on monomer A. In addition, racemic form of 4 was used in the crystallization experiment and it is not unambiguous to assign the absolute configuration of the α-carbon atom of the inhibitor. As shown in Figure S4, good models for (R)- and (S)-4 can be generated, in which the phosphonates, hydroxamates and 3,4-dichlorophenyl groups are located almost in the same sites, but the conformations of the linker carbon skeletons differ from each other. We therefore assigned 50% occupancy for each enantiomer in the final structure. While (R)-4 is used for the following discussion on inhibitor-protein interactions, (S)-4 molecule possesses essentially the same properties.
Table 4.
Data processing and refinement statistics
| Data processing | |
| Wavelength (Å) | 1.542 |
| Space group | P21 |
| Unit cell dimensions | |
| a, b, c (Å) | 67.25, 65.18, 85.41 |
| α, β, γ (°) | 90.0, 101.3, 90.0 |
| Resolution (Å) | 100–2.56(2.6–2.56) |
| Unique reflections | 22365(1144) |
| Completeness (%) | 93.9(94.5) |
| Redundancy | 3.9(3.8) |
| Rsym (%) | 13.6(51.8) |
| I/σ(I) | 11.2(2.2) |
| Refinement | |
| Resolution (Å) | 33.45–2.55(2.71–2.55) |
| Number of reflections used in working set | 21222(3505) |
| R/Rfreea (%) | 20.8/27.8 |
| Number of non-hydrogen atoms | 5866 |
| Number of solvent waters | 119 |
| Mean B-factor from Wilson plot (Å2) | 49.90 |
| Mean B-factor, protein atoms (Å2) | 52.42 |
| Mean B-factor, solvent atoms (Å2) | 44.21 |
| Mean B-factor, inhibitors (Å2) | 59.47 |
| Mean B-factor, NADPH (Å2) | 51.54 |
| Mean B-factor, Mn2+ (Å2) | 59.17 |
| Mean B-factor, SO42− (Å2) | 56.24 |
| Root mean square deviations from ideality | |
| Bond length (Å) | 0.008 |
| Bond Angle (°) | 1.3 |
A subset of the data (5%) was excluded from the refinement and used to calculate Rfree.
Close-up views of the active site of the MtDXR:4 structure and protein-ligand interactions are shown in Figures 4a and S5 respectively. The phosphonate group of 4 is located in the phosphonate binding site of 1, with all of the three O atoms forming hydrogen bonds and/or electrostatic interactions with the residues Lys219, Asn218, Ser213 and Ser177. The hydroxamate group of 4 is found to chelate Mn2+, with the distances between the two O atoms and the metal ion being 2.04 and 2.51 Å. These two features are common to all known structures of DXR:1 complexes. The 3,4-dichlorophenyl group of 4 is located in a hydrophobic cavity, surrounded by Pro265, Ser245 and Trp203. However, there is no π-π stacking interaction between the 3,4-dichlorophenyl and the indole ring of Trp203. Moreover, it is remarkable that the electron density for the residues 198–203 in the flexible loop is relatively weak with high B factor values, showing this part of the loop flips around and has no strong interactions with the inhibitor 4. On the contrary, the loop in MtDXR:1 structure is bound well near the center of the active site, with the indole ring of Trp203 covers on top of the relatively hydrophobic carbon skeleton of 1 (Fig. 4b). It can therefore be inferred that the weak interactions between Trp203 and the 3,4-dichlorophenyl ring could account for the reduced activity of compound 4 on MtDXR.
Figure 4.
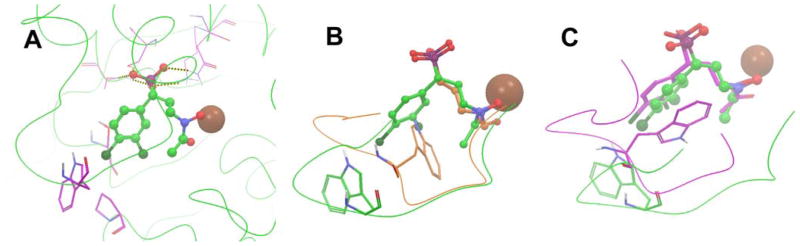
(A) The active site of the MtDXR:4 structure. The protein backbone is shown as green curved lines, 4 as a ball & stick model, and Mn2+ as a brown sphere; (B) The crystal structure of MtDXR:4 (in green) superimposed with the structure of MtDXR:1 (in orange). For clarity, only the flexible loops containing residues 198 – 208 of the two structures are shown as curved lines; (C) The crystal structure of MtDXR:4 (in green) superimposed with a modeled structure of EcDXR:4 complex (in purple). Only the flexible loops (residues 198 – 208 for MtDXR and 205 – 215 for EcDXR) of the two structures are shown as curved lines.
Next, we analyzed the binding differences of compound 4 in MtDXR and EcDXR. Although the crystal structure of EcDXR in complex with 4 has not been available, a model of how 4 binds to the protein has been generated in our previous study9c by using the crystal structure of the EcDXR:8 complex, which is also supported by experimental data. The modeled EcDXR:4 structure was aligned with the MtDXR:4 structure and, as shown in Figure 4c, the two molecules of 4 can almost be superimposed to each other and possess a similar binding pose. Nevertheless, a notable difference exists with respect to the conformation of the flexible loops of the two proteins. The EcDXR loop (in purple) is located closer to the inhibitor, with Trp211 indole ring forming a π-π stacking interaction with the 3,4-dichlorophenyl group of 4, affording a tight binding mode. This is also in line with the potent inhibitory activity of 4 (KiEcDXR = 36 nM). On the other hand, the loosely bound loop of MtDXR might be again responsible for the considerably lowered affinity of 4 to the enzyme (KiMtDXR = 1500 nM).
The exact origin for the weak interaction between Trp203 in MtDXR and compound 4 is unclear, but should be attributed to factors from both the protein and the inhibitor. From the side of the protein, the flexible loop of EcDXR has one conformationally more rigid proline residue (Pro209), while that of MtDXR contains two proline residues (Pro201 and Pro207), which could restrict the conformational change of Trp203 from having a favorable π-π stacking interaction with the 3,4-dichlorophenyl group of inhibitor 4. However, this might not be the only reason, since Trp203 is still able to move closely to 1 (Fig. 4b). From the side of the inhibitor, the relatively weak electron deficiency on 3,4-dichlorophenyl ring could be another unfavorable factor. For example, phosphonate compound 21 containing this group (Table 1) has essentially no activity against both EcDXR and MtDXR. More electron-deficient pyridine analog 8 is a good DXR inhibitor and, interestingly, shows a slightly improved activity on MtDXR over EcDXR, suggesting a pyridine containing analog of 4 could be particularly active against MtDXR.
Synthesis Aspect
Compounds 1 – 6 were prepared according to published methods8j, 9a and 39 – 42 are commercially available. Compounds 7 – 10, 14, 15, 21 and 23 were from our previous studies.9b, 9cScheme 1 shows the general synthetic methods for making the other compounds. 2-bromopyridine or bromobenzene was converted to their corresponding phosphonate diethyl ester, using either a palladium catalyzed reaction or a lithium-halogen exchange followed by treatment with diethyl chlorophosphate. The esters thus obtained were hydrolyzed with bromotrimethylsilane to afford compounds 11 and 18. Compounds 12, 16, 17, 19, 20, 22 and 24 with 1 or 2 methylene linker were synthesized from their corresponding chloride with an Arbuzov reaction, followed by bromotrimethylsilane mediated hydrolysis. For making pyridine containing phosphonates 26 – 38, a substituted 2-picoline, which was either commercially available or prepared from a bromo-substituted 2-picoline using a Suzuki coupling, was oxidized with m-chloroperbenzoic acid (m-CPBA) to give a pyridine oxide. It was then treated with Ac2O at 90 °C followed by hydrolysis (KOH) in MeOH to afford a 2-hydroxymethyl pyridine, which was reacted with thionyl chloride to give the corresponding chloride. It can then be readily converted to 26– 38, using an Arbuzov reaction followed by hydrolysis. To make compound 13 with a three-methylene linker, 2-picoline was treated with lithium diisopropylamide (LDA) followed by alkylation with diethyl 2-bromoethylphosphonate. The resulting diethyl ester was hydrolyzed with bromotrimethylsilane to give 13. The reaction of the sodium salt of tetraethyl diphosphonate with 2-methoxypyridine-5-carbaldehyde produced an α,β-unsaturated phosphonate, which subjected to hydrogenation (5% Pd/C) and hydrolysis to give compound 25. Suzuki coupling of methyl 5-bromopyridine-2-carboxylate or -2-acetate with benzeneboronic acid afforded, after hydrolysis, compounds 45 and 46. Hydroxamates 43, 44 and 47 were synthesized from their corresponding acids with an amide forming reaction with O-benzylhydroxylamine, followed by hydrogenation. Finally, compound 48 was made from sodium sulfite and 2-chloromethyl-5-phenylpyridine.
Scheme 1a.

aReagents and conditions: (i) PH(OEt)2, Pd(Ph3P)4, Ph3P, DIPEA, toluene, reflux; (vi) n-BuLi, ClP(O)(OEt)2, THF, −78 °C; (iii) TMSBr, CH3CN, rt, then MeOH/H2O; (iv) P(OEt)3, 120 °C, overnight; (v) RB(OH)2, Pd(Ph3P)4, Na2CO3, toluene/H2O (v/v 3:1), 90 °C; (vi) m-CPBA, CHCl3; (vii) Ac2O, 90 °C; (viii) KOH, MeOH; (ix) SOCl2, CH2Cl2; (x) LDA, 2-bromoethylphosphonate, THF, −78 °C; (xi) NaH, tetraethyl diphosphonate, THF, 0 °C; (xii) H2, 5% Pd/C, MeOH; (xiii) NH2OBn, HOBT, EDC, TEA, CH2Cl2; (ixv) Na2SO3, H2O reflux.
Conclusion
Our SAR/QSAR and crystallographic studies of E. coli and M. tuberculosis DXRs have resulted in several novel observations that have implications in the design of potent inhibitors. First, a total of 41 lipophilic phosphonate and related compounds were synthesized and their inhibitory activity tested against E. coli DXR, with the best compound 9 (5-phenylpyridin-2-ylmethylphosphonic acid) possessing a KiEcDXR of 420 nM. Second, SAR studies show that 1) an electron-deficient aromatic ring, especially a pyridine, and a phosphonate headgroup are essential for the inhibition; 2) one methylene linker between these two groups is optimal; and 3) certain substituents (e.g., 5-phenyl or 5-hydroxyl) on the aromatic ring provide enhanced activity. In addition, these SARs can be rationalized using the crystal structures of the EcDXR:8 and EcDXR:9 complexes. We also developed a 3-D QSAR model for EcDXR inhibition that had good predictivity. Third, the 41 compounds as well as 7 known EcDXR inhibitors with a broad range of chemical diversity were tested against M. tuberculosis DXR, in an effort to find new anti-tuberculosis agents. The vast majority of the inhibitors were found to exhibit considerably weakened activity against MtDXR, compared to that on EcDXR. In particular, fosmidomycin and its derivatives 1 – 4 with low nM Ki values against EcDXR show 5 – 40× less affinities to MtDXR (KiMtDXR: 130 – 1500 nM). Fourth, the x-ray crystal structure of MtDXR in complex with 4 is disclosed. The fosmidomycin moiety of 4 exhibits an expected binding mode, with the phosphonate located in the favored site and the hydroxamate group chelating the central metal ion. However, the flexible loop of MtDXR shows relatively weak electron density in the structure, in which Trp203 indole ring has no π-π stacking interaction with the 3,4-dichlorophenyl group of 4. This could represent a structural basis for the considerably lowered activity of 4 against MtDXR.
Experimental Section
All reagents were purchased from Alfa Aesar (Ward Hill, MA) or Aldrich (Milwaukee, WI). Compounds 1 – 6 were prepared according to published methods8j, 9a and 39 – 42 are commercially available. Compounds 7 – 10, 14, 15, 21 and 23 were from our previous studies.9b, 9c All compounds were characterized by 1H, 31P and/or 19F NMR on a Varian (Palo Alto, CA) 400-MR spectrometer. The purities were monitored by a Shimadzu Prominence HPLC with a Zorbax C18 or C8 column (4.6 × 250 mm) or 1H (at 400 MHz) absolute spin-count quantitative NMR analysis with imidazole as an internal standard. The purities of all compounds were found to be >95%.
General method A
A chloride (1 mmol) was heated with triethyl phosphite (5 mmol) at 120 °C under N2 overnight. Upon removal of excess triethyl phosphite under reduced pressure, the residue was purified with flash column chromatography to give the corresponding phosphonic acid diethyl ester.
General method B
To a solution of a phosphonic acid diethyl ester (1 mmol) in dry CH3CN (3 mL) was added slowly bromotrimethylsilane (3 mmol) at 0 °C. After stirring overnight at room temperature, the solution was evaporated to dryness and MeOH (5 mL) was added to the residue. The solvent was removed under reduced pressure again and the residue redissolved in MeOH (5 mL). Neutralization with 2 M NaOH to pH = 8 gave a disodium salt of a phosphonic acid as a white powder, which may be recrystallized in H2O/acetone if needed.
General method C
A mixture of a bromide (1 mmol), a boronic acid (1.5 mmol), Na2CO3 (424 mg, 4 mmol) and Pd(Ph3P)4 (58 mg, 5 mmol%) in toluene/H2O (v/v 3:1, 9 mL) was heated to 90 °C under N2 overnight. The product was extracted with EtOAc (3 × 10 mL) and purified with flash column chromatography.
General method D
To a solution of a 2-methylpyridine (25 mmol) in CHCl3 (100 mL) was added m-CPBA (11.2 g with 50% purity, 32.5 mmol). After stirring for 5 h, the reaction was quenched by adding saturated NaHCO3 (30 mL) and the aqueous layer extracted with CH2Cl2 (15 mL). The combined organic phases were washed with 2 N HCl (15 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give the corresponding pyridine oxide, which was used without further purification. It was then treated with acetic anhydride (50 mL) at 90 °C for 12 h and the resulting dark solution concentrated under reduced pressure. The residue was dissolved in a MeOH (30 mL) and KOH (2.1g, 15 mmol) added at 0 °C. After stirring for 3 h at room temperature, the solvent was removed and the residue treated with saturated NaHCO3 (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were dried, filtered, concentrated and purified with flash column chromatography (silica gel, hexane/EtOAc: 1/1) to give a pyridin-2-ylmethyl alcohol. To a solution of the alcohol (1 mmol) in CH2Cl2 (5 mL) was added thionyl chloride (0.22 mL, 3 mmol) solution in CH2Cl2 (2 mL) at 0 °C. The reaction was warmed to room temperature and stirred overnight. Upon removal of the solvent, the residue was treated with saturated Na2CO3 (5 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried, filtered and evaporated to give the corresponding chloride, which was used without purification, following the general methods A and B, to produce a phosphonic acid disodium salt as a white powder.
Pyridin-2-ylphosphonic acid disodium salt (11)
To a solution of 2-bromopyridine (790 mg, 5 mmol), diethyl phosphite (967 μL, 7.5 mmol) and diisopropylethylamine (DIPEA, 5 mL, 25 mmol) in toluene (40 mL) were successively added Ph3P (1.31 g, 5 mmol) and Pd(Ph3P)4 (290 mg, 5 mmol%) under N2. The reaction mixture was refluxed overnight. After removing solvent under reduced pressure, the residue was purified with column chromatography (silica gel, EtOAc) to give the corresponding phosphonate (350 mg), which was hydrolyzed, following the general method A, to give compound 11 as a white powder (320 mg, 31.5% yield). 1H NMR (400 MHz, D2O): δ 8.41 (d, J = 4.8 Hz, 1H), 7.69–7.67 (m, 2H), 7.23–7.21 (m, 1H). 31P NMR (162 MHz, D2O): δ 6.0.
2-(Pyridin-2-yl)ethylphosphonic acid disodium salt (12)
To a solution of 2-(pyridin-2-yl)ethanol (2.46 g, 20 mmol) was added slowly thionyl chloride (5.74 mL, 80 mmol) at 0 °C. The reaction mixture was heated to 80 °C for 3 h. Upon removal of excessive thionyl chloride under reduced pressure, the residue was treated with Saturated NaHCO3 (30 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and evaporated to give the crude chloride, which was used for the next step without purification. Compound 12 was obtained from the chloride following the general methods A and B as a white powder (3.8 g, 82% yield). 1H NMR (400 MHz, D2O): δ 8.29 (d, J = 4.8 Hz, 1H), 7.77–7.73 (m, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.21 (m, 1H), 2.93–2.68 (m, 2H), 1.80–1.72 (m, 2H). 31P NMR (162 MHz, D2O): δ 22.4.
3-(Pyridin-2-yl)propylphosphonic acid disodium salt (13)
To a solution of diisopropylamine (1.51 mL, 10.8 mmol) in THF (12 mL) was added dropwise n-BuLi (2.46 mL, 2.93 M, 7.2 mmol) at −10 °C. After 30 min, the solution was cooled to −78 °C and 2-picoline (558 mg, 6 mmol) was added slowly. The reaction mixture was stirred for 1 h, followed by addition of 2-bromoethylphosphonate (960 μL, 5 mmol). After 2 h, the reaction was warmed to room temperature and stirred for additional 1h. The reaction was quenched with statured NH4Cl (10 mL) and extracted with EtOAc (3 × 25 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified with column chromatography (silica gel, EtOAc) to give the corresponding diethyl phosphonate, which was hydrolyzed, following the general method A, to afford compound 13 as a white powder (516 mg, 33.4% yield). 1H NMR (400 MHz, D2O): δ 8.30 (d, J = 4.8 Hz, 1H), 7.77–7.71 (m, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.19 (m, 1H), 2.74 (t, J = 7.6 Hz, 2H), 1.82–1.75 (m, 2H), 1.41–1.32 (m, 2H). 31P NMR (162 MHz, D2O): δ 21.8.
Pyrazin-2-ylmethylphosphonic acid disodium salt (16)
To a solution of 2-methylpyrazine (1 mL, 22 mmol) in the CCl4 (80 mL) were added N-chlorosuccinimde (4.27 g, 31.5 mmol) and benzoyl peroxide (0.26 g, 1.1 mmol). The reaction mixture was stirred overnight. Upon removal of the solvent under reduced pressure, the residue was purified with column chromatography (silica gel, hexane/EtOAc: 6/1) to give the corresponding chloride, which was used following the general methods A and B, to give compound 16 as an off-white powder (1.46 g, 30.4% yield). 1H NMR (400 MHz, D2O): δ 8.47 (d, J = 1.6, 1H), 8.38 (s, 1H), 8.31 (d, J = 1.6 Hz, 1H), 3.10 (d, J = 20.4 Hz, 2H). 31P NMR (162 MHz, D2O): δ 15.9.
Pyrimidin-4-ylmethylphosphonic acid disodium salt (17)
Compound 17 was prepared from 4-methylpyrimidine, following a similar procedure as 16, as an off-white powder (158 mg, 37% yield). 1H NMR (400 MHz, D2O): δ 8.84 (m, 1H), 8.46 (d, J = 4.8 Hz, 1H), 7.44 (s, 1H), 3.03 (d, J = 20.4 Hz, 2H). 31P NMR (162 MHz, D2O): δ 13.9.
Phenylphosphonic acid disodium salt (18)
To a solution of bromobenzene (468 mg, 3 mmol) in THF (9 mL) was added n-BuLi (1.23 mL, 2.93 M, 3.6 mmol) at −78 °C. After stirring 1.5 h, diethyl chlorophosphate (776 mg, 4.5 mmol) was added. The reaction mixture was stirred for additional 3 h at the same temperature, quenched with saturated NH4Cl (10 mL) and extracted with EtOAc (3 × 10 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified with flash column chromatography (silica gel, hexane/EtOAc: 1/1) to give the diethyl phosphonate, which was hydrolyzed, following the general method B, to afford compound 18 as a white powder (340 mg, 56% overall yield). 1H NMR (400 MHz, D2O): δ 7.30–7.21 (m, 5H). 31P NMR (162 MHz, D2O): δ 17.2.
Benzylphosphonic acid disodium salt (19)
Compound 19 was prepared from benzyl chloride (250 mg, 2 mmol), following the general methods A and B, as a white powder (293 mg, 68.1% yield). 1H NMR (400 MHz, D2O): δ 7.20–7.04 (m, 5H), 2.13 (d, J = 20.0 Hz, 2H). 31P NMR (162 MHz, D2O): δ 21.6.
2-Phenylethylphosphonic acid disodium salt (20)
Compound 20 was prepared from phenylethylchloride (280 mg, 2 mmol), following the general methods A and B, as a white powder (300 mg, 65% yield). 1H NMR (400 MHz, D2O): δ 7.16–7.01 (m, 5H), 2.21 (m, 2H), 2.13 (m, 2H). 31P NMR (162 MHz, D2O): δ 19.5.
2-(2-Hydroxyphenyl)ethylphosphonic acid disodium salt (22)
Compound 22 was prepared from 2-chloroethylphenol (160 mg, 1 mmol), following the general methods A and B, as a white powder (94 mg, 40.5% yield). 1H NMR (400 MHz, D2O): δ 7.10 (d, J = 7.2 Hz, 2H), 6.99 (t, J = 7.2 Hz, 1H), 6.79 (t, J = 7.2 Hz, 1H), 6.73 (t, J = 7.2 Hz, 1H), 2.61 (m, 2H), 1.57 (m, 2H). 31P NMR (162 MHz, D2O): δ 22.5.
5-(1-Hydroxy-pyridin-2-one)methylphosphonic acid disodium salt (24)
To a solution of 6-methoxypyridine-3-carbaldehyde (274 mg, 2 mmol) in methanol (6 mL) was slowly added NaBH4 (114 mg, 3 mmol) at 0 °C. After 1 h at room temperature, the solvent was removed under reduced pressure and the reaction quenched with water and extracted with EtOAc (3 × 10 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting crude alcohol was dissolved in CH2Cl2 (8 mL) and a solution of SOCl2 (0.43 mL, 6 mmol) in CH2Cl2 (5 mL) added slowly at 0 °C. After 3 h, the solvent was removed under vacuum. The residue was treated with saturated Na2CO3 (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried, filtered and evaporated to give the corresponding chloride, which was used following the general method A to give the diethyl phosphonate. It (1 mmol) was dissolved in CHCl3 (5 mL) and m-CPBA (690 mg, 50%, 2 mmol) added. After 5 h, the reaction was quenched by adding saturated NaHCO3 (10 mL) and the aqueous layer extracted with CH2Cl2 (15 mL). The combined organic phases were washed with 2 N HCl (5 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give the crude product. It was refluxed in acetyl chloride (5 mL) for 1h. After excessive acetyl chloride was removed, the residue was washed with diethyl ether and hydrolyzed, following the general Method B, to give compound 24 as a white powder (152 mg, 61% overall yield). 1H NMR (400 MHz, D2O): δ 7.98 (s, 1H), 7.42 (d, J = 8.8 Hz, 1H), 6.63 (d, J = 8.8 Hz, 1H), 2.72 (d, J = 18.8 Hz, 2H); 31P NMR (162 MHz, D2O): δ 18.6.
2-(Pyridin-2-one-6-yl)ethylphosphonic acid disodium salt (25)
To a solution of tetraethyl diphosphonate (1.1 mL, 5.5 mmol) in THF (15 mL) was added NaH (242 mg, 6.1 mmol, 60% in oil) at 0 °C. 6-methoxypyridine-2-carbaldehyde (730 mg, 5 mmol) was added to the resulting solution. After 2 h at room temperature, the reaction mixture was quenched with saturated NH4Cl and extracted with EtOAc (3 × 15 mL). The combined organic phases were dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was hydrogenated with 5% Pd/C in methanol (15 mL) for 2h to afford the saturated ester, which was hydrolyzed, following the general method B, to afford compound 25 as a white powder (816 mg, 66% yield). 1H NMR (400 MHz, D2O): δ 7.49–7.41(m, 1H), 6.32–6.23 (m, 2H), 2.61 (m, 2H), 1.57 (m, 2H). 31P NMR (162 MHz, D2O): δ 19.3.
(4-Bromopyridin-2-yl)methylphosphonic acid disodium salt (26)
It was prepared following the general method D as a white powder in an overall 41% yield from 4-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 8.07 (d, J = 7.2 Hz, 1 H), 7.58 (s, 1 H), 7.32 (d, J = 7.2 Hz, 1 H), 2.89 (d, J = 19.6 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 14.8.
(4-Phenylpyridin-2-yl)methylphosphonic acid disodium salt (27)
It was prepared following the general method D as a white powder in an overall 51% yield from 4-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 8.30 (d, J = 4.4 Hz, 1 H), 7.72 (s, 1H), 7.70 (s, 1H), 7.66 (s, 1H), 7.45–7.39 (m, 4 H), 3.01 (d, J = 19.6 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.6.
4-(4-Fluorophenyl)pyridin-2-ylmethylphosphonic acid disodium salt (28)
It was prepared following the general method D as a white powder in an overall 37% yield from 4-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 8.25 (d, J = 7.2 Hz, 1 H), 7.74 (t, J = 8.8 Hz, 2 H), 7.62 (s, 1 H), 7.38 (m, 1 H), 7.16 (t, J = 8.8 Hz, 2 H), 2.97 (d, J = 20 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.5; 19F NMR (376 MHz, D2O): δ −116.2.
(5-Bromopyridin-2-yl)methylphosphonic acid disodium salt (29)
It was prepared following the general method D as a white powder in an overall 46% yield from 5-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 7.52–7.46 (m, 1 H), 7.32 (s, 1 H), 7.30 (s, 1 H), 2.93 (d, J = 19.6 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.3.
(5-Hydroxylpyridin-2-yl)methylphosphonic acid disodium salt (30)
It was prepared from 31 by hydrogenation with 5% Pd/C in 99% yield as a white powder. 1H NMR (400 MHz, D2O): δ 7.64 (m, 1 H), 7.30–7.28 (m, 2 H), 2.96 (d, J = 19.2 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 14.9.
(5-Benzyloxypyridin-2-yl)methylphosphonic acid disodium salt (31)
It was prepared following the general method D as a white powder in an overall 21% yield from 5-benzyloxy-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 8.01 (s, 1 H), 7.39–7.28 (m, 7 H), 5.08 (s, 2H), 2.90 (d, J = 19.6 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 16.3.
(6-Bromopyridin-2-yl)methylphosphonic acid disodium salt (32)
It was prepared following the general method D as a white powder in an overall 45% yield from 6-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 7.52–7.46 (m, 1 H), 7.32 (s, 1 H), 7.30 (s, 1 H), 2.93 (d, J = 19.6 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.3.
(6-Phenylpyridin-2-yl)methylphosphonic acid disodium salt (33)
It was prepared following the general method D as a white powder in an overall 40% yield from 6-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 7.77–7.64 (m, 3 H), 7.48–7.32 (m, 5 H), 3.03 (d, J = 20 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.6;
5-(4-Fluorophenyl)pyridin-2-ylmethylphosphonic acid disodium salt (34)
It was prepared following the general method D as a white powder in an overall 32% yield from 5-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 8.48 (s, 1H), 7.85 (d, J = 8.0 Hz, 1 H), 7.59–7.56 (m, 2 H), 7.44 (d, J = 8.8 Hz, 1 H), 7.13 (t, J = 8.8 Hz, 2 H), 2.98 (d, J = 20 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.5; 19F NMR (376 MHz, D2O): δ −115.8;
5-(4-Chlorophenyl)pyridin-2-ylmethylphosphonic acid disodium salt (35)
It was prepared following the general method D as a white powder in an overall 36% yield from 5-bromo-2- methylpyridine. 1H NMR (400 MHz, D2O): δ 8.50 (s, 1H), 7.86 (d, J = 8.0 Hz, 1 H), 7.56–7.40 (m, 5 H), 2.98 (d, J = 20 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.4;
5-(4-Methylphenyl)pyridin-2-ylmethylphosphonic acid disodium salt (36)
It was prepared following the general method D as a white powder in an overall 28% yield from 5-bromo-2- methylpyridine. 1H NMR (400 MHz, D2O): δ 8.50 (s, 1H), 7.84 (d, J = 7.2 Hz, 1 H), 7.51 (d, J = 8.0 Hz, 2 H), 7.42 (d, J = 7.2 Hz, 1 H), 7.26 (d, J = 8.0 Hz, 2 H), 2.97 (d, J = 20 Hz, 2 H), 2.24 (s, 3H); 31P NMR (162 MHz, D2O): δ 15.5;
5-(3-Methoxyphenyl)pyridin-2-ylmethylphosphonic acid disodium salt (37)
It was prepared following the general method D as a white powder in an overall 32% yield from 5-bromo-2-methylpyridine. 1H NMR (400 MHz, D2O): δ 8.56 (s, 1H), 7.84 (d, J = 8.0 Hz, 1 H), 7.38–7.35 (m, 2 H), 7.20–7.17 (m, 2H), 6.91 (d, J = 8.0 Hz, 1 H), 3.77 (s, 3H), 3.10 (d, J = 20 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 16.2;
5-(Naphthalen-2-yl)pyridin-2-ylmethylphosphonic acid disodium salt (38)
It was prepared following the general method D as a white powder in an overall 27% yield from 5-bromo-2- methylpyridine. 1H NMR (400 MHz, D2O): δ 8.57 (s, 1 H), 7.98–7.78 (m, 5 H), 7.63–7.61 (m, 1 H), 7.44–7.38 (m, 3 H), 2.97 (d, J = 20 Hz, 2 H); 31P NMR (162 MHz, D2O): δ 15.5;
N-hydroxy-pyridine-2-carboxamide (43)
To a solution of pyridine-2-carboxylic acid (411 mg, 3 mmol), NH2OBn (525 μL, 4.5 mmol) and triethylamine (1.26 mL, 9 mmol) in the CH2Cl2 (20 mL) were added HOBT (405 mg, 3mmol) and N-ethyl-N′-(3-aminopropyl)carbodiimide hydrochloride (864 mg, 4.5 mmol), respectively. The reaction mixture was stirred overnight at room temperature. The reaction was quenched with water (10 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were washed successively with saturated Na2CO3 (10 mL) and brine, dried, filtered and evaporated. The resulting oil was purified with flash column chromatography (silica gel, hexane/EtOAc: 2/1) to give the amide, which was hydrogenated with 10% Pd/C in methanol (12 mL) for 3h to afford compound 43 as a pale yellow oil (201 mg, 48.9% overall yield). 1H NMR (400 MHz, D2O): δ 8.46–8.44 (m, 1H), 7.90–7.86 (m, 1H), 7.82–7.80 (m, 1H), 7.50–7.47 (m, 1H).
N-hydroxy-(pyridin-2-yl)acetamide (44)
It was prepared from pyridin-2-ylacetic acid following the similar procedure as 112 as a colorless oil (169 mg, 37% overall yield). 1H NMR (400 MHz, D2O): δ 8.32–8.30 (m, 1H), 7.72–7.68 (m, 1H), 3.56 (s, 2H).
5-Phenylpyridine-2-carboxylic acid (45)
The methyl ester of compound 45 was prepared from 5-bromopyridine-2-carboxylate (324 mg, 1.5 mmol), following the general method C. It was dissolved in MeOH (5 mL) and 3 equivalent of KOH was added at 0 °C. After stirring 3 h at room temperature, the solvent was removed under vacuum. The residue was dissolved in H2O (1 mL), acidified with 2 M HCl, and extracted with ethyl acetate (30 mL). The organic layer was concentrated and purified with column chromatography (silica gel, hexane/EtOAc: 5/1) to give 45 as a white solid (214 mg, 72% yield). 1H NMR (400 MHz, D2O): δ 8.68 (s, 1H), 8.03–8.02 (m, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.61 (s, 1H), 7.59 (s, 1H), 7.44–7.35 (m, 3H).
(5-Phenylpyridin-2-yl)acetic acid (46)
It was prepared from 5-bromopyridine-2-acetate (350 mg, 1.5 mmol) following the similar procedure as 45, as a white solid (257 mg, 81% yield). 1H NMR (400 MHz, D2O): δ 8.82 (s, 1H), 8.60 (d, J = 7.2 Hz, 1H) 7.83 (d, J = 7.2 Hz, 1H), 7.62 (s, 1H), 7.61 (s, 1H), 7.47–7.44 (m, 3H), 4.01 (s, 2H).
N-hydroxy-(5-phenylpyridin-2-yl)acetamide (47)
It was prepared from 46 (212 mg, 1 mmol) following the similar procedure as 43, as a white solid (164 mg, 73% yield). 1H NMR (400 MHz, d6-DMSO): δ 10.73 (br, 1H), 8.86 (s, 1H), 8.75 (s, 1H), 8.00 (d, J = 8.0 Hz, 1H) 7.70 (s, 1H), 7.68 (s, 1H), 7.50–7.46 (m, 2H), 7.42–7.40 (m, 2H), 4.01 (s, 2H).
5-Phenylpyridin-2-ylmethanesulfonic acid sodium salt (48)
The mixture of 2-chloromethyl-5-phenylpyridine (203.5 mg, 1 mmol) and sodium sulfite (126 mg, 1 mmol) in the water (3 mL) was heated under reflux for 12 h. The solvent was concentrated under reduced pressure and the solid obtained was filtered and washed with diethyl ether to give 48 as a white powder (240 mg, 97%). 1H NMR (400 MHz, D2O): δ 8.76 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.73–7.48 (m, 6H), 4.37 (s, 2H).
DXR inhibition assay
E. coli DXR expression, purification and activity assay were carried out as previously reported.9b,c In brief, the inhibition assay was performed in a 96-well microplate using the purified EcDXR (10 nM), 2 mM MgCl2, 100 μM DXP, 50 μM NADPH in 50 mM HEPES buffer (pH = 7.5) containing 25 μg/mL BSA (bovine serum albumin). Inhibitors were pre-incubated with EcDXR for 10 min at 30 °C, before adding DXP to initiate the reaction. The process was monitored at 340 nm with a Beckman DTX-880 microplate reader. The initial velocities of wells containing increasing concentrations of an inhibitor were calculated and input into Prism (version 3.0, GraphPad Software, Inc., La Jolla, CA). The IC50 values were then obtained by using a standard dose response curve fitting in the software. The IC50s used for calculating Ki values were the mean values of at least three experiments with standard deviations of less than 20%. Ki values were calculated using the formula Ki = IC50/(1+[S]/Km), where [S] is the concentration of DXP (100 μM) and Km is the literature value of 99 μM.20
M. tuberculosis DXR was expressed and purified as described in the literature.18 BL21-DE3 strain (Lucigen) was transformed and cultured at 37 °C in LB medium containing ampicillin (50 μg/mL). Upon reaching an optical density of ~0.6 at 600 nm, DXR expression was induced for 3 hours by adding 0.42 mM isopropylthiogalactoside at 37 °C. Cells were then harvested and resuspended in 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl (buffer A). After addition of 1 mM phenylmethylsulfonyl fluoride and sonication at 0 °C, the lysate was centrifuged at 20,000 rpm for 20 min and the supernatant was collected and subjected to an affinity column chromatography using the Talon cobalt resin (Clontech). The resin was washed with 5 mM imidazole in buffer A and then the protein was eluted with 150 mM imidazole in buffer A. After desalting (using HiTrap, GE Healthcare) to 20 mM Tris pH 7.5, 150 mM NaCl, 2% glycerol, the protein was concentrated and stored in small aliquots at −80 °C.
The inhibition assay for MtDXR was carried out similarly as the EcDXR experiment. It was done in a 96-well microplate using the purified MtDXR (50 nM), 4 mM MgCl2, 100 μM DXP, 50 μM NADPH in 50 mM HEPES buffer (pH = 7.6) containing 25 μg/mL BSA. Compounds were incubated with MtDXR for 10 min at 30 °C before adding DXP. The initial velocities were calculated and the IC50 values for inhibitors obtained by using Prism. The IC50s used for calculating Ki values were from at least three independent experiments with standard deviations of less than 20%. Ki values were calculated using the formula Ki = IC50/(1+[S]/Km), where [S] is the concentration of DXP (100 μM) and Km is the literature value of 47.1 μM.2d
Molecular modeling
Docking studies were performed as previously reported9b,c using Schrödinger suite 2010,13 which includes all of the programs described below. Crystal structures of E. coli DXR were prepared using the module “protein preparation wizard” in Maestro 9.115 with default protein parameters: water molecules were removed, hydrogens added, inhibitors extracted, and NADPH remained in the protein structure for docking. H-bonds were then optimized and the protein was energy-minimized using OPLS-2005 force field. A receptor grid, which is large enough to contain the whole active site, was generated using Glide12 without constraints. Compounds were built, minimized using OPLS-2005 force field in Maestro and then docked into the prepared protein structure using Glide (docking parameters: standard-precision and dock flexibly).
QSAR studies were performed with default settings using Phase14 (version 3.2). All compounds were built and geometry- and energy-minimized using the OPLS-2005 force field in Maestro. Compound alignment was carried out using the “Flexible Ligand Alignment” module in Maestro with compound 8 as a template. The aligned compounds were imported into the Phase program. A partial least-squares (PLS) method was used to correlate the activities of these compounds with the 3-D structural features calculated by Phase with default settings. The QSAR model was then validated by performing five leave-5-out training/test sets.
Crystallization and structure refinement
The crystallization of MtDXR was performed according to a published procedure.18 In brief, purified MtDXR (10 mg/mL) was mixed with an equal volume of the crystallization buffer containing 2 mM MnCl2, 3 mM NADPH, 0.2 M (NH4)2SO4, 25% polyethylene glycol 3350, and 0.1 M Bis-Tris, pH 5.7. The protein crystals appear in ~1 week at room temperature, which were soaked for 1 h in the crystallization buffer containing an inhibitor (0.2 M). Data were collected to a resolution of 2.56 Å using a Rigaku FR-E+ SuperBright X-ray source at Baylor College of Medicine and were then processed using the program HKL2000.21 The initial structure was obtained by molecular replacement using the program Phaser22 with the coordinates of 2JCZ as a target. The refinement was carried out using the program CNS.23 Since the inhibitor 4 was synthesized in a racemic form, both (R)- and (S)-4 were built into the model with the occupancy of each enantiomer set to 0.5. The data collection and refinement statistics are provided in Table 4.
Supplementary Material
Acknowledgments
This work was supported by a grant (R21AI088123) from the National Institute of Allergy and Infectious Diseases (NIAID) to Y.S., grants AI049151 and AI065357 from NIAID to D.C.C. and a grant (Q1279) from Robert Welch Foundation to B.V.V.P. The recombinant M. tuberculosis DXR expression plasmid was kindly provided by Dr. T. Alwyn Jones (Uppsala University, Sweden). We thank the staff of the X-ray Crystallography Facility at Baylor College of Medicine for assistance in data collection.
ABBREVATIONS
- DXR
1-deoxy-D-xylulose-5-phosphate reductoisomerase
- EcDXR
DXR from E. coli
- MtDXR
DXR from Mycobacterium tuberculosis
- DXP
1-deoxy-D-xylulose-5-phosphate
- IPP
isopentenyl diphosphate
- DMAPP
dimethylallyl diphosphate
- MEP
2-C-methyl-D-erythritol-4-phosphate
- SAR
structure activity relationship
- QSAR
quantitative structure activity relationship
Footnotes
PDB ID CODES: The coordinates for the Mycobacterium tuberculosis DXR:4 complex have been deposited in Protein Data Bank as entry 3RAS.
Supporting Information Available. Figures S1 – S5 showing Sequence alignment for EcDXR and MtDXR, superimposed structures of EcDXR:1 and MtDXR:1, the overall structure of the MtDXR:4 complex, the electron density of inhibitor 4, and Ligplot interaction diagram for (R)-4 in MtDXR. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.See a recent review and the references cited therein: Hunter WN. The non-mevalonate pathway of isoprenoid precursor biosynthesis. J Biol Chem. 2007;282:21573–21577. doi: 10.1074/jbc.R700005200.
- 2.(a) Singh N, Cheve G, Avery MA, McCurdy CR. Targeting the methyl erythritol phosphate (MEP) pathway for novel antimalarial, antibacterial and herbicidal drug discovery: inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase (DXR) enzyme. Curr Pharm Des. 2007;13:1161–1177. doi: 10.2174/138161207780618939. [DOI] [PubMed] [Google Scholar]; (b) Rodriguez-Concepcion M. The MEP pathway: a new target for the development of herbicides, antibiotics and antimalarial drugs. Curr Pharm Des. 2004;10:2391–2400. doi: 10.2174/1381612043384006. [DOI] [PubMed] [Google Scholar]; (b) Testa CA, Brown MJ. The methylerythritol phosphate pathway and its significance as a novel drug target. Curr Pharm Biotechnol. 2003;4:248–259. doi: 10.2174/1389201033489784. [DOI] [PubMed] [Google Scholar]; (d) Dhiman RK, Schaeffer ML, Bailey AM, Testa CA, Scherman H, Crick DC. 1-Deoxy-D-xylulose 5-phosphate reductoisomerase (IspC) from Mycobacterium tuberculosis: towards understanding mycobacterial resistance to fosmidomycin. J Bacteriol. 2005;187:8395–8402. doi: 10.1128/JB.187.24.8395-8402.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuzuyama T, Shimizu T, Takahashi S, Seto H. Fosmidomycin, a specific inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett. 1998;39:7913–7916. [Google Scholar]
- 4.Mine Y, Kamimura T, Nonoyama S, Nishida M, Goto S, Kuwahara S. In vitro and in vivo antibacterial activities of FR-31564, a new phosphonic acid antibiotic. J Antibiot (Tokyo) 1980;33:36–43. doi: 10.7164/antibiotics.33.36. [DOI] [PubMed] [Google Scholar]
- 5.(a) Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]; (b) Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, Matsiegui PB, Binder R, Lell B, Wiesner J, Baranek T, Jomaa H, Kremsner PG. Fosmidomycin for malaria. Lancet. 2002;360:1941–1942. doi: 10.1016/S0140-6736(02)11860-5. [DOI] [PubMed] [Google Scholar]; (c) Borrmann S, Adegnika AA, Matsiegui PB, Issifou S, Schindler A, Mawili-Mboumba DP, Baranek T, Wiesner J, Jomaa H, Kremsner PG. Fosmidomycin-clindamycin for Plasmodium falciparum infections in African children. J Infect Dis. 2004;189:901–908. doi: 10.1086/381785. [DOI] [PubMed] [Google Scholar]; (d) Borrmann S, Lundgren I, Oyakhirome S, Impouma B, Matsiegui PB, Adegnika AA, Issifou S, Kun JF, Hutchinson D, Wiesner J, Jomaa H, Kremsner PG. Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2006;50:2713–2718. doi: 10.1128/AAC.00392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Borrmann S, Adegnika AA, Moussavou F, Oyakhirome S, Esser G, Matsiegui PB, Ramharter M, Lundgren I, Kombila M, Issifou S, Hutchinson D, Wiesner J, Jomaa H, Kremsner PG. Short-course regimens of artesunate-fosmidomycin in treatment of uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2005;49:3749–3754. doi: 10.1128/AAC.49.9.3749-3754.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Oyakhirome S, Issifou S, Pongratz P, Barondi F, Ramharter M, Kun JF, Missinou MA, Lell B, Kremsner PG. Randomized controlled trial of fosmidomycin-clindamycin versus sulfadoxine-pyrimethamine in the treatment of Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2007;51:1869–1871. doi: 10.1128/AAC.01448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sakamoto Y, Furukawa S, Ogihara H, Yamasaki M. Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli. Biosci Biotechnol Biochem. 2003;67:2030–2033. doi: 10.1271/bbb.67.2030. [DOI] [PubMed] [Google Scholar]
- 7.Brown AC, Parish T. DXR is essential in Mycobacterium tuberculosis and fosmidomycin resistance is due to a lack of uptake. BMC Microbiol. 2008;8:78–86. doi: 10.1186/1471-2180-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For development based on fosmidomycin, see Shtannikov AV, Sergeeva EE, Biketov SF, Ostrovskii DN. Evaluation of in vitro antibacterial activity of fosmidomycin and its derivatives. Antibiot Khimioter. 2007;52:3–9.Kuntz L, Tritsch D, Grosdemange-Billiard C, Hemmerlin A, Willem A, Bach TJ, Rohmer M. Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reducto-isomerase. Biochem J. 2005;386:127–135. doi: 10.1042/BJ20041378.Merckle L, de Andres-Gomez A, Dick B, Cox RJ, Godfrey CR. A fragment-based approach to understanding inhibition of 1-deoxy-D-xylulose-5-phosphate reductoisomerase. Chembiochem. 2005;6:1866–1874. doi: 10.1002/cbic.200500061.Munos JW, Pu X, Liu HW. Synthesis and analysis of a fluorinated product analogue as an inhibitor for 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Bioorg Med Chem Lett. 2008;18:3090–3094. doi: 10.1016/j.bmcl.2007.11.062.Ortmann R, Wiesner J, Silber K, Klebe G, Jomaa H, Schlitzer M. Novel deoxyxylulosephosphate-reductoisomerase inhibitors: fosmidomycin derivatives with spacious acyl residues. Arch Pharm (Weinheim) 2007;340:483–490. doi: 10.1002/ardp.200700149.Silber K, Heidler P, Kurz T, Klebe G. AFMoC enhances predictivity of 3D QSAR: a case study with DOXP-reductoisomerase. J Med Chem. 2005;48:3547–3563. doi: 10.1021/jm0491501.Woo YH, Fernandes RP, Proteau PJ. Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp. PCC6803 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Bioorg Med Chem. 2006;14:2375–2385. doi: 10.1016/j.bmc.2005.11.012.Kurz T, Schlüter K, Kaula U, Bergmann B, Walter RD, Geffken D. Synthesis and antimalarial activity of chain substituted pivaloyloxymethyl ester analogues of Fosmidomycin and FR900098. Bioorg Med Chem. 2006;14:5121–5135. doi: 10.1016/j.bmc.2006.04.018.Kurz T, Schlüter K, Pein M, Behrendt C, Bergmann B, Walter RD. Conformationally restrained aromatic analogues of fosmidomycin and FR900098. Arch Pharm Chem Life Sci. 2007;340:339–344. doi: 10.1002/ardp.200700013.Haemers T, Wiesner J, Van Poecke S, Goeman J, Henschker D, Beck E, Jomaa H, Van Calenbergh S. Synthesis of α-substituted fosmidomycin analogues as highly potent Plasmodium falciparum growth inhibitors. Bioorg Med Chem Lett. 2006;16:1888–1891. doi: 10.1016/j.bmcl.2005.12.082.Devreux V, Wiesner J, Jomaa H, Rozenski J, Van der Eycken J, Van Calenbergh S. Divergent strategy for the synthesis of α-aryl-substituted fosmidomycin analogues. J Org Chem. 2007;72:3783–3789. doi: 10.1021/jo0700981.
- 9.For non-fosmidomycin-like inhibitors, see Yajima S, Hara K, Sanders JM, Yin F, Ohsawa K, Wiesner J, Jomaa H, Oldfield E. Crystallographic structures of two bisphosphonate:1-deoxyxylulose-5-phosphate reductoisomerase complexes. J Am Chem Soc. 2004;126:10824–10825. doi: 10.1021/ja040126m.Deng L, Sundriyal S, Rubio V, Shi Z, Song Y. Coordination chemistry based approach to lipophilic inhibitors of 1-deoxy-D-xylulose-5-phosphate reductoisomerase. J Med Chem. 2009;52:6539–6542. doi: 10.1021/jm9012592.Deng L, Endo K, Kato M, Cheng G, Yajima S, Song Y. Structures of 1-deoxy-D-xylulose-5-phosphate reductoisomerase/lipophilic phosphonate complexes. ACS Med Chem Lett. 2011;2:165–170. doi: 10.1021/ml100243r.
- 10.For high-throughput screening, see Gottlin EB, Benson RE, Conary S, Antonio B, Duke K, Payne ES, Ashraf SS, Christensen DJ. High-throughput screen for inhibitors of 1-deoxy-d-xylulose 5-phosphate reductoisomerase by surrogate ligand competition. J Biomol Screen. 2003;8:332–339. doi: 10.1177/1087057103008003011.
- 11.(a) Yajima S, Nonaka T, Kuzuyama T, Seto H, Ohsawa K. Crystal structure of 1-deoxy-D-xylulose 5-phosphate reductoisomerase complexed with cofactors: implications of a flexible loop movement upon substrate binding. J Biochem. 2002;131:313–317. doi: 10.1093/oxfordjournals.jbchem.a003105. [DOI] [PubMed] [Google Scholar]; (b) Steinbacher S, Kaiser J, Eisenreich W, Huber R, Bacher A, Rohdich F. Structural basis of fosmidomycin action revealed by the complex with 2-C-methyl-D-erythritol 4-phosphate synthase (IspC). Implications for the catalytic mechanism and anti-malaria drug development. J Biol Chem. 2003;278:18401–18407. doi: 10.1074/jbc.M300993200. [DOI] [PubMed] [Google Scholar]; (c) Mac Sweeney A, Lange R, Fernandes RP, Schulz H, Dale GE, Douangamath A, Proteau PJ, Oefner C. The crystal structure of E. coli 1-deoxy-D-xylulose-5-phosphate reductoisomerase in a ternary complex with the antimalarial compound fosmidomycin and NADPH reveals a tight-binding closed enzyme conformation. J Mol Biol. 2005;345:115–127. doi: 10.1016/j.jmb.2004.10.030. [DOI] [PubMed] [Google Scholar]; (d) Yajima S, Hara K, Iino D, Sasaki Y, Kuzuyama T, Ohsawa K, Seto H. Structure of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in a quaternary complex with a magnesium ion, NADPH and the antimalarial drug fosmidomycin. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:466–470. doi: 10.1107/S1744309107024475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glide, version 5.5. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 13.Schrödinger Suite, version 2010. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 14.Phase, version 3.2. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 15.Maestro, version 9.1. Schrödinger, LLC; New York, NY: 2010. [Google Scholar]
- 16.(a) Altincicek B, Hintz M, Sanderbrand S, Wiesner J, Beck E, Jomaa H. Tools for discovery of inhibitors of the 1-deoxy-D-xylulose 5-phosphate (DXP) synthase and DXP reductoisomerase: an approach with enzymes from the pathogenic bacterium Pseudomonas aeruginosa. FEMS Microbiol Lett. 2000;190:329–333. doi: 10.1111/j.1574-6968.2000.tb09307.x. [DOI] [PubMed] [Google Scholar]; (b) Woo YH, Fernandes RP, Proteau PJ. Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp. PCC6803 1-deoxy-D-xylulose 5-phosphate reductoisomerase. Bioorg Med Chem. 2006;14:2375–2385. doi: 10.1016/j.bmc.2005.11.012. [DOI] [PubMed] [Google Scholar]; (c) Carretero-Paulet L, Ahumada I, Cunillera N, Rodriguez-Concepcion M, Ferrer A, Boronat A, Campos N. Expression and molecular analysis of the Arabidopsis DXR gene encoding 1-deoxy-D-xylulose 5-phosphate reductoisomerase, the first committed enzyme of the 2-C-methyl-D-erythritol 4-phosphate pathway. Plant Physiol. 2002;129:1581–1591. doi: 10.1104/pp.003798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Grolle S, Bringer-Meyer S, Sahm H. Isolation of the dxr gene of Zymomonas mobilis and characterization of the 1-deoxy-D-xylulose 5-phosphate reductoisomerase. FEMS Microbiol Lett. 2000;191:131–137. doi: 10.1111/j.1574-6968.2000.tb09329.x. [DOI] [PubMed] [Google Scholar]; Mueller C, Schwender J, Zeidler J, Lichtenthaler HK. Properties and inhibition of the first two enzymes of the non-mevalonate pathway of isoprenoid biosynthesis. Biochem Soc Trans. 2000;28:792–793. [PubMed] [Google Scholar]; (e) Schwender J, Muller C, Zeidler J, Lichtenthaler HK. Cloning and heterologous expression of a cDNA encoding 1-deoxy-D-xylulose-5-phosphate reductoisomerase of Arabidopsis thaliana. FEBS Lett. 1999;455:140–144. doi: 10.1016/s0014-5793(99)00849-2. [DOI] [PubMed] [Google Scholar]; (f) Rohdich F, Lauw S, Kaiser J, Feicht R, Kohler P, Bacher A, Eisenreich W. Isoprenoid biosynthesis in plants: 2C-methyl-D-erythritol-4-phosphate synthase (IspC protein) of Arabidopsis thaliana. FEBS J. 2006;273:4446–4458. doi: 10.1111/j.1742-4658.2006.05446.x. [DOI] [PubMed] [Google Scholar]
- 17.See a recent review and the references cited therein: Jassal M, Bishai WR. Extensively drug-resistant tuberculosis. Lancet Infect Dis. 2009;9:19–30. doi: 10.1016/S1473-3099(08)70260-3.
- 18.Henriksson LM, Unge T, Carlsson J, Aqvist J, Mowbray SL, Jones TA. Structures of Mycobacterium tuberculosis 1-deoxy-D-xylulose-5-phosphate reductoisomerase provide new insights into catalysis. J Biol Chem. 2007;282:19905–19916. doi: 10.1074/jbc.M701935200. [DOI] [PubMed] [Google Scholar]
- 19.Koppisch AT, Fox DT, Blagg BS, Poulter CD. E. coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry. 2002;41:236–243. doi: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- 20.Kuzuyama T, Takahashi S, Takagi M, Seto H. Characterization of 1-deoxy-D-xylulose 5-phosphate reductoisomerase, an enzyme involved in isopentenyl diphosphate biosynthesis, and identification of its catalytic amino acid residues. J Biol Chem. 2000;275:19928–19932. doi: 10.1074/jbc.M001820200. [DOI] [PubMed] [Google Scholar]
- 21.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Macromolecular Crystallography Part A, Method Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 22.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.