

Abstract
The nematode Caenorhabditis elegans is an excellent model system in which to study long-distance cell migration in vivo. This chapter describes methods used to study a subset of migratory cells in the hermaphrodite nematode, the distal tip cells. These methods take advantage of the organism’s transparent body and the expression of green fluorescent protein to observe cell migration and behavior. Additionally, the availability of nematode mutants and gene knockdown techniques that affect cell migration allow the analysis and comparison of wild-type and aberrant migratory paths. Methods for nematode growth and maintenance, strain acquisition, observation and live imaging, gene knockdown, and analysis of cell migration defects are covered.
Keywords: C. elegans, Cell migration, Mutants, RNAi, Distal tip cells, Live imaging, Green fluorescent protein
1. Introduction
Caenorhabditis elegans is a relatively simple animal that has a finite number of cells, a transparent body, and that executes larval development in only 36 h. In addition to these advantages, a subset of cells undergo long-distance migrations that are easily observed during larval stages (Table 1), making C. elegans an excellent model organism for studying cell migration. Here, we describe methods to study cell migration in C. elegans, focusing on the migration of hermaphroditic distal tip cells (DTCs). Beginning at the L2 larval stage, two DTCs migrate longitudinally away from the mid-body along the ventral basement membrane, then turn to migrate dorsally, and turn again to migrate longitudinally back toward the mid-body along the dorsal basement membrane. The migratory paths of the DTCs are reflected in the shape of the gonad arms. Wild-type DTC migration yields two U-shaped gonad arms in the hermaphrodite (Fig. 1a, b).
Table 1.
Cells that undergo long-distance migration in the Caenorhabditis elegans hermaphrodite and the gene promoters that are active in these cells
| Migratory cell(s) | Active promoter |
|---|---|
| ALM neurons | mec-4 (16) |
| CAN neurons | ceh-23 (17) |
| QL | unc-73 (18) |
| PQR neuron | osm-6 (19) |
| HSN neurons | unc-86 (20) |
| QR | unc-73 (18) |
| AQR neuron | osm-6 (19) |
| AVM neuron | mec-4 (16) |
| SDQR neuron | unc-119 (21) |
| Embryonic coelomocytes | hlh-8 (22) |
| M cell | hlh-8 (22) |
| Sex myoblasts | hlh-8 (22) |
| Gonadal distal tip cells | lag-2 (23) |
Fig. 1.
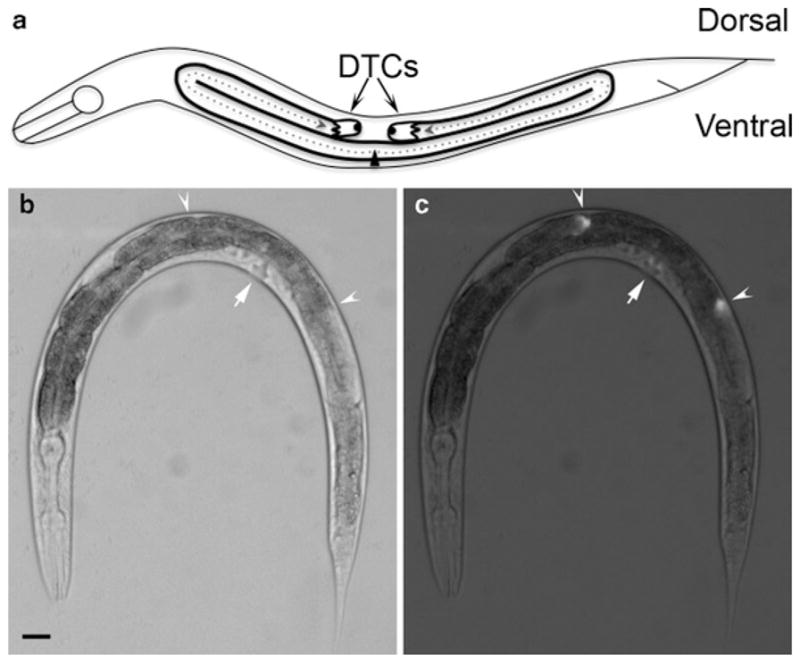
Distal tip cell migration and gonad morphology in the Caenorhabditis elegans hermaphrodite. (a) Schematic diagram of a late L4 animal. At the L2 larval stage, DTCs initiate migration on the ventral side (at the triangle, which represents the vulva) and follow the paths indicated by the dotted arrows. Migration terminates on the dorsal side opposite the vulva, yielding two U-shaped gonad arms. DIC image (b) and fluorescence superimposed on DIC image (c) of an L4 nematode carrying the lag-2p::GFP transgene. The lag-2 promoter is active in DTCs and drives the expression of GFP in these cells. Arrowheads indicate the DTCs and arrows indicate the location of the vulva. Anterior is to the left and ventral is down in all images. Scale bar represents 20 μm.
The transparency of C. elegans combined with the use of the green fluorescent protein (GFP) and other inherently fluorescent protein variants has greatly aided the visualization of migrating cells. A typical GFP transgene consists of a promoter that is active in the migrating cell of interest driving the expression of GFP (Fig. 1c), which then enables the analysis of cell migration in live animals and bypasses fixation procedures. Expression of a cellular protein fused to GFP can also be used to follow protein subcellular localization or to provide a more detailed view of cell shape changes during migration (Table 1, see Note 1).
The ease of forward and reverse genetic approaches in C. elegans allows one to dissect the functions of specific genes during cell migration. Certain genetic mutant strains exhibit cell migration defects, which, when characterized, have revealed the roles of key molecules that are required for cell movement or pathfinding (1–4). In addition, RNA interference (RNAi) is a straightforward approach to reduce the specific expression of a particular gene of interest. The availability of RNAi libraries covering ~94% of the 19,000 predicted ORFs in the C. elegans genome makes it feasible to phenocopy the absence of genes for which mutants have not yet been generated (5–8). Because RNAi can be initiated at different times during development, one can control the knockdown of a gene in a temporal fashion and, therefore, bypass earlier requirements for a gene. For example, there are many genes that are essential for embryonic development, but determining the roles for these genes at later stages of development is complicated by the lethal nature of the mutants. One can allow wild-type animals to develop past the embryonic stage and then reduce gene expression by RNAi during postembryonic development to analyze its role in cell migration. More recently, tissue-specific RNAi strains have been developed based on mutant strains that are unable to respond to RNAi treatment. In such a genetic background, the introduction of a transgene that confers RNAi sensitivity in a tissue of interest allows RNAi to affect a single type of cell or tissue (9).
Phenotypes induced by RNAi depend on the level of knockdown of gene expression and one potential disadvantage of the RNAi technique is incomplete knockdown. Genetic strains with increased sensitivity to RNAi treatment have been characterized, such as the rrf-3(pk1426) mutant strain (10) and the eri-1(mg366) mutant strain, which shows increased effects in most tissues including the nervous system (11).
In general, the study of cell migration in C. elegans relies on imaging (bright-field, differential interference contrast (DIC), fluorescence, and time-lapse video microscopies), cell-labeling (transgenic GFP expression), and genetic manipulation (mutants and RNAi). These topics will be addressed here, as the following sections will describe how to acquire wild-type, mutant, and transgenic C. elegans strains (Subheading 3.1), how to grow and maintain C. elegans in culture (Subheading 3.2), how to observe cell migration in C. elegans using a microscope (Subheading 3.3), how to immobilize nematodes to make cell migration movies (Subheading 3.4), and how to knock down the expression of specific genes using RNAi (Subheading 3.5).
2. Materials
2.1. Nematode Growth and Maintenance
Wild-type, mutant, and transgenic C. elegans strains.
Nematode growth medium (NGM) plates: 2.5 g peptone, 17 g agar, and 3 g NaCl. Add water to a total volume of 1 L and autoclave to dissolve the agar and sterilize the medium. Once the medium has cooled to 55°C, add the following sterile solutions: 1 mL of 1 M CaCl, 1 mL of 1 M MgSO4, 25 mL of 1 M KH2PO4, pH 6.0, 1 mL of 5 mg/mL cholesterol in ethanol, and 5 mL of 10, 000 units/mL nystatin suspension. Stir to mix well. Aliquot agar medium into 60-mm plastic Petri plates (see Note 2).
OP50 Escherichia coli bacteria strain.
Luria broth (LB) bacterial growth medium: 10 g tryptone, 5 g yeast extract, and 10 g NaCl. Add water to a total volume of 1 L, stir to mix, and autoclave to sterilize.
10-mL borosilicate glass pipet and pipet bulb or pipet controller.
A wire worm pick: a 1 in. length of 28-gauge platinum wire melted to the end of a borosilicate glass Pasteur pipet (see Note 3).
A metal spatula.
Alcohol burner.
95% Ethanol.
20°C incubator.
A dissecting microscope.
2.2. Observation of Worms Using Microscopy
Small 2.5-mL pipet bulb.
Borosilicate glass Pasteur pipet.
Glass coverslips (22 × 60 mm).
M9 buffer: 3 g KH2PO4, 6 g Na2HPO4, 5 g NaCl, and 1 mL of 1 M MgSO4. Add water to a total volume of 1 L. Sterilize by autoclaving.
2% Agarose in M9 buffer.
M9 buffer with NaN3: add NaN3 to M9 for a 0.08 M final concentration.
M9 buffer with 0.1% tricaine and 0.01% levamisole: Prepare 10% solutions of tricaine (made fresh every time) and levamisole: 100 mg in 1 mL of M9. Add 10 μL of 10% tricaine and 1 μL of 10% levamisole to 1 mL of M9 (see Note 4).
A microscope with fluorescence and DIC optics and at least 10× and 40× objective lenses. In addition to this equipment, video microscopy requires a 60× objective lens, a computer-controlled motorized focus-drive unit, and camera (see Note 5).
2.3. RNAi Treatment of Worms by Feeding
N2 or RNAi-sensitized worm strains, such as rrf-3 (pk1426) (10), and tissue-specific RNAi strains, such as the hypodermis-specific strain, rde-1(ne219); lin-26p::rde-1 (9).
HT115 (DE3) E. coli strain carrying the pL4440 vector. One clone should carry a control pL4440 vector, which can either be the empty vector or a vector with 500–1,000-bp fragment of GFP, depending on the nature of the experiment. The other clone should contain the pL4440 vector with a 500–1,000-bp fragment of your gene of interest.
LB bacterial growth medium with 100 μg/mL ampicillin.
60 mm NGM plates with 1 mM IPTG.
Bleach solution: 4.5 mL water, 2 mL household bleach (5% solution of NaOCl), and 0.5 mL of 10 M NaOH.
1.5-mL microcentrifuge tubes.
M9 buffer.
Bench-top microfuge.
Vortex.
Bunsen burner.
23°C incubator.
3. Methods
3.1. Acquiring Worm Strains
Wild-type, mutant, and transgenic strains can be requested from the C. elegans Genetics Center (CGC; http://www.cbs.umn.edu/CGC/) or from individual laboratories. To order strains from the CGC, there is a required annual fee and a small fee per strain requested.
3.2. Nematode Growth and Maintenance
C. elegans are grown on OP50 bacterial lawns on NGM plates. OP50 bacteria can also be obtained from the CGC.
3.2.1. Seeding NGM Plates with OP50 Bacteria
Using sterile technique, inoculate LB medium without anti-biotics with OP50.
Grow the OP50 culture for 2 days at room temperature or overnight at 37°C with shaking.
Using a sterile technique, place ~100 μL of the liquid OP50 culture in the center of an NGM plate using a sterile glass pipet.
Spread this volume of OP50 culture using the tip of the pipet (see Note 6).
Allow the bacteria to grow overnight at least.
3.2.2. Transferring Individual Worms Between NGM Plates (“Picking”)
Briefly flame the wire pick to sterilize it and wait for several seconds to let the pick cool.
Using a dissecting microscope, identify the worm you wish to transfer.
Gently slide the flat end of the pick under a single worm and lift it off the agar.
Still using the dissecting microscope, quickly transfer the animal to a new, seeded NGM plate to avoid desiccation of the animal. Gently lower the pick to the agar without disturbing the surface, and let the worm crawl onto the new plate (see Note 7).
Be sure to sterilize the pick each time to avoid bacterial/fungal contamination and cross-contamination of strains.
Incubate the worms at 20°C.
3.2.3. Transferring Large Numbers of Worms Between NGM Plates (“Chunking”)
Dip a clean metal spatula into 95% ethanol and flame to sterilize.
Use the spatula to cut a chunk of the agar from the NGM plate that contains worms.
Remove the chunk of agar and quickly transfer the chunk of agar to a fresh, seeded NGM plate.
Sterilize the spatula between each transfer to avoid bacterial/fungal contamination and cross-contamination of strains.
Incubate the worms at 20°C.
3.3. Observation and Analysis of Cell Migration Using Microscopy
3.3.1. Preparation of an Agarose Pad and Mounting Animals onto the Pad
Heat 2% agarose solution in a microwave oven to melt.
Using a small pipet bulb and a glass Pasteur pipet, drop 2 small drops of the molten agar onto a 22 × 60 mm coverslip.
Quickly place another 22 × 60 mm coverslip over the drop of agarose, but perpendicular to the first cover slip (Fig. 2).
Allow the agarose to solidify, then gently slide the bottom slide out from under the agarose, leaving the agarose pad stuck to the top coverslip. Write on the coverslip with the agarose pad on it to indicate which side the agarose pad is on.
Place 10 μL of M9 with NaN3 on the agarose pad (see Note 8).
Using a wire worm pick and a dissecting microscope, transfer animals at the desired developmental stage to the drop of M9 (see Note 9).
Transfer animals before the drop of M9 evaporates (see Note 10).
Place a coverslip slowly onto the drop of M9 (see Note 11).
Fig. 2.
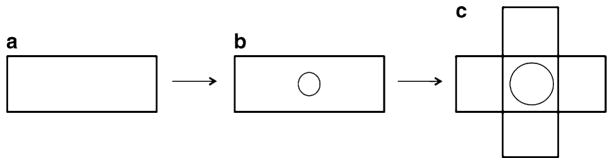
Diagram of the preparation of the agarose pad. (a) A single 22 × 60 mm coverslip. (b) The same 22 × 60 mm coverslip with two drops of molten 2% agarose on it (circle). (c) A second 22 × 60 mm coverslip placed on top of the molten 2% agarose, perpendicular to the first coverslip. The drop of agarose will spread out upon placement of the second coverslip.
3.3.2. Observing Animals Under the Compound Microscope
Place the coverslip with agarose pad and nematodes onto the stage of the microscope. Ensure that the slide has been placed such that animals are being visualized through a coverslip and not the side that has both the coverslip and agarose pad.
Under low (10×) magnification and bright field, locate and focus on an animal. To observe GFP expression in a transgenic animal, switch to the fluorescence source and switch off bright field (see Note 12).
Switch to 40× magnification to observe the cells of interest. This magnification will be needed to observe gonad shape.
Repeat steps 2 and 3 to observe other animals.
3.3.3. Analysis and Scoring of Cell Migration Defects
When scoring migration defects in either mutant or RNAi-treated animals, it is important to have observed wild-type migration of the same cell type. In the case of DTC migration, the shape of the hermaphroditic gonad reflects the DTC migratory path (Figs. 1a and 3).
In wild-type animals, the U-shaped gonad morphology reflects migration of the DTC on the ventral side, the turn from ventral to dorsal, and then migration back toward the vulva along the dorsal side (Fig. 1). Perturbations of DTC migration can result in a variety of phenotypes, including little to no DTC migration (Fig. 4c), excessive DTC migration and turning (Fig. 4d), or pathfinding defects, in which the DTC stops short on the dorsal side (Fig. 4e) or exhibits a meandering migration path, resulting in a gonad arm with a wave-like morphology (Fig. 4f). Observation and analysis of these phenotypes can give important information about potential roles of the gene of interest. For example, gon-1 is required for DTC migration, and worms lacking gon-1 expression exhibit no DTC migration (2).
It is important to score both the penetrance and the qualitative aspects of the phenotypes. Penetrance refers to the more quantitative aspect of the analysis, which is the proportion of cells that exhibit a migratory defect. This information is important to determine the severity of the migration defect and gives information about the role of the gene of interest during cell migration (see Note 13).
When scoring cell migration, it is useful to make drawings (and/or take pictures) of the cell migration pathway, and record the anatomical landmarks in relation to the migrating cells. Keeping a record of the experiment is extremely useful for future analysis.
Fig. 3.

Staging and developmental timing of the Caenorhabditis elegans hermaphrodite. (a–d) DIC images showing vulval morphology and the posterior gonadal arm at different stages of development. Arrowheads indicate the location of the vulva and arrows indicate the progression of DTC migration. Late L3 (a), early L4 (b), mid L4 (c), and late L4 (d) larval stages of N2 hermaphrodites are shown. By late L3 (a, arrow), the DTC has completed the ventral-to-dorsal turn and is starting to migrate on the dorsal side. The DTC continues to migrate in this direction throughout the L4 stage (b–d, arrows). The morphology of the vulva also changes as it develops. The vulval precursor appears as a relatively simple structure during late L3 (a, arrowhead) and progressively becomes more complex during the L4 stage (b–d, arrowheads). Anterior is to the left and ventral is down in all images. Scale bars represent 20 μm. (e) Table showing the temperature dependence of C. elegans developmental timing. Development occurs more rapidly at warmer temperatures (24).
Fig. 4.

Examples of wild-type and defective DTC migration paths and gonad morphologies. (a) Diagram showing one arm of the hermaphrodite gonad. An arrow indicates the DTC. Cuboidal cells are maturing oocytes. Ovals are fertilized embryos. (b–f) DIC images showing wild-type morphology (b) and three types of defects (c–f). Treated rrf-3(pk1426) hermaphrodites were grown on Escherichia coli HT115(DE3) carrying an empty vector (b) or RNAi targeting vectors for act-1 (c), ced-5 (d), pat-3 (e), or dyn-1 (f). Arrows indicate DTC migration paths. Anterior is to the left, ventral is down in all images except (c) and (e) in which anterior is to the right. Bars, 20 μm. Adapted with permission from Journal of Cell Science (http://www.jcs.biologists.org) and originally published in Cram et al. (2006) (doi: 10.1242/jcs.03274) (25).
3.4. Video Microscopy of Cell Migration in Nematodes
Place 15 μL of M9 with 0.1% tricaine and 0.01% levamisole on a 2% agarose pad on a coverslip (see Note 14).
Using a worm pick and a dissecting microscope, transfer animals to the drop of M9 and let sit for 5–10 min for the anesthetic to take effect.
Gently lower a coverslip on top (see Note 15).
Place the slide onto the stage of the microscope.
Locate and focus on a nematode using bright field at a low magnification (10× or 20×). Switch off bright field and turn on the fluorescence source to observe GFP expression.
Using a higher resolution objective (60×), locate and focus on the DTC or other cell of interest (see Note 12).
Switch to the camera and center the cell in the field of view.
Specify the timing interval for the pictures to be taken in both bright-field (DIC) and fluorescence channels and the total number of time points. If multiple focal planes are required, specify the top and the bottom limits for sectioning along the Z-axis, and the distance between focal planes. Depending on specific applications, a sectioning interval of 0.2–1 μm along the Z-axis is convenient for DTC visualization (see Note 16).
Analyze images pertaining to each time point separately to track temporal changes. Merge the DIC and confocal micrographs to observe changes in position of the cell within the context of the nematode body, or analyze images obtained in the confocal channel separately to track changes in cell morphology. For visualization of data in 3D, images in multiple focal planes along the same Z-axis can be represented as maximum intensity projections.
Convert the sequences of image files into movies using available software. We have used Quicktime software with success; however, the choice of software should be based on what is appropriate and available for each specific operating system.
3.5. RNAi Treatment of Worms
This RNAi feeding protocol spans 5 days, from spreading bacteria, inducing expression of double-stranded RNA, nematode egg preparation, hatching and growth on RNAi bacteria, and analysis. The HT115 (DE3) E. coli strain carrying fragments of the C. elegans genome in the pL4440 vector can be ordered from Geneservice™ (http://www.geneservice.co.uk/products/rnai/).
3.5.1. Spreading HT115 Bacteria onto IPTG NGM Plates
Using a sterile technique, inoculate LB containing ampicillin with the appropriate HT115 clone (see Note 17).
Grow the culture overnight and no longer than 18 h at 37°C while shaking (see Note 18).
Using a sterile technique, place 150 μL of the culture in the center of an IPTG containing NGM plate.
Gently move the plate in a brief circular motion to spread the culture, but not so much to have it touch the edge of the plate.
Let the liquid dry and incubate at room temperature overnight to induce expression of double-stranded RNA.
3.5.2. Egg Preparation and RNAi
Rinse a NGM plate of gravid adult hermaphrodites with water. Pipet the water across the plate a few times to loosen all worms from the plate. Then transfer the liquid to a microfuge tube.
Spin the tube at 13793×g for 10 s in a microfuge. Aspirate the supernatant, preserving the loose worm pellet at the bottom.
Add 1 mL of bleach solution to the tube (bleach solution should be made fresh).
Vortex the tube for 2 s at the maximum setting and incubate at room temperature for 3 min. Then vortex the tube again and incubate at room temperature for 2 more min.
Spin the tube at 13793×g for 10 s. Aspirate the supernatant, again preserving the pellet at the bottom.
Add 1 mL M9 buffer to rinse the pellet and spin the tube at 13,000 rpm for 15 s. Aspirate the supernatant.
Repeat step 6.
Resuspend the pellet in approximately 200 μL M9 buffer.
Aliquot between 3 and 5 μL onto a coverslip. Using a dissecting microscope, count the number of eggs on the coverslip to determine the concentration of eggs in M9 buffer.
Depending on the concentration of eggs in the buffer, aliquot the appropriate volume to transfer approximately 200 eggs per plate. Use the IPTG NGM plates that were spread with the RNAi bacteria the day before.
Place in 23°C incubator and incubate for 48–50 h.
Analyze the worms using the methods described in Subheading 3.3.
Acknowledgments
This work was supported by grants from the NIH (R01 GM059383 and NIGMS Cell Migration Consortium U54 GM064346). M.C.W. is supported by a postdoctoral fellowship from the New Jersey Commission on Cancer Research (10-2409-CCR-EO). M.M. was supported by a Predoctoral Training Grant in Genetics and Molecular Biology (T32 GM007388).
Footnotes
Transgenes exist in two forms: extrachromosomal arrays and integrated arrays. Extrachromosomal arrays (Ex in C. elegans nomenclature) consist of multiple repeats of the transgene and are not integrated into the genome. The transmission of extrachromosomal arrays is variable, and therefore selecting individuals carrying the GFP transgene to propagate the strain is necessary for maintenance. Integrated arrays (Is in C. elegans nomenclature) are transgenes that have been integrated into the genome. Once the integrated array is homozygous, all animals should express GFP.
By aliquoting 12 mL of medium in each 60-mm Petri plate, you can expect to make approximately 100 NGM plates. Also, by aliquoting an equal amount of NGM media into each plate, the depth of agar in all plates is similar, which eliminates large adjustments in focus when switching between plates to look at worms under the dissecting microscope.
When fashioning a worm pick, flatten the free end of the platinum wire. This flattened end can be used as a scoop to pick up animals and allows them to easily crawl off the pick onto the agar.
Sodium azide treatment stops nematode movement completely, but it is toxic to the animal. Tricaine and levamisole are less effective than azide at inducing paralysis, but nematodes can be rescued after prolonged anesthesia (also see Note 14). To recover anesthetized animals, apply plenty of M9 by placing the tip of the pipet next to the edge of the top coverslip, gently slide the coverslip off, and add another drop of M9 on top of the nematode of interest. Wait until the worm starts moving before placing it on a plate with food.
Spinning disk confocal microscopy reduces the danger of phototoxicity or photobleaching and is advisable for live nematode imaging experiments lasting for several hours.
When spreading the OP50 liquid culture on NGM plates using the tip of a glass pipet, be sure that the tip of the pipet is smooth and does not break the surface of the agar. Nematodes will burrow into the agar at a broken surface, making observation and recovery of these worms difficult. Also, avoid spreading the bacteria to the edge of the plate to make it less likely that worms will crawl up the side of the plate, dry out, and die.
Alternately, the pick can be used to pick up a small amount of bacteria. Worms can stick to bacteria on a pick and can be transferred to a new plate by touching the bacteria and worm to the agar, allowing the worm to crawl away.
Agarose pads help immobilize the specimen for observation.
The staging of the animal is crucial when analyzing the end point of cell migration. It is important that animals with aberrant phenotypes are compared to wild-type phenotypes at the same stage since gonad size and the position of the ventral-dorsal turn are related to overall body size. DTC migration is best scored at the late L4 stage, which is the stage in which DTCs have completed their migration (Fig. 3). Depending on which cell types are being scored, C. elegans postembryonic staging can be done by observing vulval development (Fig. 3).
If possible, move more than one animal at a time. Also, shake or quickly drag the wire pick in the M9 drop to release worms rapidly into the drop, as opposed to letting the worm swim off the pick, which is a slower process. If necessary, add another drop of M9 if you notice evaporation; however, try to keep this to a minimum to avoid concentrating the M9 buffer too much. It is important to not lose too much of the M9 volume to avoid bubbles forming between the coverslips. Bubbles can impede proper visualization of the animals.
Try to drop one side of the coverslip over the animals first and then slowly lower the coverslip from one end to the next. This can prevent bubble formation.
DTC observation can be facilitated by using the lag-2p::GFP strain. If using this strain, switch off the bright field and turn on the fluorescence source to locate the DTCs.
In some cases, the phenotype will be subtle and a more careful quantification is necessary for scoring cell migration defects. For example, a subset of defects exhibit a U-shaped path back to the dorsal mid-body, but the path is shorter than that of wild type. This means that the ventral to dorsal turn occurs at a location that is closer to the mid-body than in a wild-type animal. This difference in size can be quantified by determining the ratio of the length of the ventral migration path (from the vulva to the turn) to the length of the worm (from the vulva to either the tip of the tail or the tip of the head). The ratio between these two values can be compared between wild-type animals and genetically manipulated animals to determine whether the migratory paths of DTCs are shortened.
Tricaine plus levamisole anesthetic treatment provides an amenable and straightforward way for preventing nematode movement; however, it also slows down nematode developmental processes. For example, the DTC ventral to dorsal turn is completed in 1 h in untreated L3 larvae, but requires ~6 h in anesthetized hermaphrodites. A similar effect has been observed in neuron migration (12). The recommended concentration is optimal for keeping animals alive, but paralyzed for several hours. Nematodes can survive anesthesia for 9 h or more and be recovered for subsequent analysis. Also, nematodes can be re-anesthetized after at least 2 h of recovery. In addition to anesthesia, nematodes can be immobilized by other methods that require special equipment (cooling to 4°C or microfluidic chambers) or do not allow recovery of animals after analysis (gluing) (12–15).
In order to prevent desiccation, seal the coverslip with Vaseline around the periphery of the agarose pad, leaving one corner open for aeration.
Nematodes treated with levamisole and tricaine will exhibit slight movements that may change the focal plane of the DTC. Readjust the settings every hour if necessary. The specimen should not be illuminated in between photographic recordings to avoid heat shock and/or photobleaching.
When planning for an RNAi experiment, it is useful to have both a negative and a positive control. HT115 clones carrying an empty L4440 plasmid or with a fragment encoding GFP can be used as a negative control. When scoring DTC migration, a useful positive control is a clone carrying a fragment of the gene gon-1 in the L4440 plasmid. C. elegans treated with gon-1 RNAi exhibit DTCs that are unable to migrate, resulting in dramatically truncated gonad arms. This phenotype is easily scored using a dissecting microscope.
Overgrowth of RNAi bacterial strains for longer than 18 h results in the reduction of RNAi effect.
References
- 1.Nishiwaki K. Mutations affecting symmetrical migration of distal tip cells in Caenorhabditis elegans. Genetics. 1999;152:985–997. doi: 10.1093/genetics/152.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blelloch R, Anna-Arriola SS, Gao D, Li Y, Hodgkin J, Kimble J. The gon-1 gene is required for gonadal morphogenesis in Caenorhabditis elegans. Dev Biol. 1999;216:382–393. doi: 10.1006/dbio.1999.9491. [DOI] [PubMed] [Google Scholar]
- 3.Meighan CM, Schwarzbauer JE. Control of C. elegans hermaphrodite gonad size and shape by vab-3/Pax6-mediated regulation of integrin receptors. Genes Dev. 2007;21:1615–1620. doi: 10.1101/gad.1534807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2:131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- 5.Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- 6.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 7.Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, Vidal M. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 2004;14:2162–2168. doi: 10.1101/gr.2505604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahringer J The C. elegans Reseach Community, WormBook, editor. Reverse genetics. WormBook. 2006 Jun 6; doi: 10.1895/wormbook.1.47.1. http://wormbook.org. [DOI]
- 9.Qadota H, Inoue M, Hikita T, Koppen M, Hardin JD, Amano M, Moerman DG, Kaibuchi K. Establishment of a tissue-specific RNAi system in C. elegans. Gene. 2007;400:166–173. doi: 10.1016/j.gene.2007.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RH. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12:1317–1319. doi: 10.1016/s0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645–649. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- 12.Knobel KM, Jorgensen EM, Bastiani MJ. Growth cones stall and collapse during axon outgrowth in Caenorhabditis elegans. Development. 1999;126:4489–4498. doi: 10.1242/dev.126.20.4489. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki H, Kerr R, Bianchi L, Frokjaer-Jensen C, Slone D, Xue J, Gerstbrein B, Driscoll M, Schafer WR. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron. 2003;39:1005–1017. doi: 10.1016/j.neuron.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Rohde CB, Zeng F, Gonzalez-Rubio R, Angel M, Yanik MF. Microfluidic system for on-chip high-throughput whole-animal sorting and screening at subcellular resolution. Proc Natl Acad Sci USA. 2007;104:13891–13895. doi: 10.1073/pnas.0706513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Podbilewicz B, Gruenbaum Y. Live Imaging of Caenorhabditis elegans: Preparation of Samples. Cold Spring Harb Protoc. 2006 doi: 10.1101/pdb.ip19. [DOI] [PubMed] [Google Scholar]
- 16.Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature. 1991;349:588–593. doi: 10.1038/349588a0. [DOI] [PubMed] [Google Scholar]
- 17.Forrester WC, Perens E, Zallen JA, Garriga G. Identification of Caenorhabditis elegans genes required for neuronal differentiation and migration. Genetics. 1998;148:151–165. doi: 10.1093/genetics/148.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honigberg L, Kenyon C. Establishment of left/right asymmetry in neuroblast migration by UNC-40/DCC, UNC-73/Trio and DPY-19 proteins in C. elegans. Development. 2000;127:4655–4668. doi: 10.1242/dev.127.21.4655. [DOI] [PubMed] [Google Scholar]
- 19.Collet J, Spike CA, Lundquist EA, Shaw JE, Herman RK. Analysis of osm-6, a gene that affects sensory cilium structure and sensory neuron function in Caenorhabditis elegans. Genetics. 1998;148:187–200. doi: 10.1093/genetics/148.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finney M, Ruvkun G. The unc-86 gene product couples cell lineage and cell identity in C. elegans. Cell. 1990;63:895–905. doi: 10.1016/0092-8674(90)90493-x. [DOI] [PubMed] [Google Scholar]
- 21.Maduro M, Pilgrim D. Identification and cloning of unc-119, a gene expressed in the Caenorhabditis elegans nervous system. Genetics. 1995;141:977–988. doi: 10.1093/genetics/141.3.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harfe BD, Vaz Gomes A, Kenyon C, Liu J, Krause M, Fire A. Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes Dev. 1998;12:2623–2635. doi: 10.1101/gad.12.16.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henderson ST, Gao D, Lambie EJ, Kimble J. lag-2 may encode a signaling ligand for the GLP-1 and LIN-12 receptors of C. elegans. Development. 1994;120:2913–2924. doi: 10.1242/dev.120.10.2913. [DOI] [PubMed] [Google Scholar]
- 24.Epstein HF, Shakes DC, editors. Caenorhabditis elegans: Moderm Biological Analysis of an Organism. Vol. 48. Academic Press, Inc; San Diego: 1995. [Google Scholar]
- 25.Cram EJ, Shang H, Schwarzbauer JE. A systematic RNA interference screen reveals a cell migration gene network in C. elegans. J Cell Sci. 2006;119:4811–4818. doi: 10.1242/jcs.03274. [DOI] [PubMed] [Google Scholar]