

Abstract
Alzheimer’s disease (AD) is a progressive and fatal neurodegenerative disorder marked by memory impairment and cognitive deficits. A major component of AD pathology is the accumulation of amyloid plaques in the brain, which are comprised of amyloid beta (Aβ) peptides derived from the amyloidogenic processing of the amyloid precursor protein (AβPP) by β- and γ-secretases. In a subset of patients, inheritance of mutations in the AβPP gene is responsible for altering Aβ production, leading to early onset disease. Interestingly, many of these familial mutations lie within the transmembrane domain of the protein near the GxxxG and GxxxA dimerization motifs that are important for transmembrane interactions. As AβPP dimerization has been linked to changes in Aβ production, it is of interest to know whether familial AβPP mutations affect full-length APP dimerization. Using bimolecular fluorescence complementation (BiFC), blue native gel electrophoresis, and live cell chemical cross-linking, we found that familial Alzheimer’s disease (FAD) mutations do not affect full-length AβPP dimerization in transfected HEK293 and COS7 cells. It follows that changes in AβPP dimerization are not necessary for altered Aβ production, and in FAD mutations, changes in Aβ levels are more likely a result of alternative proteolytic processing.
Keywords: Alzheimer’s disease, amyloid-β precursor protein, familial Alzheimer’s disease, amyloid beta-peptides, protein dimerization
Introduction
The amyloid precursor protein (AβPP) is a single-pass transmembrane glycoprotein that is expressed in a wide variety of tissues [1], and in the brain, it is concentrated at the synapses of neurons [2]. The biological function of the protein is unclear, but it appears to be involved in platelet aggregation [3], metal homeostasis [4-6], cellular adhesion and cell-cell communication [7], as well as a host of neuronal processes including cellular growth, differentiation, migration, arborization, synaptic transmission, axonal transport, memory formation, and neuroprotection [2,8-17]. Proteolytic processing of AβPP by β-secretase, also known as the β site AβPP cleaving enzyme (BACE1), or memapsin-2 [18,19], followed by γ-secretase, a large multi-subunit complex comprised of presenilin (PS), nicastrin (Nct), anterior pharynx defective (Aph-1) and the presenilin enhancer (Pen-2) [20], is well established to generate amyloid beta peptides (Aβ). In Alzheimer’s disease (AD), aberrant production and aggregation of Aβ leads to formation of amyloid plaques, a pathologic feature that contributes to severe neurodegeneration.
The structure of AβPP resembles a cell surface receptor [21] and can form homodimers [21,22] as well as heterodimers with interacting ligands such as Notch [23-25] and the amyloid precursor like proteins 1 and 2 (APLP1 and 2) [7,26]. This is similar to many receptor proteins that participate in ligand-induced activation; therefore, AβPP dimers may be important in signal transduction for subsequent gene transcription regulation [27-29], but may also have other unidentified biologically significant effects. Dimerization of AβPP at the ectodomain is mainly regulated by its E1 growth factor like domain that possesses a loop region of disulphide bonds [30], its E1 metal-binding domain that coordinates metal ions such as copper and zinc, and its E2 collagen binding domain that adheres to extracellular matrix molecules such as heparin, collagen [28,31,32] and possibly N-cadherin [33]. Most notably, a dimerization site also is located at the juxtamembrane/transmembrane domain (TMD) in which there are three consecutive GxxxG motifs (Aβ residues 25-37) [34] that are followed immediately by a GxxxA motif [35]. Both GxxxG and GxxxA are common helical structural patterns important for protein-protein interactions and can be found in many proteins including Glycophorin A, from which the GxxxG motif was first identified [36,37].
Various studies have shown that AβPP dimerization may influence Aβ production but the effect on Aβ levels is debatable. The initial work reported that stable AβPP dimers formed by disulphide bonds at the juxtamembrane region (AβPP695 K624C mutation) result in an increase of Aβ production by 6-8 fold [22]. In contrast, using an FKBP/rapamycin system to induce AβPP dimers results in 50% reduction in Aβ when up to 70% of AβPP is in dimer form [38]. Most studies on the TMD GxxxG/GxxxA dimerization motif have been conducted by mutational analysis with the resulting effect of decreased Aβ levels, although the mutations themselves may affect AβPP processing such that the effects on Aβ are independent of dimerization in mutational analysis [38] and interpreting such results should be done with caution. Interestingly, mutation to isoleucine (GxxxI) seems to disrupt TMD dimerization under a ToxCAT system [34], while mutations to isoleucine and leucine (GxxxL/GxxxI), but not alanine (GxxxA), enhance dimerization at the C-terminal fragment (CTF). Thus, the effects of AβPP processing and Aβ production may also be dependent on the precise composition and orientation of the AβPP dimers [39]. Further investigations into the TMD found that some Aβ lowering NSAIDs can bind to the GxxxG dimerization motif [40]. In particular, sulindac sulfide and its derivatives destabilize the AβPP TMD dimer in a concentration dependent manner, which correlates with lowered Aβ production [41]. Additionally, introducing mutations at the GxxxG motif in AβPP mutants that cause early onset Alzheimer’s disease decrease Aβ levels and rescue the effects of the disease [42]. These data suggest that the AβPP TMD GxxxG/GxxxA dimerization motifs play a critical role in the generation of Aβ despite uncertain outcomes. Thus, we focused our attention on the TMD GxxxG/GxxxA dimerization motif.
Inheritance of familial mutations in the AβPP gene can exacerbate Aβ production or shift the Aβ42/40 ratio, leading to an early onset of Alzheimer’s disease (AD) [43]. There are many documented familial kindreds with AβPP mutations, which include the A692G Flemish [44], K670N/M671L Swedish [45], T714I Austrian [46], T714A Iranian [47], V715A German [48], I716V Florida [49], V717I London [50], V717L Indiana [51], L723P Australian [51], and the V715M French mutations [52]. Interestingly, most of these mutations are located within the transmembrane domain of AβPP, near the GxxxG and GxxxA dimerization motifs. We are interested in determining whether these familial AβPP mutations affect Aβ levels by altering AβPP dimerization state. A similar study has been conducted and found that familial Alzheimer’s disease (FAD) mutations destabilize TMD dimerization and that dimerization negatively correlated with disease onset and Aβ production. However, this study was carried out in micelles and phospholipid bilayers, and only examined the TMD segment [35]. We would like to determine whether these familial mutations affect full-length APP dimerization in vitro as this is more biologically relevant. For this investigation, we employed three different methods of analysis: bimolecular fluorescent complementation (BiFC), blue native (BN) gels, and protein cross-linking. Results of this study provide evidence that FAD AβPP mutations do not affect full-length AβPP dimerization and thereby AβPP dimerization does not appear to account for the changes in Aβ production in early onset FAD.
Materials and methods
AβPP FAD mutants constructs
A wild type (WT) APP751 insert was previously cloned into a pcDNA3 mammalian expression vector (Invitrogen) [25]. Using the WT APP751 plasmid as a template, various familial AβPP mutants were made by using designed sense and anti-sense primers (Integrated DNA technologies) via site-directed mutagenesis using the QuikChange Mutagenesis Kit (Strategene). The primer sets for the different mutants are listed as follows: Austrian T714I sense (5’ – GTGT TGTCATAGCGATAGTGATCGTCATCAC – 3’) and anti-sense (5’ – GTGATGACGATCAC TATC-GCTATGACAACAC – 3’), French V715M sense (5’ – GTTGTCATAGCGACAATGAT CGTCATCACCT – 3’) and anti-sense (5’ – AGGTGATGACGATCAT-TGTCGCTATGACAA C – 3’), and London V717I sense (5’ – ATAGCGACAGTGATCATCATCACCTT-GGTGA – 3’) and anti-sense (5’ – TCACCAAGGTG-ATGATGATCACTGTCGCTAT – 3’). The sequences of all cloned plasmids were verified at the Tufts University Core Facility DNA Sequencing Lab, Boston, MA.
AβPP-venus bimolecular fluorescence complementation (BiFC) constructs
The Venus pBiFC VN173 and pBiFC VC155 were gifts from Dr. Chang-Dang Hu from Purdue University [53]. Cloning of the Venus fragments into WT APP751 pcDNA1 was carried out previously in our lab [23]. Mutant AβPP Venus N-terminus fragment (VN) and AβPP Venus C-terminus fragment (VC) plasmids were constructed using the previously designed primers. The AβPP insert is between a HindIII and SalI site, while the VN and VC inserts are between a SalI and NotI site. A 13 amino acid linker region (STVPRARDPPVAT) is inserted between AβPP and the VN or VC inserts. The AβPP YFP plasmid was also cloned previously and shares the same cloning sites [23].
Cell culture and transfection
HEK293 and COS7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (CellGro) supplemented with 10% fetal bovine serum (FBS) (Atlantic Biologicals) and 1% penicillin-streptomycin (CellGro) at 37°C and 5% CO2. The day before transfection, cells were split using 0.25% Trypsin-EDTA into 6 well plates at a density of 1x105 cells/well. DNA was transfected into cells using Nanofect (Qiagen). Forty-eight hours post transfection, cells were lysed in lysis buffer (100mM potassium phosphate, pH7.8; 0.2% Triton X-100; Complete mini protease cocktail inhibitor tablet (Roche)). Lysates were stored at –80°C until used.
BiFC-flow cytometry
Transfected cells were washed twice with 1x PBS and then trypsinized with 500μL Trypsin-EDTA for 5min at 37°C. The trypsinized cells were then transferred to 1.5mL centrifuge tubes and centrifuged at 344g at 4°C. The supernatant was then removed and discarded, and the cells were again washed twice with 1x PBS on ice. Cells were re-suspended into 1mL of 1x PBS and transferred into 5mL Polystyrene round-bottom tubes (BD Falcon) on ice. Measurement of BiFC signal was carried out using the FACScan flow cytometer (BD Biosciences, San Jose, CA, USA) at the Boston University Core Laboratories & Facilities Flow Cytometry Core. For each sample, a total of 20,000 cells were counted and the Cell Quest Pro Software (BD Biosciences) was used to acquire data. Cells were then analyzed using the Summit Software (DakoCytomation) where cells that exhibited forward and side scatter features typical of live cells were gated.
Fluorescence confocal microscopy
Cells were grown and transfected on cover glasses in 6-well plates. 48 hours after transfection, cells were fixed in 100% ice-cold ethanol for 10min. Then, 300nM DAPI in PBS (Invitrogen) was added to the cells for nuclear staining for 10min. Samples were mounted on slides using 90% glycerol in 100mM Tris (pH 8.0) and were kept at 4°C until used. For obtaining images, the Zeiss LSM510 at the Boston University Confocal facility was used. The excitation and emission wavelengths were for Venus – 515/528nm (yellow/green) and DAPI – 372/456 (blue). To excite the fluorophores, the lasers used included the 488nm FITC laser for the Venus BiFC green emission and 405nm UV laser for DAPI emission.
Sodium dodecyl sulphate - polyacrylamide (SDS-PAGE) electrophoresis and western blotting
Protein concentration in cell lysates was measured by the Pierce Bicinchoninic Acid (BCA) Assay in accordance with manufacturer’s protocol. 10μg of total protein was denatured in Laemmli sample buffer and separated on 8% Tris-Glycine gels at 150V for 60min. Proteins were then transferred onto 0.4μm Immobilon Hybridization nitrocellulose filter membranes (Millipore). Primary antibody for western blots was 1:1000 mouse monoclonal 6E10 (Covance) against amino acids 1-17 of Aβ but also recognizing full length AβPP and sAβPPα. Secondary antibodies used for western blots were 1:5000 peroxidase labeled goat anti-mouse IgG (H+L) The Supersignal West Pico Chemiluminescent Substrate (Thermo scientific) was added to the membrane for 5min and the blots were then developed on Phoenix Research Products F-BX810 Blue X-Ray Films.
Blue native (BN) gel analysis
The Invitrogen NativePAGE Novex 4-16% Bis-Tris Gel System was used in accordance with the manufacturer’s protocol. Transfected cells were solubilized in 250μL of lysis buffer with a final concentration of 1% digitonin, and a protease inhibitor cocktail consisting of 50μg/mL PMSF, 1μg/mL Aprotitin, 1μg/mL Leupetin, 1μg/L Pepsatin in 100% ethanol on ice. Cell lysates were centrifuged at 16,873g for 5min at 4°C, and the supernatant was removed and transferred to a new microcentrifuge tube with the cell debris discarded. Lysates were stored at –80°C until used. For separation, samples were prepared in 30-40μL total volume in Millipore water, with 10μg protein and 1μL of the provided 5% G-250 sample additive. The NativeMARK unstained protein standard (Invitrogen) was used as a protein ladder. Gels were run at 150V at room temperature for about 1.5 hours using light blue cathode buffer for the first 1/3 of the run, followed by dark blue cathode buffer for the rest of the run. Following electrophoresis, gels were incubated with 0.1% SDS for 15min to facilitate transfer of proteins onto membranes. Proteins were then transferred onto Immoblion 0.4μM PVDF membranes (Millipore) at 25V for 1 hour. After transfer, the PVDF membranes were incubated in 8% acetic acid for 15min to fix proteins. Finally, the membranes were washed with methanol to rinse off the Coomassie blue stain and were used for western blot and film development as previously described.
Live cell chemical cross-linking
Live cell cross-linking was carried out in accordance with the method published by Chen et al. [23]. Transiently transfected cells in 6-well plates were washed twice with 1mL of 1x DPBS (Dulbecco’s Phosphate-Buffered Saline) and incubated with 1mL of 100μM dithiobis succinimidylpropionate (DSP), a thiol-clevable and amine-reactive cross-linker, for 30min at room temperature. After that, the cross-linking solution was removed and 1mL of 50mM Tris buffer (pH7.4) was added to quench the reaction. After quenching, cells were lysed with 250μL lysis buffer (1% Nonidet P-40, 0.1% SDS). The lysates were collected as previously described and were stored at –80°C until used.
Data analysis
Experimental data were plotted using the GraphPad Prism 5 software and were analyzed using one way analysis of variance (ANOVA) to test for statistical significance across samples. Dunnett’s test was used post-hoc to determine which specific comparison is significant. Protein expression in Western blots were assessed and normalized by densitometry using ImageJ.
Results
Development of the AβPP – venus bimolecular fluorescence complementation system (BiFC)
Previously our lab has developed the AβPP-YFP BiFC system to monitor AβPP-AβPP and AβPP-Notch interactions [23]. In this study, we employed the same technique, but using a new fluorescent protein, Venus, which is not temperature sensitive in its maturation into a fluorophore and is 13-fold brighter than YFP, making it more suitable for in vitro assays [53,54]. The method of BiFC involves splitting the Venus protein into two over-lapping fragments: one corresponding to the N-terminal end (amino acids 1-172) and one corresponding to the C-terminal end (amino acids, 155-238) [53,55]. Both fragments are then separately tagged to the C-terminal end of AβPP. When AβPP is in monomeric form, the tagged Venus fragments are not fluorescent, as they do not constitute a full Venus protein. However, upon AβPP dimerization, the fragments are brought close enough to complement each other and re-constitute into a fully fluorescent protein. The fluorescence generated can then be correlated to the amount of AβPP dimerization. A schematic of the system is depicted in Figure 1A.
Figure 1.
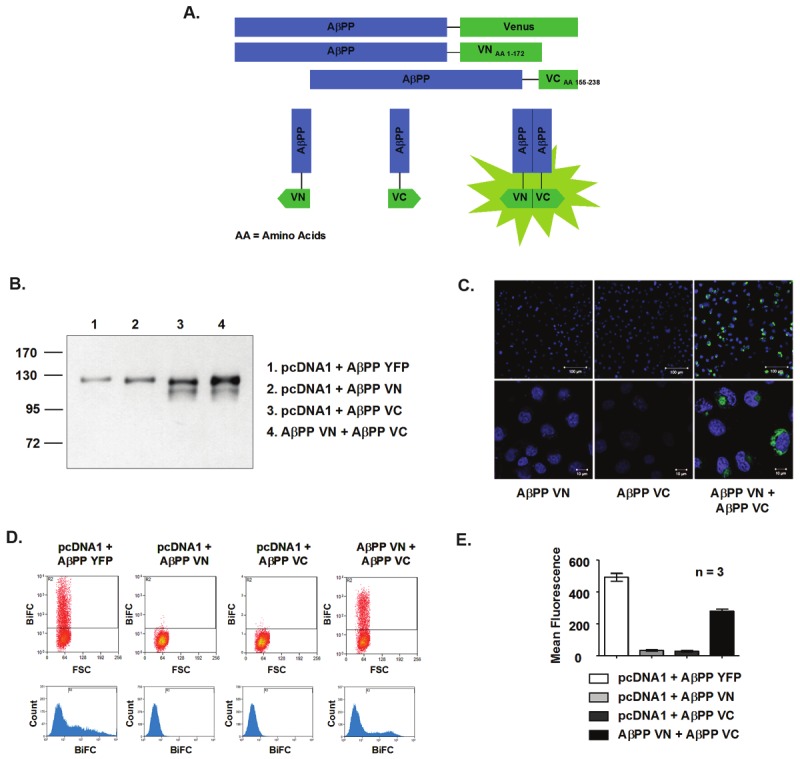
Development of the AβPP-Venus BiFC system. A. Diagram of the BiFC System. Venus is split into two fragments, VN173 (amino acids 1-172) and VC155 (amino acids 155-238), and separately tagged to the C-terminal end of AβPP. These fragments do not fluoresce when AβPP is in monomeric form. However, when AβPP dimerizes, fluorescence is seen when the two protein fragments come into the proximity of each another. B. Representative blot showing protein expression of BiFC constructs with the approximate molecular weights in HEK293 cell lysates. C. BiFC as visualized by fluorescence microscopy in COS7 cells. Blue = nuclear staining with DAPI. Green = fluorescence of BiFC. AβPP VN or AβPP VC alone does not give off fluorescence. Top Magnification = 40x, scale bar = 100μM. Bottom Magnification = 60x Oil, scale bar = 10μM. D. BiFC fluorescence detected by flow cytometry in HEK293 cells. Top: Dot plot of BiFC vs. Forward Scatter (FSc). Boxed cells in Gate R2 show the proportion of cells with fluorescence. Bottom: Representative histogram showing distribution of fluorescence intensity in total gated cells. E. Representative plot of mean fluorescence from BiFC flow cytometry. Samples of AβPP VN and AβPP VC alone emit only background signal. The AβPP YFP control shows high fluorescence, while the AβPP VN + AβPP VC sample gives off fluorescence that is significantly higher than background. Results are plotted as mean ± standard error, n = 3.
To assess protein expression, the AβPP-Venus BiFC plasmids were transiently transfected into HEK293 or COS7 cells and the cell lysates were subsequently run on SDS-PAGE gels followed by western blotting. The expected molecular weight of AβPP YFP (as well as AβPP Venus) is ~140kDa, with APP being ~110kDa and YFP/Venus being ~30kDa. AβPP VN is predicted to be ~130kDa, with VN consisting of 172 amino acids of the fluorescent protein with a molecular weight of ~21.7kDa. AβPP VC is predicted to be ~120kDa, with VC consisting of 83 amino acids of the fluorescent protein with a molecular weight of ~10.4kDa. When AβPP VN and AβPP VC are co-transfected into cells, bands corresponding to each of the different proteins should be seen. Figure 1B is a representative blot of the different BiFC constructs from HEK293 cell lysates. Notice that the molecular weights shown differ slightly from the expected molecular weights, which could be due to the properties of the gel or running conditions. Also, an extra band is seen for the AβPP VC plasmid, which may be an immature form of AβPP, AβPP without the Venus tag, or a breakdown product. Despite these results, the molecular sizes of the proteins are consistent for all other experiments, and the overall size differences between the different proteins are as expected. Accordingly, it appears that the Venus BiFC constructs can be expressed in cells and that they are expressed properly.
The AβPP-Venus BiFC system can detect AβPP dimerization qualitatively using fluorescence microscopy. As shown in Figure 1C, cells that are transiently transfected with the AβPP VN or AβPP VC fragments alone do not display fluorescence, as expected. The blue nuclear DAPI staining illustrates that cells are present despite an absence of signal from the Venus fragments. When cells are co-transfected with both AβPP VN and AβPP VC, the tagged VN and VC fragments come together to form a fully fluorescent Venus protein upon dimerization, and green fluorescence is detected.
The AβPP-Venus BiFC system can also be used to detect AβPP dimerization quantitatively using flow cytometry. In Figure 1D, gated areas in the dot plots and histograms show fluorescent cells in the AβPP YFP control as expected. Fluorescent cells are also seen in cells co-transfected with both the AβPP VN and AβPP VC plasmids (AβPP Venus BiFC), representing AβPP dimerization. In contrast, samples of AβPP VN or AβPP VC alone do not show fluorescent cells in the gated areas. Figure 1E plots the mean fluorescence for the AβPP-Venus BiFC samples for an average of 3 sets of experiments in HEK293 cells. For AβPP YFP, the mean fluorescence was 493.7 ± 24.53; for AβPP VN, 33.92 ± 2.2; for AβPP VC, 29.79 ± 3.12, and for AβPP BiFC, 282.0±11.5. Mean fluorescence represents the average amount of fluorescence in a cell, or for the BiFC samples, the average amount of AβPP dimers in a cell. Overall, the data presented provides evidence that the AβPP-Venus BiFC system developed can be used as a method to compare WT and FAD mutant AβPP dimerization.
Comparison of WT and familial Alzheimer’s disease mutant AβPP dimers using BiFC-flow cytometry
BiFC-flow cytometry was conducted for WT and FAD AβPP mutants to evaluate whether the mutations affect dimerization. Three mutants, Austrian, French, and London AβPP, were selected to exemplify mutations located near the TMD GxxxG motif and a forth mutant, K624C, was used as a positive control for increased dimerization [22]. A schematic diagram showing the location of these mutants in AβPP is shown in Figure 2. pcDNA1 and AβPP YFP were used as negative and positive controls, respectively, for fluorescence to confirm that the BiFC system is functional. Figure 3A shows representative dot plots for the various constructs. For the pcDNA1 negative control, no fluorescent cells are present in the gated area. For the AβPP YFP positive control, a large proportion of fluorescent cells are within the gate. The WT and mutant AβPP constructs all show varying amounts of fluorescent cells, indicating that they are all capable of forming dimers. In Figure 3B, the BiFC-flow cytometry data obtained from dot plots are displayed as % mean fluorescence relative to WT AβPP BiFC, with WT set at 100%. An average of three independent experiments from HEK293 cells are shown. Protein expression of the various constructs is illustrated in the representative blot in Figure 3C. Except for the K624C mutant that forms stable dimers, all FAD AβPP mutants have comparable mean fluorescence which is not significantly different from that of WT AβPP following normalization to protein expression (K624C: 205.6±15.7, *p<0.05; Austrian: 134.4±10.6; French: 109.0±21.3; London: 116.4±18.3). The results were observed in both HEK293 cells and COS7 cells and suggest that the FAD mutants do not affect dimerization. Interestingly, in COS7 cells (data not shown), no differences were seen between WT and K624C even after normalization to protein expression. No differences were seen in HEK293 prior to normalization as well. There could be various reasons for this, which will be communicated in the discussion section. To further validate these findings, we used two additional methods to monitor protein-protein interactions.
Figure 2.
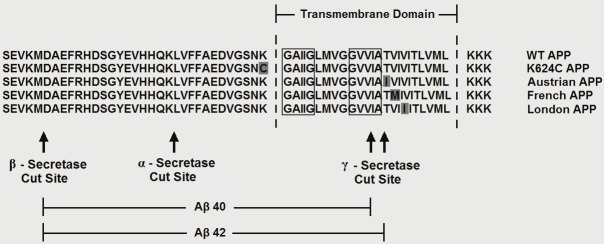
Schematic diagram showing the proximity of the FAD AβPP mutantions to the dimerization motifs in the TMD. The sequence of WT AβPP, the three FAD AβPP mutants examined as well as that of a positive control for AβPP dimerization (K624C) are shown in reference to the TMD, the GxxxG and GxxxA dimerization motifs and the secretase cut sites. The dimerization motifs are indicated with boxes and mutant amino acids are shown in grey.
Figure 3.

Comparison of WT and FAD AβPP mutant dimerization by BiFC-flow cytometry. A. Representative BiFC-flowcytometry dot plots showing the proportion of fluorescent cells or AβPP dimers of WT and familial mutant AβPP constructs,as well as a positive control for AβPP dimerization (K624C), in HEK293 cells. B. BiFC mean fluorescencedata of the various AβPP constructs are plotted as a % of WT AβPP BiFC normalized to protein expression. Resultsare expressed as mean ± standard error, n = 3. Significant differences are *=p≤0.05. C. Representative westernblot showing protein expression of the various AβPP constructs.
Comparison of WT and familial Alzheimer’s disease mutant AβPP dimers using live cell chemical cross-linking
To confirm the BiFC results, chemical cross-linking using DSP was carried out to compare the monomers and dimers found in WT and FAD AβPP mutant transfected cells. The chemical cross-linking method allows examination of the ratio of monomers to dimers, which was not quantifiable under the BiFC method, as the green fluorescence represents only the average amount of dimers presented in a cell, and not the total amount of AβPP. This ratio may only be calculated using the BiFC method if a second fluorescent tag is added to all AβPP proteins. Figure 4A and 4B show representative blots from cross-linked HEK293 and COS7 cells, respectively. The expected molecular weight of AβPP monomers is ~110kDa and APP dimers is ~220kDa in SDS-PAGE gels. Under chemical cross-linking and without the reducing agent β-mercaptoethanol (β-ME), all transiently transfected AβPP samples display monomer, dimer, and tetramer bands, with the monomers being the predominant form. This is consistent with other studies which have also found that AβPP does not form significant levels of dimers [22,34,38]. Nevertheless, the dimer band for the K624C mutant appears to have the highest intensity among all AβPP samples, which is to be expected, as it is a mutant that favors dimer formation. On the other hand, the WT and familial AβPP mutants display similar intensities of the dimer band, indicating again that WT and mutant AβPP have comparable amounts of dimers produced. In the presence of β-ME, most of the higher molecular weight forms of the protein disappeared, indicating cross-linking by disulphide bonds and confirming that the live cell cross-linking method by DSP is functional.
Figure 4.
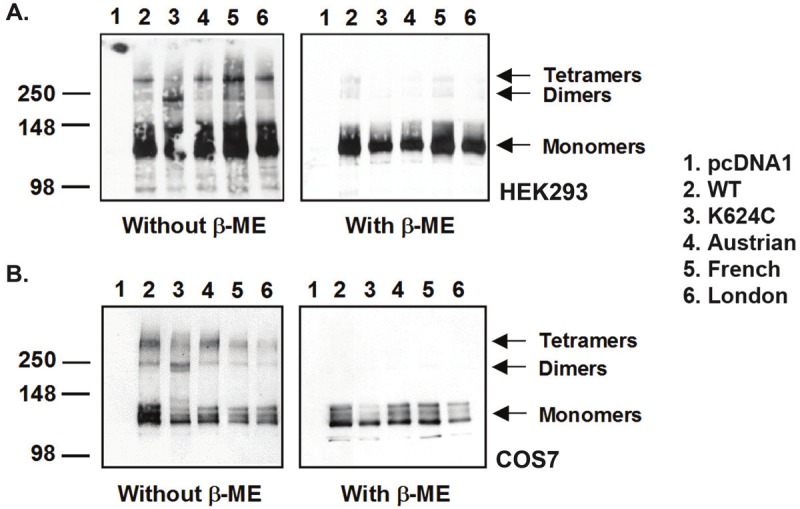
Comparison of WT and FAD AβPP mutant dimerization by live cell cross-linking. Representative cross-linking blot in A. HEK293 and B. COS7 cells. Left: Following cross-linking with DSP, AβPP exists in monomeric, dimeric, and oligomeric forms, with the monomeric form being the most predominant. Right: With reduction of disulphide bonds using β-mercaptoethanol (β-ME), most of the higher oligomeric forms of AβPP disappear, indicating that the higher oligomeric forms are derived from successful cross-linking by DSP. Except for the K624C positive control, not much difference is seen between WT and familial mutant AβPP dimer fomation.
Comparison of WT and familial Alzheimer’s disease mutant AβPP dimers using blue native (BN) gel analysis
Blue native (BN) gel experiments were also carried out to compare dimerization of WT and FAD AβPP mutants. This method examines untagged protein without chemical manipulation, and as as a result allows for better analysis of the proteins in their native physiological states. Figure 5A and 5B show representative gels from transfected HEK293 and COS7 cells, respectively. Under blue native conditions, the expected molecular weight of the APP monomer is ~300kDa and of the AβPP dimer is ~600kDa [38,56]. For pcDNA1 mock transfected cells, no AβPP band is seen on the blots. For K624C transfected cells, both monomer and dimer bands are seen, which is expected from a mutant that forms constitutive dimers. For the various WT and FAD mutant AβPP transfected cells, a predominant monomer band is seen at low exposure. At higher exposure, dimer bands are visible for the different constructs, along with bands representing higher molecular weight AβPP oligomers. This result again indicates that AβPP primarily exists in monomeric form. Further, the WT and mutant proteins again display comparable amounts of dimers as seen in the high exposure blots, which is observed in both HEK293 and COS7 cells.
Figure 5.
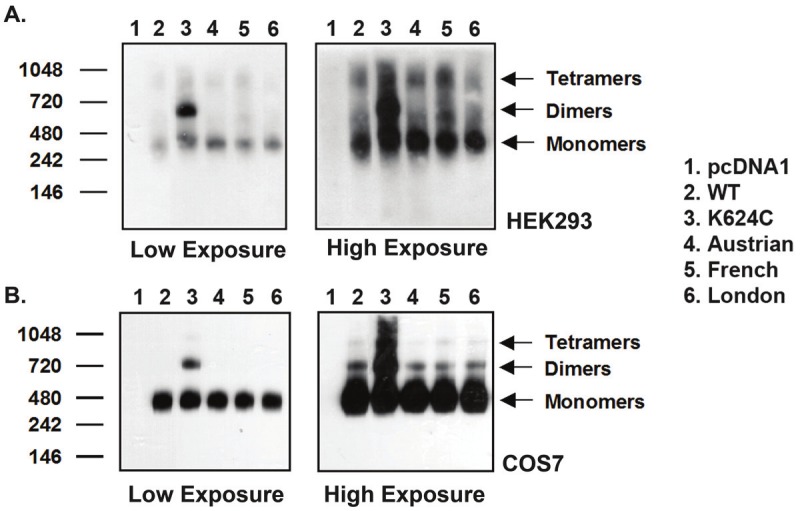
Comparison of WT and FAD AβPP mutant dimerization by blue native (BN) gel analysis. Representative BN gels in A. HEK293 and B. COS7 cells. Left: Low exposure of BN gel shows that AβPP predominantly exists in monomeric form. Right: High exposure of BN gel shows AβPP exists in monomeric, dimeric, and oligomeric forms. Except for the K624C positive control, not much difference is seen between WT and familial mutant AβPP dimer formation.
Discussion
In this study, we examined the role of AβPP dimerization on Aβ production with a focus on FAD AβPP mutants that cause altered Aβ production leading to early onset AD [43]. A number of reports in the literature propose that AβPP dimerization affects Aβ levels [22,38]. Interestingly, it was recently reported that AβPP751 forms more dimers than AβPP695 by using the BiFC system [57] and the APP751 construct was used in this study, which may allow for better comparison of APP dimers. Since most of the FAD AβPP mutations are located near the TMD GxxxG/GxxxA motif, it is likely that the mutants influence Aβ production through changes in dimerization. However, using three different methods of analysis, AβPP-Venus BiFC, live cell chemical cross-linking, and blue native gel electrophoresis, we demonstrate here that WT and mutant AβPP form very similar levels of dimers.
Various factors may affect BiFC signals which could explain the apparent lack of significant differences between the K624C APP positive control and the WT and mutant APP samples in COS7 cells and HEK293 cells prior to normalizing for protein expression. One explanation could be that the BiFC signal is due to the Venus fragments coming together independently of AβPP dimerization. As such, the Venus fragments would be driving AβPP dimerization so that the fluorescence would be the same in all samples and would only depend on the expression of the Venus proteins. We previously performed some BiFC-flow cytometry experiments with Notch2, proteins that form very few dimers, and have some indications that the BiFC fragments may drive dimerization, but not to a drastic degree that could affect the outcome of the results [25]. Another explanation may be that the fluorescent signal is saturated as the plasmids were transfected into HEK293 and COS7 cells which favor high protein expression. We believe that this is probably not the case as the AβPP YFP signal is much higher than that seen for the Venus constructs. A third explanation could be that the constitutive dimerization represented by the K624C mutation, which is located at the juxtamemrane domain of AβPP, may not be detected by the Venus fragments that are tagged at the C-terminal end of AβPP. Alternatively, the mutations at the TMD may not affect full-length AβPP dimerization, which has been reported, using the TOXR system, for the GxxxG mutant that affects AβPP dimerization at the TMD, but does not affect full-length APP dimerization [34]. Finally, it is important to note that BiFC may detect AβPP dimers, as well as higher oligomers. Currently we do not have the ability to distinguish the BiFC signals between the different forms of the protein, and this could also complicate the interpretation of our findings. However, the cross-linking and blue native gel experiments do confirm the findings of the BiFC assay, which makes the BiFC results credible.
Our results indicate that the altered Aβ production by the mutants is not simply a result of changes in dimer formation. Additionally, only a small portion of WT and mutant proteins seems to form dimers as indicated by the cross-linking and blue native gel results, so it seems unlikely that the small quantities of dimers present would drastically affect disease outcome. However, this does not rule out the possibility that AβPP dimers can contribute to changes in Aβ production. It is still possible that low level dimerization can increase Aβ through other mechanisms, such as altered AβPP processing.
Changes in Aβ levels are mainly due to changes in the amyloidogenic processing of AβPP. Various factors can lead to altered processing of the protein, with dimerization being one possible contributor. β-secretase or BACE is known to form enzymatically functional dimers under native conditions [58], which have been shown to immunohistochemically co-localize with AβPP [59]. Similarly, presenilin, the catalytic core of γ-secretase, also forms dimers [60] that are catalytically active [61]. However, there is still no direct evidence showing that BACE and presenilin dimers process AβPP dimers or have a preference for dimeric AβPP. Yet, one study has shown that the addition of a synthetic loop peptide that prevents AβPP ectodomain dimerization at the GFLD region also decreases Aβ levels. Since the relative decrease in Aβ corresponded to a decrease in AβPPsβ levels generated from β-secretase processing, this study suggests that the loop peptide may have impaired AβPP dimerization, which in turn impaired β-secretase cleavages [30]. Also, it is generally accepted that following α- and β-secretase cleavages, γ-secretase sequentially cleaves the remaining AβPP C-terminal fragment (CTF) at multiple sites starting at the C-terminus from amino acids 715 to 710 to generate Aβ peptides of various lengths from Aβ 39 to 42 [18,34]. It has been postulated that when AβPP is in monomeric form, the sequential cleavage by γ-secretase is unperturbed and proceeds freely along the length of AβPP to produce shorter Aβ species such as Aβ38. However, if AβPP is dimerized at the transmembrane domain, steric hindrance may reduce the ability of the γ-secretase complex in cleaving AβPP into smaller Aβ species [34]. In this sense, increased dimerization would increase the levels of toxic Aβ40 and Aβ42, while decreasing the levels of the less toxic Aβ forms such as Aβ38 [34]. As such, it seems that AβPP dimerization may also affect γ-secretase processing.
In terms of the FAD AβPP mutants, the mutations themselves may confer an additional contributory role in altering AβPP processing. For example, mutations at the GxxxG motif may interfere with AβPP substrate recognition by the secretases. Indeed, molecular modeling has revealed that mutant amino acid residues in FAD AβPP mutants occupy the protein interface differently than WT AβPP such that the mutant amino acid side chain protrudes out of the polypeptide chain [42]. Also, solid state NMR spectroscopy and replica exchange molecular dynamic (REMD) simulations have shown that mutations at the GxxxG motif prevent unraveling of the transmembrane helix to form coils. Changing from a helix to coil conformation is necessary for γ-secretase to process AβPP, and deviation from this step could alter Aβ levels [62,63]. Also, REMD has shown that the γ-secretase cleavage site in GxxxG mutants is shifted towards the center of the membrane, which may lead to additional mismatch interactions between the protease and the protein, further altering AβPP processing and Aβ production [62]. Based on the findings in the literature and our own results, we propose that examining the FAD AβPP mutations and their specific interactions with β- and γ- secretases, rather than examining their effect on dimerization, may be the key to understanding their role in Aβ formation.
Acknowledgments
We thank Earl Gillespie for the initial cloning of the WT AβPP VN and VC fragments and Dr. Sun Young Oh for the design of the FAD mutant AβPP primers for mutagenesis. This work was supported by a grant from the Alzheimer’s Association.
References
- 1.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci U S A. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aydin D, Weyer SW, Muller UC. Functions of the APP gene family in the nervous system: insights from mouse models. Exp Brain Res. 2012;217:423–34. doi: 10.1007/s00221-011-2861-2. [DOI] [PubMed] [Google Scholar]
- 3.Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P, et al. The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J Biol Chem. 1990;265:15977–15983. [PubMed] [Google Scholar]
- 4.Bellingham SA, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, Cappai R, Camakaris J. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem. 2004;91:423–428. doi: 10.1111/j.1471-4159.2004.02731.x. [DOI] [PubMed] [Google Scholar]
- 5.Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 7.Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Muller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klar A, Baldassare M, Jessell TM. F-spondin: a gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell. 1992;69:95–110. doi: 10.1016/0092-8674(92)90121-r. [DOI] [PubMed] [Google Scholar]
- 9.Kwak YD, Brannen CL, Qu T, Kim HM, Dong X, Soba P, Majumdar A, Kaplan A, Beyreuther K, Sugaya K. Amyloid precursor protein regulates differentiation of human neural stem cells. Stem Cells Dev. 2006;15:381–389. doi: 10.1089/scd.2006.15.381. [DOI] [PubMed] [Google Scholar]
- 10.Young-Pearse TL, Bai J, Chang R, Zheng JB, Lo-Turco JJ, Selkoe DJ. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007;27:14459–14469. doi: 10.1523/JNEUROSCI.4701-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qiu WQ, Ferreira A, Miller C, Koo EH, Selkoe DJ. Cell-surface beta-amyloid precursor protein stimulates neurite outgrowth of hippocampal neurons in an isoform-dependent manner. J Neurosci. 1995;15:2157–2167. doi: 10.1523/JNEUROSCI.15-03-02157.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA. Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. EMBO J. 2005;24:2944–2955. doi: 10.1038/sj.emboj.7600757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 14.Meziane H, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, Ungerer A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci U S A. 1998;95:12683–12688. doi: 10.1073/pnas.95.21.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gralle M, Botelho MG, Wouters FS. Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem. 2009;284:15016–15025. doi: 10.1074/jbc.M808755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006;26:7212–7221. doi: 10.1523/JNEUROSCI.1450-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 18.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 19.Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 21.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 22.Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer’s disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 23.Chen CD, Oh SY, Hinman JD, Abraham CR. Visualization of APP dimerization and APP-Notch2 heterodimerization in living cells using bimolecular fluorescence complementation. J Neurochem. 2006;97:30–43. doi: 10.1111/j.1471-4159.2006.03705.x. [DOI] [PubMed] [Google Scholar]
- 24.Oh SY, Chen CD, Abraham CR. Cell-type dependent modulation of Notch signaling by the amyloid precursor protein. J Neurochem. 2010;113:262–274. doi: 10.1111/j.1471-4159.2010.06603.x. [DOI] [PubMed] [Google Scholar]
- 25.Oh SY, Ellenstein A, Chen CD, Hinman JD, Berg EA, Costello CE, Yamin R, Neve RL, Abraham CR. Amyloid precursor protein interacts with notch receptors. J Neurosci Res. 2005;82:32–42. doi: 10.1002/jnr.20625. [DOI] [PubMed] [Google Scholar]
- 26.Kaden D, Voigt P, Munter LM, Bobowski KD, Schaefer M, Multhaup G. Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J Cell Sci. 2009;122:368–377. doi: 10.1242/jcs.034058. [DOI] [PubMed] [Google Scholar]
- 27.Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 28.Dahms SO, Hoefgen S, Roeser D, Schlott B, Guhrs KH, Than ME. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2010;107:5381–5386. doi: 10.1073/pnas.0911326107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004;117:4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- 30.Kaden D, Munter LM, Joshi M, Treiber C, Weise C, Bethge T, Voigt P, Schaefer M, Beyermann M, Reif B, Multhaup G. Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect beta-secretase cleavage of APP. J Biol Chem. 2008;283:7271–7279. doi: 10.1074/jbc.M708046200. [DOI] [PubMed] [Google Scholar]
- 31.Beher D, Hesse L, Masters CL, Multhaup G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem. 1996;271:1613–1620. doi: 10.1074/jbc.271.3.1613. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 33.Asada-Utsugi M, Uemura K, Noda Y, Kuzuya A, Maesako M, Ando K, Kubota M, Watanabe K, Takahashi M, Kihara T, Shimohama S, Takahashi R, Berezovska O, Kinoshita A. N-cadherin enhances APP dimerization at the extracellular domain and modulates Abeta production. J Neurochem. 2011;119:354–363. doi: 10.1111/j.1471-4159.2011.07364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorman PM, Kim S, Guo M, Melnyk RA, McLaurin J, Fraser PE, Bowie JU, Chakrabartty A. Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer’s disease mutants. BMC Neurosci. 2008;9:17. doi: 10.1186/1471-2202-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doura AK, Kobus FJ, Dubrovsky L, Hibbard E, Fleming KG. Sequence context modulates the stability of a GxxxG-mediated transmembrane helix-helix dimer. J Mol Biol. 2004;341:991–998. doi: 10.1016/j.jmb.2004.06.042. [DOI] [PubMed] [Google Scholar]
- 37.Brosig B, Langosch D. The dimerization motif of the glycophorin A transmembrane segment in membranes: importance of glycine residues. Protein Sci. 1998;7:1052–1056. doi: 10.1002/pro.5560070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eggert S, Midthune B, Cottrell B, Koo EH. Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J Biol Chem. 2009;284:28943–28952. doi: 10.1074/jbc.M109.038646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kienlen-Campard P, Tasiaux B, Van Hees J, Li M, Huysseune S, Sato T, Fei JZ, Aimoto S, Courtoy PJ, Smith SO, Constantinescu SN, Octave JN. Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J Biol Chem. 2008;283:7733–7744. doi: 10.1074/jbc.M707142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, Cottrell BA, Torpey J, Rosenberry TL, Fauq A, Wolfe MS, Schmidt B, Walsh DM, Koo EH, Golde TE. Substrate-targeting gamma-secretase modulators. Nature. 2008;453:925–929. doi: 10.1038/nature07055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Richter L, Munter LM, Ness J, Hildebrand PW, Dasari M, Unterreitmeier S, Bulic B, Beyermann M, Gust R, Reif B, Weggen S, Langosch D, Multhaup G. Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc Natl Acad Sci U S A. 2010;107:14597–14602. doi: 10.1073/pnas.1003026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munter LM, Botev A, Richter L, Hildebrand PW, Althoff V, Weise C, Kaden D, Multhaup G. Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J Biol Chem. 2010;285:21636–21643. doi: 10.1074/jbc.M109.088005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 44.Hendriks L, van Duijn CM, Cras P, Cruts M, Van Hul W, van Harskamp F, Warren A, McInnis MG, Antonarakis SE, Martin JJ, et al. Presenile dementia and cerebral haemorrhage linked to a mutation at codon 692 of the beta-amyloid precursor protein gene. Nat Genet. 1992;1:218–221. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 45.Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- 46.Kumar-Singh S, De Jonghe C, Cruts M, Kleinert R, Wang R, Mercken M, De Strooper B, Vanderstichele H, Lofgren A, Vanderhoeven I, Backhovens H, Vanmechelen E, Kroisel PM, Van Broeckhoven C. Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated A beta(42) in Alzheimer’s disease. Hum Mol Genet. 2000;9:2589–2598. doi: 10.1093/hmg/9.18.2589. [DOI] [PubMed] [Google Scholar]
- 47.Pasalar P, Najmabadi H, Noorian AR, Moghimi B, Jannati A, Soltanzadeh A, Krefft T, Crook R, Hardy J. An Iranian family with Alzheimer’s disease caused by a novel APP mutation (Thr714Ala) Neurology. 2002;58:1574–1575. doi: 10.1212/wnl.58.10.1574. [DOI] [PubMed] [Google Scholar]
- 48.Cruts M, Dermaut B, Rademakers R, Van den Broeck M, Stogbauer F, Van Broeckhoven C. Novel APP mutation V715A associated with presenile Alzheimer’s disease in a German family. J Neurol. 2003;250:1374–1375. doi: 10.1007/s00415-003-0182-5. [DOI] [PubMed] [Google Scholar]
- 49.Eckman CB, Mehta ND, Crook R, Perez-tur J, Prihar G, Pfeiffer E, Graff-Radford N, Hinder P, Yager D, Zenk B, Refolo LM, Prada CM, Younkin SG, Hutton M, Hardy J. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of A beta 42(43) Hum Mol Genet. 1997;6:2087–2089. doi: 10.1093/hmg/6.12.2087. [DOI] [PubMed] [Google Scholar]
- 50.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 51.Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science. 1991;254:97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- 52.Ancolio K, Dumanchin C, Barelli H, Warter JM, Brice A, Campion D, Frebourg T, Checler F. Unusual phenotypic alteration of beta amyloid precursor protein (betaAPP) maturation by a new Val-715 --> Met betaAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 1999;96:4119–4124. doi: 10.1073/pnas.96.7.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shyu YJ, Liu H, Deng X, Hu CD. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 2006;40:61–66. doi: 10.2144/000112036. [DOI] [PubMed] [Google Scholar]
- 54.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 55.Kerppola TK. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc. 2006;1:1278–1286. doi: 10.1038/nprot.2006.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, Wolfe MS. Active gamma-secretase complexes contain only one of each component. J Biol Chem. 2007;282:33985–33993. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- 57.Ben Khalifa N, Tyteca D, Courtoy PJ, Renauld JC, Constantinescu SN, Octave JN, Kienlen-Campard P. Contribution of Kunitz protease inhibitor and transmembrane domains to amyloid precursor protein homodimerization. Neurodegener Dis. 2012;10:92–95. doi: 10.1159/000335225. [DOI] [PubMed] [Google Scholar]
- 58.Westmeyer GG, Willem M, Lichtenthaler SF, Lurman G, Multhaup G, Assfalg-Machleidt I, Reiss K, Saftig P, Haass C. Dimerization of beta-site beta-amyloid precursor protein-cleaving enzyme. J Biol Chem. 2004;279:53205–53212. doi: 10.1074/jbc.M410378200. [DOI] [PubMed] [Google Scholar]
- 59.Schmechel A, Strauss M, Schlicksupp A, Pipkorn R, Haass C, Bayer TA, Multhaup G. Human BACE forms dimers and colocalizes with APP. J Biol Chem. 2004;279:39710–39717. doi: 10.1074/jbc.M402785200. [DOI] [PubMed] [Google Scholar]
- 60.Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, Lewis HD, Saxena MT, De Strooper B, Coonrod A, Tomita T, Iwatsubo T, Moore CL, Goate A, Wolfe MS, Shearman M, Kopan R. A presenilin dimer at the core of the gamma-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci U S A. 2003;100:13075–13080. doi: 10.1073/pnas.1735338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cervantes S, Saura CA, Pomares E, Gonzalez-Duarte R, Marfany G. Functional implications of the presenilin dimerization: reconstitution of gamma-secretase activity by assembly of a catalytic site at the dimer interface of two catalytically inactive presenilins. J Biol Chem. 2004;279:36519–36529. doi: 10.1074/jbc.M404832200. [DOI] [PubMed] [Google Scholar]
- 62.Miyashita N, Straub JE, Thirumalai D, Sugita Y. Transmembrane structures of amyloid precursor protein dimer predicted by replica-exchange molecular dynamics simulations. J Am Chem Soc. 2009;131:3438–3439. doi: 10.1021/ja809227c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sato T, Tang TC, Reubins G, Fei JZ, Fujimoto T, Kienlen-Campard P, Constantinescu SN, Octave JN, Aimoto S, Smith SO. A helix-to-coil transition at the epsilon-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc Natl Acad Sci U S A. 2009;106:1421–1426. doi: 10.1073/pnas.0812261106. [DOI] [PMC free article] [PubMed] [Google Scholar]