

Abstract
The G protein coupled receptors CB1 and CB2 are targets for the psychoactive constituents of cannabis, chief among them Δ9-THC. They are also key components of the multifunctional endogenous cannabinoid signaling system. CB1 and CB2 receptors modulate a wide variety of physiological systems including analgesia, memory, mood, reward, appetite and immunity. Identification and characterization of selective CB1 and CB2 receptor agonists and antagonists will facilitate understanding the precise physiological and pathophysiological roles of cannabinoid receptors in these systems. This is particularly necessary in the case of CB2 because these receptors are sparsely expressed and problematic to detect using traditional immunocytochemical approaches.
1-Propyl-2-methyl-3-(1-naphthoyl)indole (JWH015) is an aminoalkylindole that has been employed as a “CB2-selective” agonist in more than 40 published papers. However, we have found that JWH015 potently and efficaciously activates CB1 receptors in neurons. Using murine autaptic hippocampal neurons, which express CB1, but not CB2 receptors, we find that JWH015 inhibits excitatory postsynaptic currents with an EC50 of 216 nM. JWH015 inhibition is absent in neurons from CB1−/− cultures and is reversed by the CB1 antagonist, SR141716 [200 nM]. Furthermore, JWH015 partially occludes CB1-mediated DSE (~35% remaining), an action reversed by the CB2 antagonist, AM630 [1 and 3 μM], suggesting that high concentrations of AM630 also antagonize CB1 receptors.
We conclude that while JWH015 is a CB2-preferring agonist, it also activates CB1 receptors at experimentally encountered concentrations. Thus, CB1 agonism of JWH015 needs to be considered in the design and interpretation of experiments that use JWH015 to probe CB2-signaling.
Keywords: JWH015, Cannabinoid, CB1, CB2, Marijuana, AM630
1. Introduction
The endocannabinoid system has many roles within the body. Its functions are mediated via endogenous cannabinoids, including anandamide (AEA [1]) and 2-arachidonoylglycerol (2-AG [2]), binding to the well-characterized metabotropic cannabinoid receptors, CB1 and CB2 [3,4]. Cannabinoid receptors are best known as the endogenous targets of the psychoactive ingredients of marijuana and hashish, chief among them Δ9-THC [5]. These G protein-coupled receptors are widely distributed throughout the body and have been found to modulate diverse physiological systems including analgesia, memory, mood, reward, appetite, and immunity [6].
CB1 is richly expressed in the CNS [7,8] and is the chief mediator of the psychoactive effects of marijuana and hashish. Because of its prominent role in the psychoactive effects of THC, CB1 has received more attention than CB2. However, CB2 has been the object of growing interest as a potential therapeutic target, particularly for pain, inflammation, and osteoporosis [9]. CB2 is widely expressed in the immune system and is known to modulate some inflammatory responses [10–14]. As compared to CB1, CB2 is expressed at low levels in the healthy brain and has been proposed as a promising pharmacological target, insofar as CB2 activation is hypothesized to be less likely to cause adverse psychoactivity. However, to fully characterize the therapeutic potential of CB2 receptors it is essential to employ appropriately selective CB2 agonists and antagonists.
Many synthetic cannabinoids have been developed, with varying degrees of selectivity for the two cannabinoid receptors [15,16]. However the pharmacology of cannabinoid receptor ligands—endogenous, exogenous (e.g. derived from cannabis), and synthetic—has proved complex. Some nominally CB2-selective agonists have come into widespread use without a full consideration of their selectivity in a functional context. One of these is 1-propyl-2-methyl-3-(1-naphthoyl)indole (JWH015, Fig. 1A), an aminoalkylindole that has been reported to be 12–24 times more selective for CB2 than for CB1 [17–19].
Fig. 1.

JWH015 potently and efficaciously inhibits excitatory neurotransmission via CB1. (A) Structure of JWH015. (B) Sample EPSC time–course showing inhibition by JWH015 [2 μM] and reversal by the CB1 antagonist SR141716 (SR1) [200 nM]. Inset shows sample EPSC traces at time-points indicated by A, B, and C. (C) Concentration–response curve for JWH015 in wild-type (squares), GPR55 knock-out neurons (diamond), and CB1 knock-out neurons (triangle).
Since first described, JWH015 has been used as a CB2-selective agonist in more than 40 published articles. Initial characterization reported a Ki of 13.8 nM at the CB2 receptor, and a Ki of 336 nM at the CB1 receptor [17]. This offers a ~25-fold selectivity for CB2 over CB1, though a subsequent study reported only a 12-fold selectivity [19]. Regardless, a 12- to 24-fold selectivity is relatively slender margin, especially when CB1 is found at very high levels and may efficaciously signal at low occupancy. For example, CB1-signaling can be observed at receptor occupancy ranging from 4 to 14% [20]. This narrow selectivity range raises the possibility that some reported effects of JWH015 have in fact occurred via CB1, especially when employing higher concentrations or doses of the drug. But how efficacious and potent is JWH015 in an endogenous neuronal CB1 signaling system? In autaptic hippocampal neurons, CB1 activation is coupled to inhibition of calcium channels and neurotransmitter release [21–23]. These neurons express a robust CB1-dependent endogenous cannabinoid signaling system [22,24,25] including depolarization-induced suppression of excitation (DSE) [26,27]. DSE is a well-described 2-AG/CB1 receptor-dependent signaling mechanism characterized by a transient decrease in excitatory post-synaptic current (EPSC) size, with subsequent recovery back to baseline over tens of seconds.
Using autaptic hippocampal cultures we explored the action of JWH015 at CB1. Neurons in these cultures express CB1 receptors, but lack CB2 receptors, and express robust DSE [22]. Thus, they serve as a useful model for the study of the selectivity of CB1 signaling in a controlled neuronal environment. Using this system we found that JWH015 is an efficacious and relatively potent CB1 receptor agonist, similarly, the CB2-preferring antagonist, AM630, has appreciable antagonistic activity at CB1 receptors. Thus, both compounds should be used with caution as “CB2-selective” agents.
2. Experimental procedures
2.1. Culture preparation
All procedures used in this study were approved by the Animal Care Committees of Indiana University and conform to the guidelines of the National Institutes of Health on the Care and Use of Animals. Experiments were designed in such a way as to minimize the number of animals used and their suffering. Mouse hippocampal neurons isolated from the CA1 to CA3 region were cultured on microislands as described previously [28,29]. Neurons were obtained from animals (age postnatal days 0–2, killed via rapid decapitation) and plated onto a feeder layer of hippocampal astrocytes that had been laid down previously [30]. Cultures were grown in high-glucose (20 mM) medium containing 10% horse serum, without mitotic inhibitors and used for recordings after 8 days in culture and for no more than 3 h after removal from culture medium. All drugs were tested on cells from at least two different preparations.
2.2. Electrophysiology
When a single neuron is grown on a small island of permissive substrate, it forms synapses—or “autapses”—onto itself. All experiments were performed on isolated autaptic neurons. Whole-cell voltage-clamp recordings from autaptic neurons were carried out at room temperature using an Axopatch 200A amplifier (Molecular Devices, Sunnyvale, CA). The extracellular solution contained 119 mM NaCl, 5 mM KCl, 2.5 mM CaCl2, 1.5 mM MgCl2, 30 mM glucose, and 20 mM HEPES. Continuous flow of solution through the bath chamber (2 ml/min) ensured rapid drug application and clearance. Drugs were typically prepared as stock, then diluted into extracellular solution at their final concentration and used on the same day. Recording pipettes of 1.8–3 Mohm were filled with 121.5 mM potassium gluconate, 17.5 mM KCl, 9 mM NaCl, 1 mM MgCl2, 10 mM HEPES, 0.2 mM EGTA, 2 mM MgATP, and 0.5 mM LiGTP. Access resistance and holding current were monitored, and only cells with both stable access resistance and holding current were included for data analysis. Conventional stimulus protocol: the membrane potential was held at 70 mV and excitatory post-synaptic currents (EPSCs) were evoked every 20 s by triggering an unclamped action current with a 1.0-ms depolarizing step. The resultant evoked waveform consisted of a brief stimulus artifact and a large downward spike representing inward sodium currents, followed by the slower excitatory postsynaptic current. The size of the recorded EPSCs was calculated by integrating the evoked current to yield a charge value (in picocoulombs). Calculating the charge value in this manner yields an indirect measure of the amount of neurotransmitter released while minimizing the effects of cable distortion on currents generated far from the site of the recording electrode (the soma). Data were acquired at a sampling rate of 5 kHz.
2.3. DSE stimuli
After establishing a 20-s 0.5-Hz baseline, DSE was evoked by depolarizing to 0 mV for 0.05–10 s, followed by resumption of a 0.5-Hz stimulus protocol for 10–80 s. This allowed EPSCs to recover to baseline values before the next DSE stimulus.
2.4. Statistics
Values are expressed as mean ± s.e.m. Statistical analysis and dose–response curves were generated using GraphPad Prism version 4.0a for Macintosh, GraphPad Software, San Diego, CA, USA. Statistical tests used are indicated in the corresponding figure.
3. Results
3.1. JWH015 activates CB1 to inhibit excitatory postsynaptic currents
Using autaptic culture hippocampal neurons, we tested the ability of JWH015 to inhibit excitatory neurotransmission via CB1 receptors. We found that 2 μM JWH015 strongly inhibited excitatory postsynaptic currents (EPSCs; Fig. 1B and C; relative EPSC charge (1.0 = no inhibition): 0.46 ± 0.06, n = 19). This inhibition is the same as the maximal inhibition observed during DSE in the same population of neurons (Fig. 4a), JWH015 inhibition of EPSCs was CB1-mediated, as it was fully reversed by the CB1 antagonist SR141716 (200 nM; Fig. 1B; 1.0 = no inhibition); relative EPSC charge after SR141716: 0.86 ± 0.08, n = 5, p > 0.05). Furthermore, JWH015 had no effect on EPSCs in neurons cultured from mice lacking CB1 receptors (Fig. 1C, Relative EPSC charge with JWH015 (2 μM): 1.01 ± 0.04, n = 5). JWH015 has been reported to act at GPR55 [31] however JWH015 yielded strong inhibition of EPSCs in GPR55−/− neurons, indicating that the effect of JWH015 in these neurons does not depend on the presence of GPR55 (Fig. 1C, relative EPSC charge with JWH015 (3 μM): 0.53 ± 0.05, n = 8). To examine the potency of JWH015 we tested a range of concentrations (Fig. 1C) and determined that the EC50 of EPSC inhibition by JWH015 was 216 nM (95% CI: 199–238 nM). In addition, 2 μM JWH015 occluded about 70% of the DSE evoked by a 3 s depolarization, demonstrating potent competition with endogenous 2-AG (Fig. 4C).
Fig. 4.

AM630 attenuates and JWH015 occludes DSE. (A) Depolarization–response curve shows relative EPSC inhibition with increasing durations of depolarization, under control conditions and following AM630-treatment [1 μM] and [3 μM] in wild-type neurons. There is statistically significant difference between each treatment and the control at time points of 500 ms (except for 1 μM), 1 s, 3 s and 10 s. (B) Bar graph shows that AM630 increases the duration of depolarization required for a half maximal response. The treatments differ significantly from control. (C) Bar graph shows percent DSE from a 3 s stimulus that remains after treatment with JWH015 [2 μM] or AM630 [3 μM].
3.2. JWH015 acts pre-synaptically
To further confirm that the site of JWH015 action in our system was acting at presynaptic CB1 receptors we evaluated the effect of JWH015 on the paired-pulse ratio. Paired pulse-ratios (PPRs) were determined by giving two 1 ms depolarizing pulses in rapid succession (60 ms interstimulus interval) and measuring the amplitudes of the two EPSCs. The peak amplitude of the second EPSC divided by that of the first yields the PPR ratio. An increase in the PPR following drug application is consistent with a presynaptic site of action. Conversely, an unchanged PPR value suggests the drug is acting postsynaptically.
Inhibition of neurotransmission by CB1 receptor agonists is presynaptic in autaptic hippocampal neurons [21]. Therefore, CB1 activation by JWH015 would be expected to increase the paired pulse ratio. Consistent with this, we found that application of 2 μM JWH015 statistically significantly increases the paired-pulse ratio (Fig. 2A, ratio of 2nd response/1st response before JWH015: 0.88 ± 0.05; after JWH015: 1.04 ± 0.06, n = 13, p < 0.05, paired t-test).
Fig. 2.
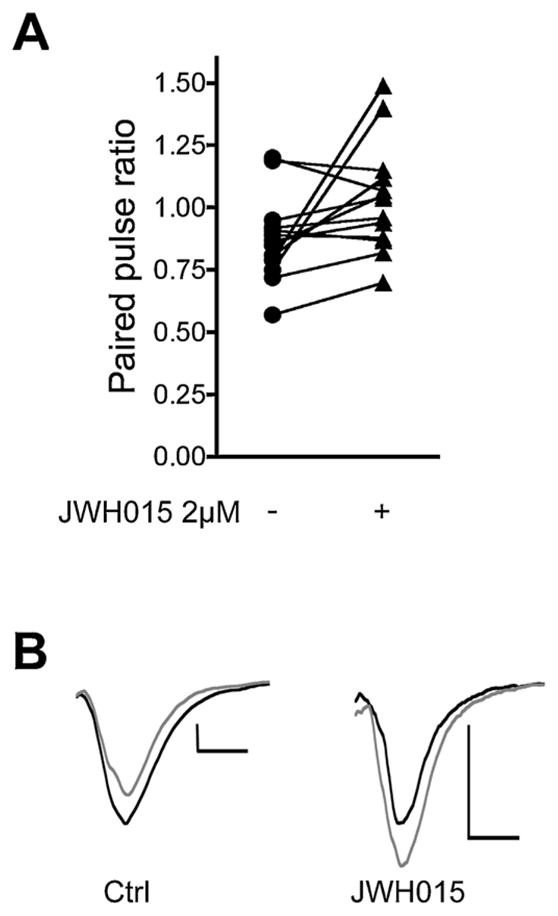
JWH015 inhibits EPSCs via a presynaptic site of action. (A) Graph shows paired-pulse ratios under control conditions (circles) and following treatment with JWH015 (triangles). (B) Sample EPSC pairs under control and after JWH015 treatment. Scale bars: 1 nA, 5 ms.
3.3. AM630 antagonizes CB1
Many investigators recognize that the limited selectivity of JWH015 is problematic. Consequently, AM630 is often used as a CB2 receptor antagonist to establish involvement of CB2 receptors. Based on binding studies, AM630 is 165 times more selective for CB2 over CB1. However with a Ki of 5.15 μM [32], one would still expect significant occupancy of CB1 receptors by AM630 at low-micromolar concentrations, as are often used [9,33,34]. Thus, we next determined if AM630 attenuated activation of CB1 by JWH015.
In neurons cultured from CB1−/− mice, a 3-s depolarization did not elicit DSE and application of 10 μM AM630 did not significantly change EPSC size (Fig. 3C, relative to the baseline EPSC charge with 3 s DSE: 0.91 ± 0.02, n = 5; 10 μM AM630: 0.97 ± 0.03, n = 5, p > 0.05 for both).
Fig. 3.

The “CB2-selective” antagonist AM630 also antagonizes CB1. (A) Sample EPSC time–course showing inhibition by JWH015 [2 μM] and reversal by the CB2-preferring antagonist AM630 [10 μM]. Inset shows sample EPSC traces at time-points indicated by A, B, and C. (B) Bar graph showing relative EPSC charge after treatment with JWH015 [2 μM] either on its own or with SR141716 [200 nM], AM630 [2 μM] or AM630 [10 μM]. *p < 0.05, **p < 0.01, one-way ANOVA vs. JWH015. (C) Bar graph showing relative EPSC charge after DSE (3-s depolarization) and/or treatment with AM630 [10 μM] in CB1−/− or wild-type mice. There are no statistically significant differences between the treatments.
We found that 10 μM AM630 strongly attenuated JWH015 inhibition of EPSCs in WT autaptic neurons (Fig. 3B, relative EPSC charge with JWH015 (2 μM): 0.46 ± 0.05, n = 19; 2 μM AM630 + 10 μM JWH015: 0.79 ± 0.09, n = 7, p < 0.05). Importantly, our results suggest that this concentration of AM630 also likely blocks endogenous CB1 signaling. Therefore we tested whether 3 μM AM630 reduced DSE. Application of 3 μM AM630 attenuated DSE (Fig. 4, relative to the baseline EPSC charge with 3 s DSE: 0.56 ± 0.19, n = 13; and with 3 s DSE + 3 μM AM630: 0.94 ± 0.02, n = 5, p < 0.05). Moreover, application of 1 μM AM630 also attenuated DSE (relative to the baseline EPSC charge with 3 s DSE + 1 μM AM630: 0.80 ± 0.08, n = 5). AM630 alone did not alter EPSC amplitude (Fig. 3C, Relative EPSC charge with AM630: 1.01 ± 0.10, n = 5, p > 0.05). AM630 increased the duration of depolarization required to elicit an equivalent magnitude of DSE (Fig. 4B). AM630 also occluded DSE: after 3 μM AM630 only 11.7 ± 4.3% DSE remained; JWH015 also partially occluded DSE: after 2 μM JWH015 only 32 ± 9.9% DSE remained (Fig. 4C).
3.4. JWH015 causes little CB1 receptor desensitization
Prolonged agonist exposure desensitizes CB1 receptors through multiples processes [35,36]. For example, we have previously shown that WIN55212-2 [100 nM] applied overnight strongly desensitizes CB1 receptors in autaptic neurons [22]. Thus, we wanted to determine whether JWH015 similarly desensitized CB1 receptors.
To test this we incubated cultured neurons with 100 nM JWH015 overnight. This produced a statistically non-significant desensitization of DSE (Fig. 5). Interestingly, even overnight incubation with 1 μM JWH015 produced statistically significant CB1 receptor desensitization only at a single duration of depolarization (3 s) (Fig. 5; relative EPSC charge with DSE 3 s: 0.61 ± 0.05, n = 13; after overnight 1 μM JWH015: 0.80 ± 0.04, n = 10, p < 0.05). We conclude that at concentrations where JWH015 significantly activates neuronal CB1 receptors to suppress synaptic transmission, it induces minimal CB1 receptor desensitization. This contrasts with WIN55212-2, which efficaciously inhibits synaptic transmission with an EC50 of 28 nM (data not shown), while overnight incubation with 100 nM of WIN55212-2 causes profound desensitization.
Fig. 5.
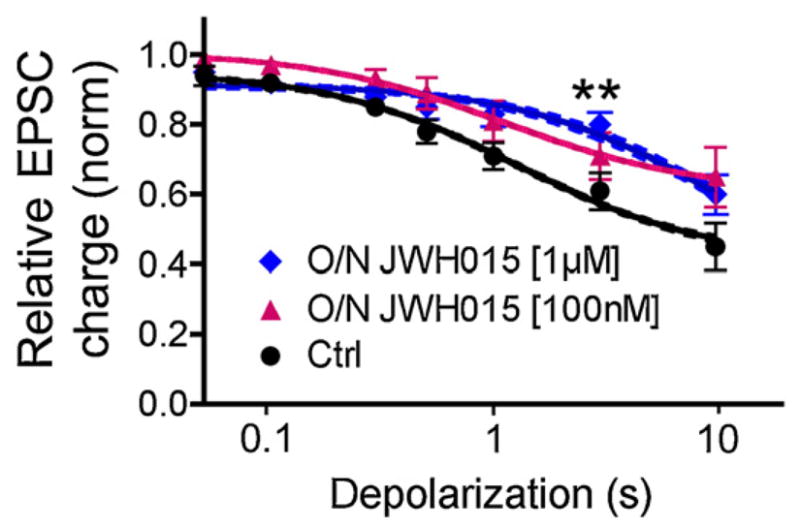
JWH015 induces little CB1 receptor desensitization. Depolarization–response curve showing relative EPSC inhibition after increasing durations of depolarization—under control conditions and after over-night treatment with 100 nM JWH015 (triangles) or 1 μM JWH015 (diamonds). The only condition where there was a statistically significant difference from control was with 1 μM JWH015 pretreatment and a depolarization of 3 s (p < 0.05 two-way ANOVA, Bonferroni post hoc test).
4. Discussion
JWH015 is a synthetic cannabinoid agonist that was synthesized by John Huffman as one of a series of aminoalkylindole analogs [19]. This compound generated excitement as it was one of the first CB2-preferring agonists to be identified, and has become the CB2 agonist of choice for many investigators. The importance of pharmacological tools like JWH015 to understand the role of CB2 receptors in physiological systems is amplified as CB2 receptor expression remains difficult to assess and the commonly used CB2 knockout mice lines have drawbacks [10,37]. In the early literature that followed the initial description of these cannabimimetic indoles, JWH015 was described as having Ki = 164 nM at CB1 [38]. Subsequently, an in vitro study determined the Ki value to be 336 nM, which is the affinity generally accepted by scientific community e.g. [17,38]. The rather modest selectivity of JWH015 for CB2 over CB1 is a potential cause for concern. With the ongoing use of JWH015 as a nominal CB2 selective agonist it was therefore imperative to examine the action of JWH015 in a well-characterized cannabinoid signaling system with robust levels of CB1 receptor expression.
We found that JWH015 is an efficacious and relatively potent agonist at CB1, inhibiting neurotransmitter release in a concentration-dependent manner, with an EC50 of 216 nM. It is also notable that AM630 substantially reversed JWH015-induced inhibition of excitatory postsynaptic currents as well as DSE (which is likely mediated by 2-AG) [22], at concentrations as low as 1 μM. AM630 alone does not affect EPSC amplitude, but serves as an antagonist at CB1. These findings raise concerns about the ongoing use of JWH015 as a CB2-selective agonist, even in combination with AM630. We conclude that one must exercise caution in the use of these drugs as well as in the interpretation of previous studies. This is especially important to note when reviewing studies using JWH015 without an antagonist (e.g. [39–41]), and those that used AM630 at micromolar concentrations (e.g. [42]). It should also be noted that JWH015 has also been implicated as an agonist at GPR55, another cannabinoid-like G protein coupled receptor [31], a receptor that does not appear to play a functional role in signaling in excitatory autaptic hippocampal neurons (unpublished observations).
A substantial number of studies have used AM630 and/or JWH015 to identify CB2-mediated processes. However, a healthy skepticism must be employed when using “receptor-specific” compounds since the knowledge pool is so shallow for many of these compounds. Close attention to drug concentrations and proper controls are needed to avoid to avoid drawing incorrect conclusions, particularly when high doses or concentrations of the drugs are used, since at higher concentrations there is a significant likelihood of “specific” compounds engaging additional, unsuspected targets.
In addition, the use of CB1−/− controls is important to ensure CB2 specificity of observed effects. For instance, the widely used CB1 receptor antagonist SR141716 has been shown to act at GPR55 receptors [31,43,44]. Our use of CB1−/− offered concrete support for our hypothesis that JWH015 acts via CB1. One should also consider the possibility of using model systems lacking CB1 when examining the effects of CB2 agonists in vitro, such as microglia cultured from CB1 null mice. In addition, there is still an obvious need for the development of alternative CB2 selective agonists/antagonists. Those promoting receptor-specific compounds should perform, as much as practical, a full characterization of the compounds to establish the specificity of the compounds in “real world” pharmacology. However, even if this is done, significant responsibility remains with the investigator to ensure that the conclusions drawn in the study are appropriately conservative given the likely limited specificity of the compounds used, particularly when compounds from a single chemical class are used.
In summary, our data indicate that the nominally CB2-selective agonist JWH015 potently and efficaciously activates endogenous neuronal CB1 receptors. Our results suggest that caution is warranted in the use of JWH015, particularly at concentrations in excess of 100 nM. Also the use of AM630 at micromolar concentrations should be viewed cautiously as this concentration of AM630 can block CB1-mediated responses. It is possible that previous studies using JWH015 and AM630 that implicated CB2 receptors in specific physiological or behavioral responses will need to be reconsidered in light of the present findings.
Abbreviations
- 2-AG
2-arachidonoylglycerol
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- JWH015
(2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- Δ9-THC
tetrahydrocannabinol
- SR141716 (aka Rimon-abant)
5-(4-chlorophenyl)-1-(2′,4-dichloro-phenyl)-4-methyl-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide
- AM630
6-iodopravadoline
- DSE
depolarization-induced suppression of excitation
- EPSC
excitatory postsynaptic current
- GPCR
G protein-coupled receptors
- PPR
paired pulse-ratio
References
- 1.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–9. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 2.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–8. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 3.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned CDNA. Nature. 1990;346:561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 4.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 5.Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. Journal of the American Chemical Society. 1964;86:1646–7. [Google Scholar]
- 6.Howlett AC. Pharmacology of cannabinoid receptors. Annual Review of Pharmacology and Toxicology. 1995;35:607–34. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 7.Matsuda LA, Bonner TI, Lolait SJ. Localization of cannabinoid receptor MRNA in rat brain. Journal of Comparative Neurology. 1993;327:535–50. doi: 10.1002/cne.903270406. [DOI] [PubMed] [Google Scholar]
- 8.Tsou K, Brown S, Sanudo-Pena MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid cb1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 9.Sophocleous A, Landao-Bassonga E, Van’t Hof RJ, Idris AI, Ralston SH. The type 2 cannabinoid receptor regulates bone mass and ovariectomy-induced bone loss by affecting osteoblast differentiation and bone formation. Endocrinology. 2011;152:2141–9. doi: 10.1210/en.2010-0930. [DOI] [PubMed] [Google Scholar]
- 10.Atwood BK, Mackie K. Cb2: a cannabinoid receptor with an identity crisis. British Journal of Pharmacology. 2010;160:467–79. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luongo L, Palazzo E, Tambaro S, Giordano C, Gatta L, Scafuro MA, et al. 1-(2′,4′-dichlorophenyl)-6-methyl-n-cyclohexylamine-1,4-dihydroindeno[1,2-c]pyraz ole-3-carboxamide, a novel cb2 agonist, alleviates neuropathic pain through functional microglial changes in mice. Neurobiology of Disease. 2010;37:177–85. doi: 10.1016/j.nbd.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 12.Racz I, Nadal X, Alferink J, Banos JE, Rehnelt J, Martin M, et al. Crucial role of cb(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. Journal of Neuroscience. 2008;28:12125–35. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merighi S, Gessi S, Varani K, Fazzi D, Mirandola P, Borea PA. Cannabinoid cb(2) receptor attenuates morphine-induced inflammatory responses in activated microglial cells. British Journal of Pharmacology. 2012 doi: 10.1111/j.1476-5381.2012.01948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merighi S, Gessi S, Varani K, Simioni C, Fazzi D, Mirandola P, et al. Cannabinoid cb(2) receptors modulate erk-1/2 kinase signalling and no release in microglial cells stimulated with bacterial lipopolysaccharide. British Journal of Pharmacology. 2012;165:1773–88. doi: 10.1111/j.1476-5381.2011.01673.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Huffman JW. The search for selective ligands for the cb2 receptor. Current Pharmaceutical Design. 2000;6:1323–37. doi: 10.2174/1381612003399347. [DOI] [PubMed] [Google Scholar]
- 16.Huffman JW, Lu J, Dai D, Kitaygorodskiy A, Wiley JL, Martin BR. Synthesis and pharmacology of a hybrid cannabinoid. Bioorganic and Medicinal Chemistry. 2000;8:439–47. doi: 10.1016/s0968-0896(99)00305-3. [DOI] [PubMed] [Google Scholar]
- 17.Aung MM, Griffin G, Huffman JW, Wu M, Keel C, Yang B, et al. Influence of the n-1 alkyl chain length of cannabimimetic indoles upon cb(1) and cb(2) receptor binding. Drug and Alcohol Dependence. 2000;60:133–40. doi: 10.1016/s0376-8716(99)00152-0. [DOI] [PubMed] [Google Scholar]
- 18.Huffman JW. Cb2 receptor ligands. Mini Reviews in Medicinal Chemistry. 2005;5:641–9. doi: 10.2174/1389557054368844. [DOI] [PubMed] [Google Scholar]
- 19.Huffman JW, Zengin G, Wu MJ, Lu J, Hynd G, Bushell K, et al. Structure-activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid cb(1) and cb(2) receptors: steric and electronic effects of naphthoyl substituents. New highly selective cb(2) receptor agonists. Bioorganic and Medicinal Chemistry. 2005;13:89–112. doi: 10.1016/j.bmc.2004.09.050. [DOI] [PubMed] [Google Scholar]
- 20.Dhawan J, Deng H, Gatley SJ, Makriyannis A, Akinfeleye T, Bruneus M, et al. Evaluation of the in vivo receptor occupancy for the behavioral effects of cannabinoids using a radiolabeled cannabinoid receptor agonist, r-[125/131i]am2233. Synapse. 2006;60:93–101. doi: 10.1002/syn.20277. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. Journal of Neurophysiology. 1999;82:1286–94. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 22.Straiker A, Mackie K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. Journal of Physiology. 2005;569:501–17. doi: 10.1113/jphysiol.2005.091918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit q-type calcium currents in att20 cells transfected with rat brain cannabinoid receptor. Journal of Neuroscience. 1995;15:6552–61. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Straiker A, Mackie K. Metabotropic suppression of excitation in murine autaptic hippocampal neurons. Journal of Physiology. 2007;578:773–85. doi: 10.1113/jphysiol.2006.117499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kellogg R, Mackie K, Straiker A. Cannabinoid cb1 receptor-dependent long-term depression in autaptic excitatory neurons. Journal of Neurophysiology. 2009;102:1160–71. doi: 10.1152/jn.00266.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoshida T, Hashimoto K, Zimmer A, Maejima T, Araishi K, Kano M. The cannabinoid cb1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar purkinje cells. Journal of Neuroscience. 2002;22:1690–7. doi: 10.1523/JNEUROSCI.22-05-01690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–75. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- 28.Furshpan EJ, MacLeish PR, O’Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in micro-cultures: Evidence for cholinergic, adrenergic, and dual-function neurons. Proceedings of the National Academy of Sciences of the United States of America. 1976;73:4225–9. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:7834–8. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levison SW, McCarthy KD. Characterization and partial purification of aim: a plasma protein that induces rat cerebral type 2 astroglia from bipotential glial progenitors. Journal of Neurochemistry. 1991;57:782–94. doi: 10.1111/j.1471-4159.1991.tb08220.x. [DOI] [PubMed] [Google Scholar]
- 31.Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. Gpr55 is a cannabinoid receptor that increases intracellular calcium and inhibits m current. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2699–704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at cb1 and cb2 cannabinoid receptors of l759633, l759656, and am630. British Journal of Pharmacology. 1999;126:665–72. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin XH, Yuece B, Li YY, Feng YJ, Feng JY, Yu LY, et al. A novel cb receptor gpr55 and its ligands are involved in regulation of gut movement in rodents. Neurogastroenterology and Motility. 2011;23:862–e342. doi: 10.1111/j.1365-2982.2011.01742.x. [DOI] [PubMed] [Google Scholar]
- 34.Keown OP, Winterburn TJ, Wainwright CL, Macrury SM, Neilson I, Barrett F, et al. 2-arachidonyl glycerol activates platelets via conversion to arachidonic acid and not by direct activation of cannabinoid receptors. British Journal of Clinical Pharmacology. 2010;70:180–8. doi: 10.1111/j.1365-2125.2010.03697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the cb1 cannabinoid receptor. Journal of Neurochemistry. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- 36.Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, et al. Distinct domains of the cb1 cannabinoid receptor mediate desensitization and internalization. Journal of Neuroscience. 1999;19:3773–80. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buckley NE. The peripheral cannabinoid receptor knockout mice: an update. British Journal of Pharmacology. 2008;153:309–18. doi: 10.1038/sj.bjp.0707527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huffman J. Design, synthesis and pharmacology of cannabimimetic indoles. Bioorganic and Medicinal Chemistry Letters. 1994;4:4. [Google Scholar]
- 39.Lepicier P, Lagneux C, Sirois MG, Lamontagne D. Endothelial cb1-receptors limit infarct size through no formation in rat isolated hearts. Life Sciences. 2007;81:1373–80. doi: 10.1016/j.lfs.2007.08.042. [DOI] [PubMed] [Google Scholar]
- 40.Trevaskis NL, Charman WN, Porter CJ. Targeted drug delivery to lymphocytes: a route to site-specific immunomodulation. Molecular Pharmaceutics. 2010;7:2297–309. doi: 10.1021/mp100259a. [DOI] [PubMed] [Google Scholar]
- 41.Wei Y, Wang X, Wang L. Presence and regulation of cannabinoid receptors in human retinal pigment epithelial cells. Molecular Vision. 2009;15:1243–51. [PMC free article] [PubMed] [Google Scholar]
- 42.Lombard C, Nagarkatti M, Nagarkatti P. Cb2 cannabinoid receptor agonist, jwh-015, triggers apoptosis in immune cells: potential role for cb2-selective ligands as immunosuppressive agents. Clinical Immunology. 2007;122:259–70. doi: 10.1016/j.clim.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Henstridge CM, Balenga NA, Schroder R, Kargl JK, Platzer W, Martini L, et al. Gpr55 ligands promote receptor coupling to multiple signalling pathways. British Journal of Pharmacology. 2010;160:604–14. doi: 10.1111/j.1476-5381.2009.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, et al. Modulation of l-alpha-lysophosphatidylinositol/gpr55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. Journal of Biological Chemistry. 2012;287:91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]