

Abstract
While the mechanisms employed by metastatic cancer cells to migrate remain poorly understood, it has been widely accepted that metastatic cancer cells can invade the tumor stroma by degrading the extracellular matrix (ECM) with matrix metalloproteinases (MMPs). Although MMP inhibitors showed early promise in preventing metastasis in animal models, they have largely failed clinically. Recently, studies have shown that some cancer cells can use proteolysis to mechanically rearrange their ECM to form tube-like “microtracks” which other cells can follow without using MMPs themselves. We speculate that this mode of migration in the secondary cells may be one example of migration which can occur without endogenous protease activity in the secondary cells. Here we present a technique to study this migration in a 3D, collagen-based environment which mimics the size and topography of the tracks produced by proteolytically active cancer cells. Using time-lapse phase-contrast microscopy, we find that these microtracks permit the rapid and persistent migration of noninvasive MCF10A mammary epithelial cells, which are unable to otherwise migrate in 3D collagen. Additionally, while highly metastatic MDAMB231 breast cancer cells are able to invade a 3D collagen matrix, seeding within the patterned microtracks induced significantly increased cell migration speed, which was not decreased by pharmacological MMP inhibition. Together, these data suggest that microtracks within a 3D ECM may facilitate the migration of cells in an MMP-independent fashion, and may reveal novel insight into the clinical challenges facing MMP inhibitors.
Keywords: microtracks, micropatterning, collagen matrix, cancer metastasis, cell migration
Introduction
During metastatic invasion, cancer cells escape from the primary tumor site and migrate through the body to secondary locations, forming new tumors. Although this process is the cause of over 90% of cancer-related fatalities1, the precise nature of the mechanisms governing this behavior remains elusive. Additionally, tumor cell migration has been shown to be adaptable to both inherent and induced changes in their microenvironment2,3, a phenomenon which remains difficult to accurately study in vivo or in vitro4.
As tumors develop, they induce changes in the peritumoral microenvironment5. Fibroblasts and other stromal cells deposit increased amounts of fibrillar collagen6, collagen crosslinking by lysyl oxidases and tissue transglutaminases is upregulated5,7, and increased tension generated on the extracellular matrix (ECM)8 leads to collagen fiber bundling and alignment perpendicular to the tumor front9. This alignment has been shown to drive cancer cell invasion, as cells will migrate away from the tumor along these aligned fibers10,11. Moreover, ‘leading’ cancer cells have been shown to invade the ECM using proteolytic degradation12–15. These cells cleave collagen fibers using matrix metalloproteinases (MMPs) and reorganize them into parallel bundles, creating a tube-like ‘microtrack’13,16. Other ‘following’ cancer cells can in turn migrate through these open environments, either individually13,14 or as collective strands17,18, which can eventually widen the microtrack further. While proteases are required to form these channels initially, cells that subsequently follow these cleared microtracks do not need to further degrade the matrix. Therefore this mode of migration in the secondary cancer cells may be one example of true protease-independent migration which exists in vivo, and we speculate that it may also help to explain the general ineffectiveness of clinical MMP inhibitors19, if they are administered after initial invasion has begun and microtracks have already been formed.
While this type of migration behavior has been noted in several different cancer subtypes, including carcinomas and fibrosarcomas13,14,20, it remains difficult to mimic the process in vitro. Microfabrication techniques have recently become popular for studying microtrack motility, analyzing tumor cell behavior within the confines of rigid PDMS or silicon channels21–25. These studies have shown that cancer cells will readily deform their cytoskeleton in order to squeeze through highly restrictive channels, to a greater extent than non-cancerous cell types22. Additionally, several studies have demonstrated that cell migration speed is dramatically affected by channel width; once the channel width is significantly wider than the cell body width, to the point that the cell does not sense confinement, the cells will revert to a less persistent motile state, similar to that observed on unconstrained 2D environments24,25. While these studies have yielded useful insight into tumor cell behavior within physically confined spaces, the relevance of these findings to physiological conditions is uncertain. Specifically, PDMS substrates generally have a Young’s modulus of ~ 1000 kPa26, while the native microenvironment in which these cancer cells are found tends to be < 1 kPa27,28. Stiffness has been found to have a profound effect on tumor cell physiology, directly mediating cell spreading, adhesion, migration, and force generation5,27,29. Therefore, the stiff nature of the PDMS channels may induce changes in the cells’ cytoskeletal organization and function that are not necessarily recapitulated in vivo.
Recently, a study by Pathak & Kumar has partially overcome this limitation by patterning microchannels using polyacrylamide, a soft, viscoelastic hydrogel whose mechanical properties can be precisely tuned23. Using a stiffness range of 400 Pa to 120 kPa, they find that migration speed is increased with increasing stiffness under mechanical confinement in 10 μm wide channels. Additionally, they attribute this behavior to an increased induction of polarity in actin stress fibers and traction forces in cells seeded in these narrow channels23. While this technique allows for the separation of the individual effects caused by physiologically-relevant matrix stiffness and physical confinement, it does not recapitulate the subcellular fibrillar architecture of the ECM that can be sensed by tumor cells9. Moreover, polyacrylamide cannot be degraded or remodeled by the cell. Alternatively, Ilina et al. have recently used two-photon laser microsurgery to create defined microtracks within dense 3D collagen matrices to study collective cell migration in tumor spheroids17. By utilizing a collagen matrix, this technique allows the cancer cells to proteolytically degrade and reorganize the collagen fibers. However, the effects of laser ablation on the collagen fiber structure are unclear.
In this study we present a technique in which cancer cell motility can be studied in a physiologically relevant, 3D environment which mimics the complex architecture of the native peritumoral ECM. Using micromolding technologies30, microtracks can be accurately and reproducibly patterned across a range of collagen densities, and can mimic the structure of native tracks produced by proteolytically active cancer cells. Our data indicate that these microtracks permit the rapid and persistent migration of epithelial cells which cannot invade a 3D collagen matrix. Additionally, highly metastatic breast carcinoma cells, which can significantly invade a 3D matrix, have enhanced motility within the collagen microtracks. Interestingly, this migration is not slowed upon MMP inhibition, suggesting that migration within previously established microtracks can continue to occur in the absence of MMP’s. This result may partially explain the lack of success of MMP inhibitors in clinical trials19.
Materials and methods
Cell culture and reagents
MDAMB231 highly metastatic breast adenocarcinoma cells (American Type Culture Collection (ATCC), Rockville, MD) were maintained in Minimum Essential Medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Invitrogen). GFP-expressing MDAMB231 cells (Cell BioLabs, San Diego, CA) were maintained in Dulbecco’s Modified Eagle Medium supplemented with 2 mM L-glutamine, 10% fetal bovine serum, 0.1 mM MEM non-essential amino acids, and 1% penicillin-streptomycin (Invitrogen). MCF10A mammary epithelial cells (ATCC) were maintained in Dulbecco’s Modified Eagle Media supplemented with 5% horse serum, 20 ng/mL EGF (Invitrogen), 10 μg/mL insulin, 0.5 μg/mL hydrocortisone, 100 ng/mL cholera toxin (Sigma-Aldrich, St. Louis, MO), and 1% penicillin-streptomycin (Invitrogen)31. All cells were cultured at 37°C and 5% CO2. For migration studies, GM6001 and the functionally inactive structural analog to GM6001 (N-t-butoxycarbonyl-L-leucyl-L-tryptophan methylamide, labelled here NC-GM6001) (Millipore, Billerica, MA) were applied to cells at a concentration of 20 μM. BB2516 (Sigma-Aldrich) was applied to cells at a concentration of 200 μM32. A commercial protease inhibitor cocktail (Cytoskeleton, Denver, CO) was applied containing working concentrations of 1 μM pepstatin A, 1.5 μM leupeptin, and 1 mM benzamidine, and 0.4 mM na-p-tosyl-L-arginine methyl ester (TAME).
RNA interference and qRT-PCR
MDAMB231 cells at 70% confluency on tissue culture plastic were transfected with 2 nM non-targeting (control) siRNA or siRNA targeting MMP14 (MT1-MMP) using Lipofectamine 2000 (2 μg/mL, Invitrogen). The non-targeting sequence was 5′-UUC CUCUCCACGCGCAGUACAUUUA-3′. The sequence targeting MMP14 was 5′-CCUACGAGAGGAAGGAUGGCAAAUU-3′. Both the control siRNA and the siRNA targeting MMP14 (accession number NM_004995.2) were synthesized by Invitrogen. siRNA knockdown was confirmed with qRT-PCR.
Total RNA was isolated three days post-transfection using RNeasy® Plus Mini Kit (Qiagen, Valencia, CA) and was converted to cDNA by first combining 1 μg total RNA, 4 μM random primers (Invitrogen), and 0.5 mM dNTP mix (New England BioLabs, Ipswich, MA), and incubating for 5 minutes at 70°C. 1X reverse transcription buffer, 2U RNase inhibitor, and 10 U M-MuLV reverse transcriptase (New England BioLabs) were added and the mixture was incubated at 42°C for 1 hour and 90°C for 10 minutes in a iCycler Thermal Cycler (Bio-Rad, Hercules, CA). 2U RNase H (New England BioLabs) was added to the mixture and incubated for 20 minutes at room temperature prior to storage at −20°C. qRT-PCR was performed with 1 μg of cDNA and 0.4 μM of specific primers against MMP14 (Forward: 5′-TGTGACGGGAACTTTGACACCG-3′; Reverse: 5′-ACGCTG CCCTTGAAACTGTGGC-3′, Integrated DNA Technologies, Coralville, IA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Forward: 5′-CATGAGAAGTATGACAACAGCCT-3′; Reverse: 5′-AGTCCTTCCACGATACCAAAGT-3′) using 1X iQ SYBR Green Supermix (Bio-Rad) on a MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad).
Microscopy
Phase contrast imaging was performed in a custom temperature, humidity, and CO2-controlled stage of a Zeiss Axio Observer Z1m inverted phase contrast microscope equipped with a Hamamatsu ORCA-ER camera and operated by AxioVision software (v. 4.8.1.0). Confocal fluorescence and reflectance imaging was performed with a Zeiss LSM700 confocal microscope on a Zeiss AxioObserver Z1 inverted stand equipped with a long working distance water-immersion C-Apochromat 40x/1.1 NA Zeiss objective and operated by Zen software (v. 2010, Carl Zeiss MicroImaging GmbH, Jena, Germany). Confocal reflectance images were obtained as previously described33,34.
Microtrack fabrication
Silicon master fabrication and imaging
Standard 100 mm-diameter silicon wafers containing wells measuring 10 x 300 and 10 x 1100 μm2, spaced 500 μm apart were coated with photoresist (SPR 220-3.0 series) and etched to a depth of 20 and 30 μm as previously described35. Wafers were imaged using a Leica 440 scanning electron microscope to confirm feature dimensions. These molds were used to cast poly(dimethylsiloxane) (PDMS, Ellsworth Adhesives, Germantown, WI) stamps using a standard 1:10 ratio of crosslinking agent to monomer.
Preparation of collagen microtracks
All collagen matrices were prepared using collagen extracted from rat tail tendons (Pel-Freez Biologicals, Rogers, AR) without pepsin as previously described33. A 10 mg/mL stock solution was diluted to the desired collagen concentration (1.5 – 5 mg/mL) with ice-cold culture media and neutralized to pH 7.0 with 1N NaOH. PDMS stamps were rendered non-adhesive by coating with a 1% solution of bovine serum albumin (BSA, Sigma-Aldrich) in PBS at room temperature for 30 minutes. Modified stamps were inverted on a drop of diluted neutralized rat tail type I collagen between two thin PDMS spacers (~1.25 mm thick) and allowed to polymerize for 90 minutes at 37°C in a method similar to that described by Nelson et al.30. To facilitate confocal fluorescence or reflectance imaging, microtracks were prepared on 43 x 50 mm glass coverslips, with 22 x 22 mm coverslips adhered with vacuum grease for spacers. Small glass coverslips were removed after polymerization and microtracks were mounted onto custom made chambers to create a 35 mm diameter well surrounding the patterned microtracks. For cellular experiments, after stamps were removed, a dilute suspension of cells was applied (~80,000 cells/mL) and allowed to settle within the collagen tracks for 2–3 minutes at 37°C. Excess cells were carefully removed by rinsing with ice cold culture medium. A second thin layer of collagen, polymerized onto an 18 mm2 diameter round glass coverslip, was inverted onto the tracks to serve as a lid. Microtracks were incubated with collagen lids for 5 minutes at 37°C before being immersed in culture medium.
Microtrack dimension analysis
Patterned microtracks
The dimensions of the patterned tracks were quantified from confocal reflectance images. 1 μm z-stacks were acquired of each track, and analyzed using ImageJ software (v. 1.44p, National Institute of Health, Bethesda, MD). The center z slice was determined and a grid of horizontal and vertical lines spaced 10μm apart was overlaid onto the image. The line drawing function was used to measure the length across the track every 4 μm (2 measurements per track) and the width every 20 μm (15 measurements per track). To measure the depth of the track, the orthogonal view function in the Zen software was used to visualize the track in the x-z plane. This image was exported and analyzed in ImageJ using a similar grid, with measurements taken every 20 μm (15 measurements per track). Dimensions were quantified for n > 45 tracks per collagen concentration and are reported as Mean ± SD.
Naturally formed microtracks in 3D collagen matrices
A 10 mg/mL type I collagen stock solution was diluted to a 1.5 mg/mL solution with ice-cold culture media and neutralized to pH 7.0 with 1N NaOH. GFP-MDAMB231 cells were seeded into the collagen matrices in a glass bottom 24-well plate (MatTek, Ashland, MA) at low density (50,000 cells/mL) to minimize cell-cell interactions. 0.5 mL solution volume was added to a 24 well plate and incubated for 1 hr at 37°C for collagen polymerization. Gels were overlaid with 0.5 mL pre-warmed culture media and incubated for 48 hours. Gels were fixed with 3.7% (v/v) formaldehyde (JT Baker, Phillipsburg, NJ) in phosphate buffered saline (PBS) and imaged with confocal fluorescence and reflectance.
The dimensions of the natural tracks formed by the cells were quantified from confocal reflectance z-stacks with 1 μm slices using ImageJ software. Z stacks were rotated such that the track was primarily horizontal and a grid of 10 x 10 μm squares was overlaid onto the image. The width of the track was measured every 10 μm. Widths were quantified for n > 25 tracks.
Cellular studies
Fluorescent staining
Cells seeded in collagen microtracks or 3D collagen matrices were allowed to spread overnight and were fixed with 3.7% (v/v) formaldehyde in PBS and permeabilized with 1% (v/v) Triton (JT Baker) in PBS. Samples were blocked with PBS/0.02% (v/v) Tween (JT Baker) with 3% (w/v) BSA and incubated with a diluted 1:100 Alexa Fluor 594-conjugated phalloidin (Invitrogen) in PBS to localize filamentous actin. Nuclei were stained with a 1:100 dilution of 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) in purified deionized water.
Cell migration studies and morphology analysis
Cells were either seeded into 1.5 mg/mL type I collagen gels or into 3 mg/mL collagen microtracks at low density as described above. Gels and microtracks were incubated for 10 hours prior to phase contrast time-lapse imaging. Cells were imaged every 10 minutes for 8 hours. Dividing cells and cells that did not move from their original location after 4 hours were excluded from analysis. Detailed information about these cells can be found in Table 1. ImageJ software was used to determine the area, perimeter, aspect ratio (length of long axis / length of short axis), and circularity, defined as (4πArea/Perimeter2), where a circle has a value of 1, and a straight line has a value of 034,36. Cell centroid position based on cell outlines and was used to calculate the mean-square displacement (<d2>)37. For both the 3D migration within the collagen gels and 2D migration studies on tissue culture plastic, cell migration speed (S) was determined by fitting the <d2> and the time interval (t) to the persistent random walk equation:
| (1) |
where P is the persistence time, using nonlinear least squares regression analysis as previously described38. Because cells traveling in collagen microtracks do not travel in a random walk, but can only migrate in two directions, speed was calculated by dividing the final displacement by time, for cells migrating in one direction for a length of 100 minutes. Maximum invasion distance was quantified for all cells in both the 3D gels and the collagen microtracks by determining the displacement of the cell from its initial position at each time point up to 100 minutes. The maximum value calculated was considered the maximum invasion distance. 100 minutes was selected as the cutoff point to minimize the number of cells which migrated out of the field of view in the microtracks.
Table 1.
Analysis of behavior of MCF10A and MDAMB231 cells within 3D collagen matrices and patterned microtracks.
| % Cells Engaging in Behavior | na | |||||
|---|---|---|---|---|---|---|
| Motile | Nonmotile | Dividing | Dead | |||
| MCF10A | 3D Matrix | 24 | 66 | 10 | 0 | 124 |
| Tracks | 81 | 14 | 3 | 3 | 73 | |
| MDAMB231 | 3D Matrix | 56 | 21 | 21 | 2 | 92 |
| Tracks | 77 | 9 | 9 | 5 | 64 | |
Represents total number of cells observed
Statistical analysis
Data were compared using analysis of variance with a post-hoc Tukey’s HSD test or Student’s t test where appropriate using JMP software (v.9, SAS, Car, NC). Statistical significance was considered with p < 0.05.
Results
Patterning of collagen microtracks
It has been shown previously that some subsets of cancer cells are capable of proteolytically degrading and reorganizing ECM fibers in such a way that a stable cleared pathway through a 3D matrix is generated (Fig. 1A)13,16. Our goal was to re-create these pathways in vitro in a defined and controllable manner to create a system that could then be used to explicitly study this unique method of cancer cell migration (Fig. 1B). To accomplish this, we adapted a method pioneered by Nelson et al.30 that uses a PDMS stamp cast from a silicon master to micromold a neutralized collagen solution into wells with defined dimensions (Fig. 1C).
Fig. 1.

Schematic of method for patterning collagen microtracks. (A) A cancer cell migrating in a 3D matrix cleaves extracellular matrix fibers using proteases, creating an open pathway at the rear of the cell body. (B) Within a patterned 3D track, cancer cells can migrate through the matrix without the need for proteolytic activity. (C) Microtracks are created in a collagen gel using a PDMS stamp to micromold a neutralized collagen solution.
To determine the relevant dimensions to use for patterning microfabricated tracks for our in vitro system, natural tracks formed by GFP-labeled MDAMB231 metastatic breast cancer cells embedded within collagen matrices were analyzed. After 48 hours of migration, the cells were fixed and natural tracks were imaged using confocal microscopy (Fig. 2A). The widths of the natural tracks were analyzed (Fig. 2B) and found to have an average width of 13.7 μm (Fig. 2B, red dotted line). These tracks were widely heterogeneous, both within a single track (average SD = 2.74 μm) and across tracks created by different cells (SD = 4.3 μm). Standard photolithographic techniques were used to etch wells into a silicon wafer with a width of 10 μm (Fig. 3A), a width calculated in over 50% of the tracks analyzed. PDMS stamps cast from this wafer were then used to imprint wells into a collagen gel, creating an array of thin, shallow wells that could be readily imaged using conventional phase contrast microscopy for time lapse studies (Fig. 3B). Additionally, because of the inherent differences in the refractive index of the collagen fibers and the spaces between them, the microstructure of the patterned tracks could be directly imaged using confocal reflectance microscopy (Fig. 3C)33,34,39.
Fig. 2.
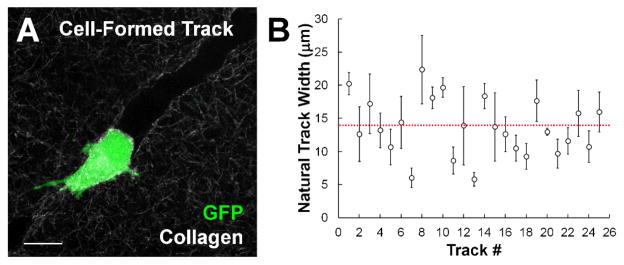
Metastatic cancer cells create heterogeneous tracks within 3D collagen matrices. (A) GFP-labeled MDAMB231 metastatic breast carcinoma cell creates a proteolytic track within a type I collagen matrix. Scale bar = 20 μm. (B) Plot of track widths in naturally formed tracks indicate a wide range of widths observed across different tracks, as well as significant deviation within a single microtrack. Each point represents one track (Mean ± SD). Red dashed line indicates overall mean across all tracks analyzed.
Fig. 3.

Imaging of silicon master and collagen microtracks. (A) SEM image of silicon master etched with wells measuring 300 x 10 x 25 μm (w, l, d). (B) Phase image of patterned collagen microtracks. (C) Confocal reflectance image of collagen fibrils around the tracks. Scale bars = 100μm.
Patterned collagen microtracks mimic natural tracks formed by cancer cells
By subsequently capping the polymerized collagen wells with an additional thin layer of polymerized collagen, an open pathway or ‘microtrack’ entirely encased in collagen was successfully created in vitro (Fig. 4A). The dimensions of the patterned tracks were characterized by measuring their length, width, and depth over 1.5 – 5 mg/mL collagen. Concentrations lower than 1.5 mg/mL and above 5 mg/mL became more difficult to consistently replicate using this method of collagen polymerization. Lower concentrations of collagen led to poorly formed tracks, as the collagen lids appeared to sink into the track body, while higher concentrations polymerized too quickly and had observable defects. Within this range, the track length across collagen concentrations was the parameter with the highest fidelity compared to the original pattern of the silicon master, with only a slight increase in length noted in the 5 mg/mL collagen condition (Fig. 4B). Our analysis indicated that, while all tracks were patterned with stamps of equal width (10 μm) and depth (20 μm), the resulting width increased with collagen density (Fig. 4C), and the resulting depth decreased with collagen density (Fig. 4D). While the achieved widths were similar to the width of the pattern, the achieved depths were around half the depth of the original pattern. However, the variation within a given track was fairly low. The average deviations in the length, width, and depth within a single microtrack were 0.88 μm, 1.38 μm, and 1.47 μm (equivalent to 0.3%, 10%, and 13% difference).
Fig. 4.

Microtracks can be reproducibly patterned with a range of collagen densities. (A) Confocal reflectance image reflecting the measured dimensions of patterned collagen tracks. Scale bar = 50 μm. Length (B), width (C), and depth (D) are quantified across tracks patterned in 1.5 – 5 mg/mL type I collagen. Mean ± SD; * indicates p < 0.05; n > 45 tracks, 2 independent experiments; dotted lines indicate length and width of original patterned silicon master.
Notably, the deviation in width across all of the patterned microtracks (11.12 ± 1.29 μm – 15.83 ± 3.89 μm) encompasses over 90% of the range of widths created naturally by the metastatic breast cancer cells (Fig. 2B). Additionally, the architecture of the native tracks was largely similar to the architecture observed in the patterned tracks (Fig. 5). The dimensions of the channel were not altered by the presence of cells. Importantly, the metastatic cancer cells were able to actively remodel their environment by attaching to, exerting tension on, and subsequently bundling surrounding collagen fibers (white arrows, Fig. 5) during a relatively short period of migration through the 3D microtrack. Notably, the cell within this microtrack is fully bounded by collagen, as indicated by the cross-section and side-view of the track. We observed that over the course of migration through the patterned microtrack, the cell would occasionally pull away slightly from one or more walls, without overt preference for either the bottom of the track or the lid, but would rarely lose contact completely with the walls. Additionally, we noted that the degree of collagen contraction dynamically changes over the course of the cell’s migration, as it forms protrusions and pulls against the track walls.
Fig. 5.
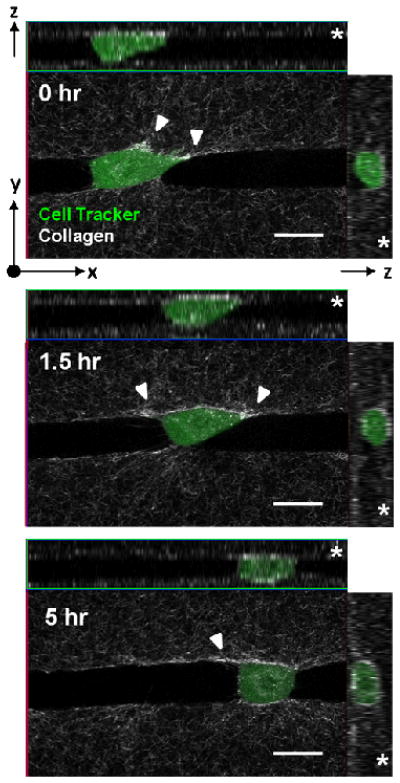
Patterned microtracks accurately reproduce tracks formed naturally by cancer cells. Cell-tracker labeled MDAMB231 cell within a patterned collagen track which has a fibrillar architecture significantly resembling that of the native proteolytic track. Cross-sectional slices indicate that cell is fully bounded by collagen (z arrow, right), and that a clear pathway is available for the cell to travel through (z arrow, top). Asterisks indicates the ‘lid’ side of the track. Over a period of 5 hours, the cell is able to move over 50 μm from its initial position, while remodeling the extracellular matrix by bundling collagen fibers (white arrows). Scale bars = 20 μm.
Next, the cell morphology of MDAMB231 cancer cells within a 3D matrix and a patterned microtrack was compared. Our data shows that seeding cells within the patterned track did not significantly affect their spread area (Fig. 6A) or perimeter (Fig. 6B). Additionally, similar patterns of aspect ratio and circularity are observed in both 3D matrices and patterned tracks (Fig. 6C). Similar results were observed in MCF10A mammary epithelial cells (data not shown). There was also no observable difference in actin organization in MDAMB231 cells seeded within 3D collagen or patterned tracks. In both cases, the actin was primarily cortical, with a notable lack of stress fibers within the cell (Fig. 6D–E).
Fig. 6.

The actin cytoskeleton of cells in microtracks is similar to that in a 3D matrix. No significant difference (p > 0.05) is observed in MDAMB231 cell area (A) or perimeter (B) when plated within a 3D collagen matrix or microtrack environment. Mean ± SD; n > 30 cells, 2 independent experiments. Cells in 3D matrices and tracks also have similar distributions of aspect ratio and circularity (C). Actin structure of the MDAMB231 cell within a 3D matrix (D) and within a patterned collagen track (E) is largely cortical. Fluorescent staining of actin fibers (left), confocal reflectance imaging of the collagen fibers (middle), and merge image with additional DAPI staining (right). View in channel is a single slice of the x–y plane. Scale bar = 20 μm.
Collagen microtracks facilitate migration in invasive and non-invasive cell types
Because tracks can provide an uninterrupted path for cells to follow17, we first examined the effect of the nature of the collagen environment on cell migration of a non-invasive mammary epithelial cell line (MCF10A). When seeded within a 3D collagen matrix, these cells are able to spread and extend multiple protrusions into the matrix, but are largely unable to propel themselves through the matrix (Fig. 7A). Within 100 minutes, MCF10A cells are only able to penetrate less than 25 μm away from their initial position (Fig. 7B). However, when seeded within the patterned collagen microtracks, MCF10A cells are able to quickly and persistently migrate through the matrix (Fig. 7C, see also Electronic Supplementary Information, Video 1). Within 100 minutes of initiating migration, these cells are able to move >150 μm away from their initial position (Fig. 7D), more than six times the distance they were able to achieve in a 3D matrix. As expected from these observations, the speed and invasion distance of MCF10A cells is significantly increased within the collagen tracks, compared to within 3D collagen matrices (Fig. 7E–F). Moreover, the percentage of motile cells in the tracks is increased by over three-fold, compared to cells in 3D collagen (Table 1).
Fig. 7.

Tracks facilitate single cell migration in 3D collagen matrix in non-invasive cells. (A) Time lapse phase contrast images of MCF10A mammary epithelial cells migrating in a 3D collagen matrix show relatively little net cell displacement. (B) Rose plot shows trajectories of MCF10A cells migrating within a fully 3D type I collagen matrix over 100 minutes. (C) Time lapse phase contrast images of MCF10A cells within a patterned 3D collagen track demonstrates the ability of these cells to significantly invade the track. (D) Rose plot shows trajectories of MCF10A cells migrating within collagen tracks for 100 minutes. Note that the trajectories have been arbitrarily rotated for a more direct visual comparison. (E) Quantification of cell migration speed and (F) maximum invasion distance indicates that MCF10A cells are able to migrate more quickly and travel a greater distance within the patterned collagen microtracks than within a 3D collagen matrix. Scale bars = 100 μm. Mean ± SD; * indicates p < 0.05; n > 30 cells, 2 independent experiments.
The migration behavior of a cell type which is consistently found to be highly invasive in a variety of extracellular matrices4,13, the MDAMB231 metastatic breast cancer cells, was also examined. These cells were able to efficiently migrate within 3D collagen, permeating between 50 and 100 μm into a collagen matrix within 100 minutes (Fig. 8A). As expected, these cells were also able to migrate persistently within collagen tracks (Fig. 8B). Additionally, the percentage of motile cells was also increased by nearly 40% in the microtracks (Table 1).
Fig. 8.

Inhibiting MMP activity increases cancer cell migration through collagen microtracks. (A) Rose plot shows trajectories of MDAMB231 metastatic breast cancer cells migrating within a fully 3D collagen matrix and (B) in collagen tracks over 100 minutes. (C) Rose plots show that treatment with GM6001, a broad spectrum MMP inhibitor, decreases invasion in 3D, but (D) increases invasion within the collagen tracks. Note that the trajectories in (B) and (D) have been arbitrarily rotated for a more direct visual comparison to (A) and (C). (E) Quantification of speed indicates that treatment with GM6001 has no effect on speed in a fully 3D matrix, but increases the speed of cells within the microtracks. Additionally, treatment with a structural analog of GM6001 (NC-GM6001) has no significant effect on speed in 3D matrices or microtracks. Mean + SD; * indicates p < 0.05; n > 30 cells, 3 independent experiments. (F) GM6001 treatment of cells migrating on 2D glass substrates has no effect on cell speed. Mean + SD; n > 40 cells, 1 independent experiment. (G) Treatment of cells with other broad spectrum inhibitors (BB2516 and a commercial protease inhibitor cocktail) has no effect on cell speed in the tracks. Mean + SD; n > 60 cells, 2 independent experiments. (H) Similarly, inhibiting MT1-MMP with siRNA significantly decreases speed in 3D matrices, but has no effect within tracks. Mean + SD; * indicates p < 0.05; n > 30 cells, 2 independent experiments.
Because previous studies have indicated that MDAMB231 cells require MMP activity to migrate4, we treated these cells with GM6001, a commonly used broad spectrum MMP inhibitor4. We found that within a 3D matrix, treatment with GM6001 appeared to slightly decrease the invasion of the MDAMB231 cells (Fig. 8C). Surprisingly, treating cells seeded within collagen microtracks with GM6001 significantly increased the invasive capacity of the MDAMB231 cells (Fig. 8D). When we quantified this data, we found that there was no statistically significant difference in cell speed with GM6001 treatment in 3D (Fig. 8E). On the contrary, treatment with GM6001 significantly increased the speed (Fig. 8E) of the MDAMB231 cells seeded within the track microenvironment while having no effect on cell migration on a rigid, 2D glass substrate (Fig. 8F). To further eliminate the possibility that this increase in speed is the result of off-target effects of GM6001, a functionally inactive structural analog of GM6001 (NC-GM6001) was applied to the cells in both 3D matrices and patterned tracks. Cells treated with NC-GM6001 displayed speeds that were not statistically different from the speed of control cells in either 3D collagen matrices or patterned channels (Fig. 8E).
To determine whether this observed increase in speed upon MMP inhibition was specific to this particular inhibitor, we treated cells with another commonly used broad spectrum MMP inhibitor which has been used clinically, BB251613,32,40, as well as a commercial protease inhibitor cocktail (PIC). Interestingly, these data do not show the same trend of increasing speed with MMP inhibition observed with GM6001 (Fig. 8G). However, migration speed is not statistically different from control cells, indicating that treatment with these MMP inhibitors does not negatively affect cancer cell migration in the collagen microtracks.
Finally, because most exogenous MMP inhibitors act on a broad range of substrates and their effects can be difficult to interpret, we chose to specifically focus on a single MMP. MT1-MMP has been repeatedly found to be the primary driver of cancer cell invasion in 3D4,12. Therefore, we specifically examined its role in migration within our 3D microtracks. We transfected the highly metastatic MDAMB231 breast cancer cells with siRNA targeting MT1-MMP and confirmed a 75% knockdown via qRT-PCR. These cells were seeded into either 3D collagen matrices or within the patterned collagen microtracks. Our results indicate that MT1-MMP knockdown significantly decreased speed within 3D matrices, but had no effect on migration speed within the tracks (Fig. 8H). These results support our findings with the broad spectrum MMP inhibitors, confirming that migration within the patterned microtracks can occur independently of MMP activity.
Discussion
In this study we demonstrate the use of patterned collagen microtracks to successfully mimic the natural tracks formed by cancer cells. Our method for studying microtrack migration in single cells is easy to perform using standard laboratory micromolding techniques (Fig. 1C). We show that tracks can be reproducibly patterned in collagen gels (Figs. 3–4), and using confocal reflectance microscopy, we show that the structure of the microtracks is very similar to the architecture of naturally created proteolytic tracks (Figs. 2, 5). These tracks increase both the migration speed and invasion distance of invasive MDAMB231 breast cancer cells and non-invasive MCF10A mammary epithelial cells compared to migration speed within 3D collagen matrices (Figs. 7, 8).
In this study we focus on mammary epithelial cells and breast carcinoma cells, primarily because the breast microenvironment contains many ‘track-like’ structures that our patterned collagen mimics13,41. Initially, epithelial cells in the breast exist in duct structures, and tumor cells can migrate along these structures during carcinoma in situ before invasion actually occurs. Invasion begins when the basement membrane surrounding these ducts is degraded, allowing cells to penetrate beyond the duct, making first contact with the surrounding ECM fibers, and thus initiating invasion41. At this point the leading cancer cells can create new tracks into the surrounding tissue, as described previously16. The structure of the mammary gland microenvironment is highly heterogeneous, with regions of loosely connected fibers and regions of densely bundled and aligned fibers that develop throughout neoplastic progression41,42. Despite the increase in collagen density, there remain significant inter-fiber clefts in the peritumoral region which serve as a network of microtracks through which cancer cells have been observed to invade. Additionally, stromal carcinoma associated fibroblasts have also been shown to create proteolytic tracks that tumor cells can subsequently use to invade14.
Although we have focused our study here on the breast microenvironment, these tracks could be used to mimic other physiologically relevant microenvironments. We use fibrillar type I collagen because it is the most dominant structural protein found in the breast and its effects on tumor cells have been extensively studied in a variety of contexts5. However, the techniques described here could be readily adapted for use with other structural protein compositions, such as laminin or fibronectin, to mimic other regions of the body. For example, there is considerable evidence that glioma cells in the brain can invade using similar track-like structures. Interstitial migration in the brain has been shown to occur along blood vessels, myelinated fibers, and white matter tracts41. These microtracks could prove to be a useful method for studying these methods of tumor migration as well.
One of the most interesting and surprising results of this study was the effect of the broad spectrum MMP inhibitor GM6001 on the migration of metastatic MDAMB231 breast cancer cells within the confined collagen tracks. While treatment with GM6001 inhibited cell migration slightly within a 3D matrix, it had the opposite effect on cells seeded in the patterned microtracks (Fig. 8). We observed a significant increase in the migration speed of MDAMB231 cells treated with GM6001 within the microtracks. Because GM6001 is known to have significant off-target effects43, we treated cells with a functionally inactive structural analog (NC-GM6001)44. We did not observe a similar result with cells treated with this negative control, nor did treatment with GM6001 affect cell migration speed in 2D (Fig. 8F), suggesting that this result does not necessarily stem from an off-target effect of the drug. However, we acknowledge that the structural analog may not account for 100% of the off-target effects of GM6001. Moreover, our subsequent studies with additional broad spectrum inhibitors BB2516 and PIC, as well as our MT1-MMP siRNA knockdown studies suggest that this increased cell speed may not be a global phenomenon with respect to MMP inhibition, but may instead be specific to this inhibitor. GM6001 acts on over 20 different targets45, some of which are also inhibited to various extents by BB2516 and/or PIC, and therefore it is difficult to determine if one of these individual targets may be specifically responsible for this surprising result. Additionally, broad spectrum MMP inhibitors have been shown to induce activation of select MMP’s under certain conditions. In particular, BB2516 and GM6001 have both been shown to lead to enhanced pro-MMP2 activation when tissue inhibitor of metalloproteinase 2 (TIMP2) is present46,47. Therefore, the complex and varied functions of these inhibitors can be difficult to interpret directly.
There are several different mechanisms that could potentially explain the increased speed observed with GM6001 treatment, if it is not the result of a simple off-target effect. It is possible that one or more of the MMP’s expressed by MDAMB231 cells may expose specific cryptic binding sites48,49 on the collagen fibers during proteolytic degradation. These additional binding sites may lead to increased cell adhesion and subsequent decreased motility. If these cryptic sites remain unexposed when the specific MMP’s responsible are inhibited under GM6001 treatment, then this could explain the resulting increase in cell speed. Importantly, we have ruled out MT1-MMP as the culprit for this behavior, as our siRNA knockdown studies did not yield a similar increase in cell speed; rather, there was no observed change in speed compared to controls.
Additionally, we speculate that the increased migration speed of cancer cells under GM6001 treatment could potentially be the result of altered receptor shedding. Receptor shedding has been a noted phenomenon for many years50, but is still not thoroughly understood. Receptor shedding occurs when the extracellular region, or ectodomain, of cellular membrane proteins is cleaved by a molecule known as a ‘sheddase’51. One key class of sheddases are the ADAM metalloproteinase disintegrins, although other MMP’s have been shown to induce shedding behavior as well51. Interestingly, ectodomain cleavage can either activate or inactivate membrane-bound receptors, depending on the specific receptor52. It is estimated that 2–4% of proteins on the cell surface are subjected to ectodomain shedding53. Ectodomain shedding can induce paracrine signalling through the release of growth factors52. Friedl et al. have shown that MV3 melanoma cells may routinely shed α2β1 integrins at cellular detachment sites in 3D matrices, releasing adhesions and promoting cell migration, and suggest that this behavior may be a potential prerequisite for the release of adhesive junctions54,55. It is possible then that GM6001 may enhance the shedding of integrins or other adhesion molecules, which would subsequently decrease the adhesion of the MDAMB231 cells to the walls of the channels and potentially lead to increased migration speed. Interestingly, BB2516 is often used to inhibit the ADAM proteinases which are responsible for many receptor shedding events51. Therefore, this may potentially help explain the differences observed between GM6001 and BB2516 treatment. Additionally, GM6001 could be altering the matrix deposition or matrix reorganization activities of the cell. Further experiments will be required to determine the precise mechanisms governing this phenomenon.
Regardless of the specific mechanism by which GM6001 increases cell speed in the microtracks, we observe no impairment in cell migration through the 3D microtracks under several different modes of MMP inhibition, suggesting that this behavior occurs in an MMP-independent fashion. These data lead us to speculate that perhaps one reason that MMP inhibitors fail when administered to patients19 could be because cancer cells are able to continue migrating within pre-formed tracks regardless of current MMP activity. Therefore, if the cancer cells have already begun forming these structures at the time that treatment is initiated, then this could explain why the MMP inhibitor does not have a significant effect on invasion.
Conclusions
We have presented a straightforward and easily adaptable platform for studying tumor cell motility in patterned collagen microtracks which mimic the complex architecture of the native tumor microenvironment. We have shown that collagen tracks of single cell width can be reproducibly patterned over a range of collagen densities, and that these tracks qualitatively and quantitatively mimic native proteolytic collagen microtracks. Additionally, these tracks facilitate the migration of both non-invasive and invasive cell types in a 3D environment in an MMP-independent fashion, perhaps revealing novel insight into the clinical challenges currently facing MMP inhibitors.
Supplementary Material
Video 1: File Name: KraningRush_ESI_Video1
Descriptive Title: Mammary Epithelial Cell Migrating in 3D Collagen Channel
Caption: MCF10A mammary epithelial cells quickly and persistently migrate within a 3D type I collagen channel over 14 hours.
Acknowledgments
The authors would like to acknowledge Jonathan Charest for his technical help in fabricating the silicon masters used in this study. The authors would also like to acknowledge the use of equipment and resources at the Cornell NanoScale Science and Technology Facility (CNF), the Cornell Nanobiotechnology Center (NBTC), and the Cornell Center for Materials Research (CCMR). This work was supported by the Cornell Center on the Microenvironment & Metastasis through Award Number U54CA143876 from the National Cancer Institute and a National Science Foundation – National Institute of Health Physical and Engineering Sciences in Oncology (PESO) award (Award number 1233827) to CAR. This work was also supported by National Science Foundation Graduate Research Fellowships to CMK and SPC.
Footnotes
Electronic Supplementary Information (ESI) available: [One supplemental video]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Christofori G. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 2.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 3.Friedl P, Wolf K. J Cell Biol. 2010;188:11–19. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabeh F, Shimizu-Hirota R, Weiss SJ. J Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, White JG, Keely PJ. BMC Med. 2008;6:11. doi: 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mehta K, Fok J, Miller FR, Koul D, Sahin AA. Clin Cancer Res. 2004;10:8068–8076. doi: 10.1158/1078-0432.CCR-04-1107. [DOI] [PubMed] [Google Scholar]
- 8.Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schroder E, Zhou J, Brunton VG, Barker N, Clevers H, Sansom OJ, Anderson KI, Weaver VM, Olson MF. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Provenzano PP, Inman DR, Eliceiri KW, Trier SM, Keely PJ. Biophys J. 2008;95:5374–5384. doi: 10.1529/biophysj.108.133116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang W, Wyckoff JB, Frohlich VC, Oleynikov Y, Huttelmaier S, Zavadil J, Cermak L, Bottinger EP, Singer RH, White JG, Segall JE, Condeelis JS. Cancer Res. 2002;62:6278–6288. [PubMed] [Google Scholar]
- 12.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Cell. 2003;114:33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 13.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. Nat Cell Biol. 2007;9:893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 14.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Nat Cell Biol. 2007;9:1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 15.Sabeh F, Ota I, Holmbeck K, Birkedal-Hansen H, Soloway P, Balbin M, Lopez-Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. J Cell Biol. 2004;167:769–781. doi: 10.1083/jcb.200408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedl P, Wolf K. Cancer Res. 2008;68:7247–7249. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 17.Ilina O, Bakker GJ, Vasaturo A, Hofmann RM, Friedl P. Phys Biol. 2011;8:015010. doi: 10.1088/1478-3975/8/1/015010. [DOI] [PubMed] [Google Scholar]
- 18.Alcaraz J, Mori H, Ghajar CM, Brownfield D, Galgoczy R, Bissell MJ. Integr Biol (Camb) 2011;3:1153–1166. doi: 10.1039/c1ib00073j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coussens LM, Fingleton B, Matrisian LM. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 20.Friedl P, Wolf K. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 21.Rolli CG, Seufferlein T, Kemkemer R, Spatz JP. PLoS One. 2010;5:e8726. doi: 10.1371/journal.pone.0008726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mak M, Reinhart-King CA, Erickson D. PLoS One. 2011;6:e20825. doi: 10.1371/journal.pone.0020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pathak A, Kumar S. Proc Natl Acad Sci U S A. 2012;109:10334–10339. doi: 10.1073/pnas.1118073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balzer EM, Tong Z, Paul CD, Hung WC, Stroka KM, Boggs AE, Martin SS, Konstantopoulos K. FASEB J. 2012 doi: 10.1096/fj.12-211441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Irimia D, Toner M. Integr Biol (Camb) 2009;1:506–512. doi: 10.1039/b908595e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ochsner M, Dusseiller MR, Grandin HM, Luna-Morris S, Textor M, Vogel V, Smith ML. Lab Chip. 2007;7:1074–1077. doi: 10.1039/b704449f. [DOI] [PubMed] [Google Scholar]
- 27.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 28.Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. Integr Biol (Camb) 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraning-Rush CM, Califano JP, Reinhart-King CA. PLoS One. 2012;7:e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson CM, Inman JL, Bissell MJ. Nat Protoc. 2008;3:674–678. doi: 10.1038/nprot.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Debnath J, Muthuswamy SK, Brugge JS. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 32.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P. J Cell Biol. 2003;160:267–277. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carey SP, Kraning-Rush CM, Williams RM, Reinhart-King CA. Biomaterials. 2012 doi: 10.1016/j.biomaterials.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kraning-Rush CM, Carey SP, Califano JP, Smith BN, Reinhart-King CA. Phys Biol. 2011;8:015009. doi: 10.1088/1478-3975/8/1/015009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charest JM, Califano JP, Carey SP, Reinhart-King CA. Macromol Biosci. 2012;12:12–20. doi: 10.1002/mabi.201100264. [DOI] [PubMed] [Google Scholar]
- 36.Cornhill JF, Levesque MJ, Herderick EE, Nerem RM, Kilman JW, Vasko JS. Atherosclerosis. 1980;35:321–337. doi: 10.1016/0021-9150(80)90130-6. [DOI] [PubMed] [Google Scholar]
- 37.Stokes CL, Lauffenburger DA, Williams SK. J Cell Sci. 1991;99(Pt 2):419–430. doi: 10.1242/jcs.99.2.419. [DOI] [PubMed] [Google Scholar]
- 38.Reinhart-King CA, Dembo M, Hammer DA. Biophys J. 2008;95:6044–6051. doi: 10.1529/biophysj.107.127662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kraning-Rush CM, Carey SP, Califano JP, Reinhart-King CA. Quantifying traction stresses in adherent cells. In: Arkin A, Asthagiri AR, editors. Comp Methods Cell Biol. Elsevier; New York: 2012. In press. [DOI] [PubMed] [Google Scholar]
- 40.Bramhall SR, Rosemurgy A, Brown PD, Bowry C, Buckels JA. J Clin Oncol. 2001;19:3447–3455. doi: 10.1200/JCO.2001.19.15.3447. [DOI] [PubMed] [Google Scholar]
- 41.Gritsenko PG, Ilina O, Friedl P. J Pathol. 2012;226:185–199. doi: 10.1002/path.3031. [DOI] [PubMed] [Google Scholar]
- 42.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobsen FE, Buczynski MW, Dennis EA, Cohen SM. Chembiochem. 2008;9:2087–2095. doi: 10.1002/cbic.200800148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azuma H, Inamoto T, Sakamoto T, Kiyama S, Ubai T, Shinohara Y, Maemura K, Tsuji M, Segawa N, Masuda H, Takahara K, Katsuoka Y, Watanabe M. Cancer Res. 2003;63:8090–8096. [PubMed] [Google Scholar]
- 45.Levy DE, Lapierre F, Liang W, Ye W, Lange CW, Li X, Grobelny D, Casabonne M, Tyrrell D, Holme K, Nadzan A, Galardy RE. J Med Chem. 1998;41:199–223. doi: 10.1021/jm970494j. [DOI] [PubMed] [Google Scholar]
- 46.Toth M, Bernardo MM, Gervasi DC, Soloway PD, Wang Z, Bigg HF, Overall CM, DeClerck YA, Tschesche H, Cher ML, Brown S, Mobashery S, Fridman R. J Biol Chem. 2000;275:41415–41423. doi: 10.1074/jbc.M006871200. [DOI] [PubMed] [Google Scholar]
- 47.Ikejiri M, Bernardo MM, Bonfil RD, Toth M, Chang M, Fridman R, Mobashery S. J Biol Chem. 2005;280:33992–34002. doi: 10.1074/jbc.M504303200. [DOI] [PubMed] [Google Scholar]
- 48.Ricard-Blum S, Ballut L. Front Biosci. 2011;16:674–697. doi: 10.2741/3712. [DOI] [PubMed] [Google Scholar]
- 49.Rundhaug JE. J Cell Mol Med. 2005;9:267–285. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Black PH. N Engl J Med. 1980;303:1415–1416. doi: 10.1056/NEJM198012113032411. [DOI] [PubMed] [Google Scholar]
- 51.Huovila AP, Turner AJ, Pelto-Huikko M, Karkkainen I, Ortiz RM. Trends Biochem Sci. 2005;30:413–422. doi: 10.1016/j.tibs.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 52.Blobel CP. Nat Rev Mol Cell Biol. 2005;6:32–43. doi: 10.1038/nrm1548. [DOI] [PubMed] [Google Scholar]
- 53.Arribas J, Massague J. J Cell Biol. 1995;128:433–441. doi: 10.1083/jcb.128.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Friedl P, Maaser K, Klein CE, Niggemann B, Krohne G, Zanker KS. Cancer Res. 1997;57:2061–2070. [PubMed] [Google Scholar]
- 55.Huttenlocher A, Sandborg RR, Horwitz AF. Curr Opin Cell Biol. 1995;7:697–706. doi: 10.1016/0955-0674(95)80112-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1: File Name: KraningRush_ESI_Video1
Descriptive Title: Mammary Epithelial Cell Migrating in 3D Collagen Channel
Caption: MCF10A mammary epithelial cells quickly and persistently migrate within a 3D type I collagen channel over 14 hours.