

Abstract
Förster resonant energy transfer (FRET) is extensively used to probe macromolecular interactions and conformation changes. The established FRET lifetime analysis method measures the FRET process through its effect on the donor lifetime. In this paper we present a method that directly probes the time-resolved FRET signal with frequency domain Fourier lifetime excitation-emission matrix (FLEEM) measurements. FLEEM separates fluorescent signals by their different phonon energy pathways from excitation to emission. The FRET process generates a unique signal channel that is initiated by donor excitation but ends with acceptor emission. Time-resolved analysis of the FRET EEM channel allows direct measurements on the FRET process, unaffected by free fluorophores that might be present in the sample. Together with time-resolved analysis on non-FRET channels, i.e. donor and acceptor EEM channels, time resolved EEM analysis allows precise quantification of FRET in the presence of free fluorophores. The method is extended to three-color FRET processes, where quantification with traditional methods remains challenging because of the significantly increased complexity in the three-way FRET interactions. We demonstrate the time-resolved EEM analysis method with quantification of three-color FRET in incompletely hybridized triple-labeled DNA oligonucleotides. Quantitative measurements of the three-color FRET process in triple-labeled dsDNA are obtained in the presence of free single-labeled ssDNA and double-labeled dsDNA. The results establish a quantification method for studying multi-color FRET between multiple macromolecules in biochemical equilibrium.
OCIS codes: (170.6280) Spectroscopy, fluorescence and luminescence; (170.2520) Fluorescence microscopy
1. Introduction
First established by Theodor Förster in the 1940s [1], Förster resonant energy transfer (FRET) is widely used as a fluorescence spectroscopy method to measure distances between fluorophores on the nanometer scale. FRET occurs when an excited donor fluorophore transfers its energy to an adjacent ground-state acceptor fluorophore through dipole coupling. This process depends strongly on the distance between molecules in the 1-10 nm range, and can therefore be exploited as a “spectroscopic ruler” [2]. With recent advances in fluorescence proteins, organic dyes and instrumentation, FRET has found an ever increasing range of applications in biological studies, ranging from tracking protein-protein interactions in cellular processes [3], probing DNA/RNA regulations and dynamics [4], to high-throughput drug screening [5].
Quantification methods of two-color FRET are well established [6, 7]. Fluorescent signals from a FRET system can be represented in terms of excitation-emission matrix (EEM) channels, which are characterized by their individual exciters (which fluorophore absorbs the excitation photon) and emitters (which fluorophore emits the fluorescence photon). For two-color FRET, three possible EEM channels exist: excitation of the donor and its subsequent fluorescence emission (donor EEM channel); excitation of the acceptor and its subsequent emission (acceptor EEM channel); and excitation of the donor, which excites the acceptor via FRET, followed by emission from the acceptor (FRET EEM channel). To quantify the absolute FRET efficiency, the current standard practice is to measure the lifetime of the donor EEM channel, in which the quenching effect of FRET causes a lifetime decrease. While it has long been known that the signal in the FRET EEM channel follows the excited-state reaction model [8, 9], few studies have directly measured the FRET EEM channel to extract information about the FRET process [10, 11]. In this paper, we present a frequency domain lifetime method that performs time-resolved analysis directly on the FRET EEM channel, as well as EEM channels of the donor and acceptor.
The advantage of analyzing FRET in a time-resolved EEM representation is two-fold:
First, when quenched donor and unquenched donor coexist, the time-resolved EEM approach provides more sensitive and more accurate quantification on the FRET efficiency and the percentage of quenched donor molecules among all donor molecules. Free fluorophores often coexist with FRET complexes in a chemical equilibrium in biological studies. Even in FRET sensors that are genetically linked together, FRET-inactive free fluorophores may still exist due to differential photobleaching between different fluorescent proteins or incomplete maturation of fusion proteins. When the donor population is a mixture of quenched and unquenched donors, the decay behavior of the donor EEM channel follows the multi-exponential decay model, in which the unquenched donor lifetime can be pre-calibrated but the quenched donor lifetime is unknown. Because two exponential decay functions with different decay constants are not orthogonal to each other, an accurate fitting of the multi-exponential decay model with unknown lifetimes requires a large separation between decay constants [12, 13], a balanced contribution of all decay components, as well as relatively high signal-to-noise ratio. These conditions might not be achievable in cellular imaging studies where the difference between quenched and unquenched donor lifetimes could be small due to a low FRET transfer rate, quenched donors could be the minority, and the fluorescent signal could be weak. As we experimentally proved in this paper (Table 2), even in the ideal case of a FRET standard solution, when the FRET quenching effect on the donor is modest, or when donor concentration is overabundant, the quenched donor lifetime obtained from direct multi-exponential fitting of the donor EEM channel becomes less accurate, or fails to converge. On the contrary, the FRET EEM channel involves only donor molecules that participate in FRET. The quenched donor lifetime measured from the FRET EEM channel is not affected by unquenched donor concentration, and its accuracy remains the same regardless the strength of FRET effect or the amount of unquenched donor.
Second, the time-resolved EEM representation of FRET can be utilized in investigation of multi-color FRET processes involving three or more fluorophore molecules. Quantification of multi-color FRET could greatly facilitate the understanding of complex cellular processes, which almost always involve multiple components through networks of dynamic interactions. In recent years, several techniques have been developed to enable two-pair or three-color FRET in both in vitro studies [14, 15] and in vivo imaging in live cells [16, 17]. These methods are ratiometric-based or at most partially lifetime-based [18]. The key challenge in quantifying multi-color FRET is the significantly increased complexity due to possible multi-way exciter-to-emitter photon-pathways. These pathways have different combinations of excitation and emission wavelengths, and are naturally separated in an EEM into different spectral channels. By analyzing each individual EEM spectral channel, the complex multi-way interactions in a multi-color FRET process can be better quantified.
In this paper, we combine the above two advantages of time-resolved EEM to quantitatively interpret multi-color FRET signal from a mixture of FRET complexes and free labels. The EEM measurements are based on Fourier lifetime excitation-emission matrix spectroscopy (FLEEM), a frequency domain lifetime technique we previously developed, which performs fluorescence intensity and lifetime measurements in all EEM channels simultaneously [19]. We demonstrate that time-resolved analysis on the EEM can extract FRET distances between fluorophores in a mixture of triple-, double- and single-labeled structures, without the need of selective photo bleaching or sample purification. Percentages of different molecular species are also obtained simultaneously.
The capability of the FLEEM spectroscopy and time-resolved EEM analysis was tested with a three-color FRET standard formed by hybridizing three fluorescently labeled single-strand DNA (ssDNA) oligonucleotides. Incomplete hybridization of the three ssDNA produced a mixture of triple-labeled and double-labeled double-strand DNA (dsDNA), as well as un-hybridized single-labeled ssDNA. FLEEM measures all EEM channels in parallel. Through time-resolved EEM analysis, we extracted distances between fluorophores in the triple-labeled dsDNA in the mixture. Percentages of fluorophores in triple-labeled, double-labeled and single-labeled DNA were simultaneously quantified. The distance measurement results were consistent with the oligonucleotide design and control experiments with two-color FRET.
The FLEEM spectroscopy is compatible with live cell confocal fluorescence imaging. Results presented in this paper establish the theoretical framework and the quantification algorithm for future multi-color FRET imaging studies through FLEEM.
2. Quantitative multi-color FRET analysis with time-resolved excitation-emission matrix
The key challenge in multi-color FRET is the large number of exciter-to-emitter photon pathways present in a multi-labeled FRET complex. FRET allows photon energy to migrate from an exciter fluorophore to a red-shifted emitter fluorophore. As the number of fluorophores N increases, the number of possible exciter-to-emitter combinations increases as N(N + 1)/2. As shown in Fig. 1(a) , in a three-color FRET process between fluorescein, Cy3 and Cy5, 6 different photon pathways are possible: fluorescein excitation-emission; Cy3 excitation-emission; Cy5 excitation-emission; fluorescein excitation-Cy3 emission; fluorescein excitation-Cy5 emission; and Cy3 excitation-Cy5 emission.
Fig. 1.

Excitation emission matrix (EEM) representation of three-color FRET between fluorescein, Cy3 and Cy5. (a) Photon pathways in a three-color FRET process. Six possible exciter-to-emitter photon pathways are present. (b) EEM representation of the three-color FRET as a function of both excitation and emission wavelengths. Different photon pathways occupy different regions of the EEM. For each photon pathway, the excitation spectrum follows the exciter, and the emission spectrum follows the emitter.
In multi-color FRET, different photon pathways represent different energy migration processes, and have different time-resolved responses. Comprehensive multi-color FRET analysis requires characterization of all photon pathways. In this section, we present a theoretical model that uses time-resolved EEM to analyze all photon pathways. Section 2.1 introduces the EEM representation of photon pathways in a multi-color FRET system, followed by section 2.2 that discusses how to apply spectral bleedthrough correction on the measured EEM to recover the ideal EEM, in which each spectral channel represent a unique exciter-to-emitter photon pathway. Section 2.3 discusses the frequency domain responses of ideal EEM channels representing different photon pathways, and section 2.4 discusses how to use frequency-domain time-resolved EEM information to quantify multi-color FRET. The EEM-based quantification method allows the quenched donor lifetimes and the molar percentages of different FRET complexes being extracted from a single time-resolved EEM measurement.
2.1 Excitation-emission matrix
Exciter-to-emitter photon pathways of a multi-color FRET sample can be represented by an excitation-emission matrix (EEM), as shown in Fig. 1(b), which is a two-variable function of both excitation and emission wavelengths. Due to the conservation of energy, the EEM is a triangular matrix. Different photon pathways are associated with different combinations of excitation and emission wavelengths, and therefore occupy different regions (spectral channels) of the EEM. Direct excitation-emissions of individual fluorophores occupy diagonal channels on the EEM, while FRET signals occupy spectral channels with larger Stokes shifts, located above the diagonal channels. Measurements on each EEM spectral channel reflect the properties of the photon pathway it represents. A direct excitation-emission channel of a fluorophore species measures the ensemble average of the fluorophores in both quenched states when they serve as donors in FRET complexes, and unquenched states as free fluorophores or acceptors in FRET complexes. A FRET EEM channel is present only if energy transfer is active between the specific exciter-emitter pair, i.e. the exciter and the emitter form molecular complexes. As discussed in section 2.3, the fluorescence decay behavior of the FRET EEM channel is different from the direct excitation-emission channels of individual fluorophores, which follow the single- or multi-exponential decay model.
2.2 Spectral bleedthrough correction of EEM
In an ideal EEM, different exciter-to-emitter photon pathways are completely separated into different EEM channels. However as clearly shown in Fig. 1(b), excitation and emission spectral overlapping between different fluorophores cause spectral bleedthrough between different EEM channels. For instance, the FRET channel between fluorescein and Cy3 (excitation ~488 nm, emission ~580 nm) has three components: signals generated by fluorescein-Cy3 FRET, excitation bleedthrough of Cy3, and emission bleedthrough of fluorescein. Spectral bleedthrough correction is therefore needed in order to recover the ideal EEM.
Spectral bleedthrough correction is a key component for all multi-color FRET studies, and has been discussed in detail in both cell imaging [16] and single molecule detection [20]. The majority of these studies present correction methods as a series of linear equations, which can be unified as a single matrix equation in the form of EEM spectral bleedthrough correction [21, 22]:
Under the framework of EEM, the relationship between an ideal EEM I and the experimentally measured EEM I′ can be written as
| (1) |
where BEm is the emission bleedthrough matrix, and BEx is the excitation bleedthrough matrix [21]. The measured EEM I′ is an m-by-n matrix, where m is the number of emission spectral channels, and n is the number of excitation spectral channels in the measurement. For a three-color FRET process, I is a 3-by-3 matrix. The emission bleedthrough matrix is an m-by-3 matrix and the excitation bleedthrough matrix is a 3-by-n matrix. The bleedthrough correction procedure aims to recover I from I′ with pre-calibrated BEm and BEx. For three-color FRET, a minimum of three excitation and emission channels are needed to completely characterize the FRET process. In such case, Eq. (1) becomes
| (2) |
where subscripts i and j denote the excitation and emission spectral channels. is the emission bleedthrough of the ith fluorophore in the jth emission channel, and is the excitation bleedthrough of the jth fluorophore by the ith excitation source. Because the excitation and emission spectra of the fluorophores do not significantly change in a FRET process, the excitation and emission bleedthrough matrices can be calibrated with single labeled samples. The spectral bleedthrough can then be corrected through simple matrix manipulation:
| (3) |
The spectral bleedthrough correction is a linear process. Therefore all linear quantities in the EEM form can be corrected as in Eq. (3). These include but are not limited to fluorescence intensity, time-resolved fluorescence decay measurements in the time domain Iij(t), and complex fluorescence responses in the frequency domain [21, 22].
2.3 Time-resolved analysis of EEM channels
Fluorescence lifetime analysis is deemed as the most robust method for quantifying FRET interactions [7]. The purpose of time-resolved EEM analysis is to measure quenched donor lifetimes and percentages of quenched donors in their total population, and to provide quantitative measurements of multi-color FRET.
2.3.1 Two-color FRET signal
After spectral bleedthrough correction, each spectral channel in the ideal EEM represents a single photon pathway. We first consider the case of a two-color FRET complex, in which three EEM channels exist: donor, acceptor and FRET. The donor or acceptor EEM channel contains signals generated by direct excitation-emission of the donor or acceptor, which follow typical exponential decay responses. The FRET channel of a two-color system contains signal generated by a one-step transfer from the donor to the acceptor. The decay behavior of one-step FRET signal follows the same model as excited-state reaction [8, 9], which can be calculated from donor and acceptor decay rate equations in the time domain [10] or in the frequency domain [8, 9]. Alternatively, the FRET process can be modeled with the signal processing theory.
Fluorescence lifetime measurements use either the time domain or the frequency domain methods [12]. In both methods, an excitation source with a specific functional form is used to excite the fluorophores, and the response of the fluorophore IFluo(t) is measured. When a fluorophore is excited by an excitation source I(t), the resulting fluorescence emission response is the convolution between I(t) and the impulse response function of the fluorophore R(t).
| (4) |
R(t) is usually modeled with single or multi-exponential decay functions. In time domain fluorescence lifetime measurements, a pulsed light source (a delta function in time) is used to excite the fluorophores, and the subsequent fluorescence emission is measured in the time domain,
| (5) |
where u(t) is the unit step function. In frequency domain lifetime methods, the intensity of the excitation source is sinusoidally modulated at a frequency ω (a delta function in the frequency domain). The fluorescence emission is then measured in the frequency domain,
| (6) |
where FT refers to the Fourier transform, and is the Fourier transform of the response function R(t).
When FRET occurs, acceptor molecules are excited by energy transfer from excited donor molecules, therefore the donor excited state population serves as the excitation source of acceptor molecules. Since the donor excited state population is directly proportional to its fluorescence emission, the FRET sensitized emission from a two-color FRET complex can be expressed as
| (7) |
where is the donor excited state population, and is the total decay rate of the donor when it is quenched by FRET. k12 is the energy transfer rate from the donor (denoted by subscript “1”) to the acceptor (denoted by subscript “2”). The subscript i in the decay rates and the response functions denotes the index of the fluorophore, and the superscript m…n denoted the molecular complex that the fluorophore is in. Note that the donor response function here is the response of donor in the presence of FRET, with a fluorescence lifetime already shortened by energy transfer to the acceptor. In time domain, the functional form of the time-resolved FRET signal is the difference of two exponential decay functions [10]. In frequency domain, the convolution in Eq. (7) becomes multiplication in the Fourier space, and the FRET signal is given by
| (8) |
2.3.2 Multi-color FRET signal
The major difference between two-color and multi-color FRET is, in multi-color FRET complexes, beside one-step FRET pathways similar to the FRET pathway in two-color FRET complex, it is possible to have multi-step FRET cascade signal in multi-color FRET complex. For example a two-step FRET in a fluorescein-Cy3-Cy5 complex can transfer photon energy from the initial donor (fluorescein, denoted by subscript “1”) to an intermediate acceptor (Cy3, denoted by subscript “2”), which can then serve as the intermediate donor of the final acceptor (Cy5, denoted by subscript “3”). Equation (8) can be generalized to describe an N-step FRET signal,
| (9) |
The frequency domain fluorescence lifetime response of the N-step FRET is a product of individual frequency domain lifetime responses of all fluorophores participating in the chain energy transfer.
The FRET channel frequency responses in Eqs. (8) and (9) hold true for any functional forms of individual fluorophore lifetime response function R1,…,N, which is not limited to single exponential decay. In this paper, organic dyes conjugated to ssDNA are used to test the time-resolved EEM analysis, and their decay models are well documented as single exponential. Thus, single exponential decay models are assumed for all R1…N in subsequent discussions. If multi-exponential decay or other models are more appropriate, the method can be easily adapted to accommodate for such more complex responses by: 1) for a participating fluorophore that serves as the initial or intermediate donor, if its FRET-free state follows the multi-exponential decay model, the quenched lifetime response function R should also follow the multi-exponential decay model, in which all decay lifetime constants are shortened by FRET; and 2) the FRET EEM channel response is the product of several multi-exponential decay functions that represents all participating fluorophores.
2.4 Time-resolved EEM analysis of multi-color FRET samples
Information from different spectral channels in an EEM can be combined to quantify FRET in a complex fluorescent sample. To avoid potential pitfalls in multi-parameter model fitting, instead of fitting all EEM channels in a single step, the time-resolved analysis of the EEM adopts a channel-by-channel stepping analysis format to fit one EEM channel at a time. Each analyzing step benefits from lifetime information obtained from all previous steps, and involves only at most one unknown lifetime parameter.
2.4.1 Channel-by-channel EEM analysis of two-color FRET sample
The frequency-domain time-resolved EEM of pure two-color FRET complex is
| (10) |
When free fluorophores are present, the EEM of the mixture becomes
| (11) |
where C12, C1, and C2 are concentrations of double-labeled complex, free donor and free acceptor labels. In the acceptor EEM channel , the signal comes from direct excitation on the accept and is unrelated to FRET, thus the lifetime of the acceptor EEM channel signal is not affected by FRET, . In consequence, free and FRET-active acceptor cannot be distinguished by time-resolved analysis.
Lifetimes of free and FRET-active donors are different because of their different decay rates,
| (12) |
The donor EEM channel measures the ensemble average of all donor molecules in both quenched and unquenched states. Double exponential decay fitting of the donor channel alone, in theory, could extract decay rates of quenched and unquenched donor, and percentages of the two donor states. However this approach requires high signal quality, balanced contribution from both lifetime components and a large separation between the two lifetime components, which might not be available under certain experimental conditions. In contrast, the FRET EEM channel involves only quenched donor, and are not affected by unquenched donor molecules. The bleedthrough corrected FRET channel can be used to extract lifetime information of quenched donor based on Eq. (8), regardless the amount of unquenched donor in the mixture or the strength of FRET transfer.
The complete analysis of spectral bleedthrough corrected EEM of a two-color FRET mixture is carried out in three steps, with each step only involving at most single lifetime parameter fitting on one EEM channel:
-
(1)
Determine the acceptor lifetime or lifetimes if the acceptor decay follows a multi-exponential model. Since the fluorescence lifetime(s) of the acceptor is not affected by FRET, the acceptor lifetime(s) can be quantified through either the acceptor EEM channel of the mixture, or calibrated with a pure acceptor standard. The acceptor lifetime(s) obtained from the mixture can serve as an internal control for monitoring instrument drifting, which may occur in future time-lapse imaging studies.
-
(2)
The fluorescence decay in FRET channel is a convolution of acceptor and donor fluorescence decay as in Eq. (8). With the acceptor lifetime(s) known, frequency response of the FRET channel signal only has one unknown lifetime model: the decay of quenched donor. If the free donor follows the single exponential decay model, the quenched donor will also be a single exponential decay, whose decay rate is raised by the FRET transfer rate. In case the FRET-free donor follows a multi-exponential decay model, the quenched donor will also be a multi-exponential decay model, whose decay rates are uniformly raised by the FRET transfer rate for all donor lifetime components. By analyzing the response of the bleedthrough-corrected FRET channel with Eq. (8), lifetime(s) of quenched donor can be extracted.
-
(3)
When free donor molecules are present, the donor EEM channel fluorescence decay is a multi-exponential decay containing both quenched and unquenched donors with different lifetimes. Once lifetime(s) of quenched donor is known, the donor EEM channel response can be fitted with the multi-exponential decay model that contains two known lifetime models: the unquenched donor and the quenched donor. The unquenched donor decay model is calibrated with a pure donor sample. The quenched donor decay model is obtained in Step 2. The fitting does not need to fit any lifetime constant, and only needs to find the percentage of quenched donor.
Unlike the donor EEM channel, which contains both quenched and unquenched donor signals, the FRET EEM channel contains only contributions from quenched donors, whose response is convolved with the acceptor fluorescence response. It is therefore more sensitive to changes in FRET effects when a sample has high concentration of unquenched donors. Table 2 compared the performance of the time-resolved EEM analysis with the performance of double exponential fitting on the donor EEM channel with fixed free donor lifetime(s). When the two methods were used to process the same data set, time-resolved EEM analysis method was more robust when the quenched donor was minority or the absolute FRET efficiency was not high.
2.4.2 EEM analysis of two-color FRET sample
In the case of three-color FRET, the frequency response EEM for a pure triple-labeled FRET sample is a 3-by-3 matrix
| (13) |
Three energy transfer rates are present in Eq. (13), k12, k23 and k13, which relates to distances from the initial donor (fluorescein) to the intermediate acceptor (Cy3), the intermediate acceptor to the final acceptor (Cy5) and the initial donor directly to the final acceptor, respectively.
The EEM become more complicated when the sample is mixture of free labels or double-labeled complexes due to incomplete labeling or reaction:
| (14) |
If all FRET processes are present, when the initial donor decays as single exponential, the EEM channel of the initial donor () of the mixture could contain as much as four components with different fluorescence decay rates. For example, lifetimes of fluorescein in four different molecule forms (fluorescein-Cy3-Cy5, fluorescein-Cy3, fluorescein-Cy5 and free fluorescein) are all different, making precise lifetime analysis impossible. To make three-color FRET practical for structural measurements in the presence of free fluorophores and incomplete complexes, FRET constructs needs to be designed in a way that k13 is negligible, i.e. there is no directy FRET between fluorescein and Cy5, so that the number of unknown decay components in the fluorescein EEM channel can be minimized. In our experimental design, k13 was kept negligible by placing fluorescein and Cy5 far apart on the oligonucleotide sequence. This reduced lifetime components in the fluorescein EEM channel () to two (unquenched or quenched by Cy3), and allowed quantitative analysis of the three-color FRET complex in the presence of incomplete complexes or free labels.
Similar to time-resolved EEM analysis of two-color FRET, in three-color FRET, a channel-by-channel analysis of bleedthrough corrected EEM is used to avoid multi-component lifetime fittings. The EEM analysis of three-color FRET proceeds as illustrated in Fig. 2 .The color codes indicate the different fluorescence decay models for individual EEM channels.
Fig. 2.
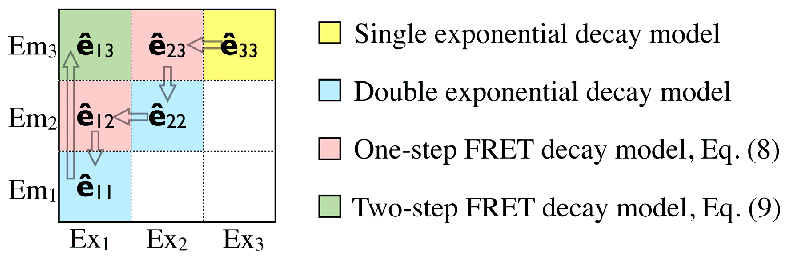
Time-resolved EEM analysis sequence of three-color FRET. The analysis is performed on a channel-by-channel basis, with each channel involving only at most one unknown lifetime parameter. EEM channels in the illustration are color-coded by their decay models. The analysis first obtains the longest wavelength acceptor (fluorophore No. 3) lifetime τ3 in ê33, then finds quenched lifetime of fluorophore No. 2 in FRET channel ê23. The percentage of quenched fluorophore, P2 is then calculated from fluorophore 2 EEM channel ê22. The FRET channel ê12 is next, which yields the quenched lifetime of fluorophore 1. The percentage of quenched fluorophore No. 1, P1 is extracted from the EEM channel ê11, and finally the FRET channel ê13 serves as a verification of the time-resolved EEM analysis.
-
(1)
Determine the lifetime of Cy5 (), the reddest fluorophore from a pure Cy5-labeled sample or, from the Cy5 EEM channel . The lifetime of Cy5 remains unchanged because Cy5 serves as acceptor in all FRET complexes.
-
(2)
EEM channel contains the signal generated by Cy3-Cy5 FRET. Channel can be fitted with the one-step FRET model (Eq. (8)) with known acceptor lifetime . The fitting yields , the lifetime of Cy3 when it is quenched by Cy5.
-
(3)
EEM channel contains fluorescence decays from both quenched and unquenched Cy3. A double exponential decay lifetime fitting with known lifetimes of quenched Cy3 (solved in Step 2) and unquenched Cy3 (measured from pure Cy3-labeled samples) can be used to obtain the percentage of quenched Cy3 (P2). The percentage of quenched Cy3 measured in this channel is the ensemble average of all possible molecular forms that contains Cy3, i.e. all molecules containing Cy3-Cy5 vs. all molecules containing Cy3.
-
(4)
EEM channel contains FRET signals between fluorescein and Cy3, whose response is the product of the donor (quenched fluorescein) and acceptor (Cy3) responses. Two different molecular complexes: double labeled complexes fluorescein-Cy5, and triple-labeled complexes fluorescein-Cy3-Cy5, can both generate fluorescein-Cy3 FRET signal. Cy3 has different fluorescence lifetimes in these two kinds of complexes, a quenched lifetime in triple-labeled complexes, and an unquenched lifetime in double-labeled complexes. Thus in the FRET model (Eq. (8)), the acceptor (Cy3) response becomes a double-exponential decay with two lifetime components (solved in Step 2) and (measured from pure Cy3-labeled sample). Binding between fluorescein-labeled ssDNA and Cy3 labeled ssDNA is not interfered by the presence or absence of Cy5-labeled ssDNA. Thus the concentration ratio between the two complexes (fluorescien-Cy3 vs. fluorescien-Cy3-Cy5) is the same as the concentration ratio between quenched vs unquenched Cy3 in all molecular forms (solved in Step 3). The acceptor (Cy3) frequency response needed in Eq. (8) for fitting channel is identical to the response of the Cy3 EEM channel (). The only unknown in channel is the decay of quenched donor (fluorescein). Fitting the response of channel with Eq. (8) yields the lifetime of quenched fluorescein .
-
(5)
EEM channel contains fluorescence decays from quenched and unquenched fluorescein. A double exponential decay lifetime model with known lifetimes of quenched fluorescein (solved in Step 4) and unquenched fluorescein (measured from single labeled samples) obtains the percentage of quenched vs. all fluorescein (P1).
-
(6)
Finally EEM channel contains signal from a two-step FRET process, (fluorescein to Cy3 then Cy3 to Cy5), and potentially signal from one-step FRET directly from fluorescein to Cy5. We compare the two-step FRET model in Eq. (9), which is a product of frequency responses of three participating fluorophores (quenched fluorescein, quenched Cy3 and final acceptor Cy5 ), against the measured signal in channel . This step serves as verification on whether one-step FRET occurs between fluorescein and Cy5. If one-step FRET is present between fluorescein and Cy5, channel 13 will deviate from the two-step FRET model.
Through time resolved EEM analysis, quenched and unquenched lifetimes of fluorescein and Cy3 are obtained. These can be used to calculate distances from fluorescein to Cy3, and Cy3 to Cy5.
P1, the percentage of quenched fluorescein, and P2, the percentage of quenched Cy3 are also determined during the same process, which are related to concentrations of triple-labeled, double-labeled complex and free labels by
| (15) |
and
| (16) |
Because the acceptor’s spectral and lifetime properties are not affected by FRET, fluorescence analysis cannot distinguish free and FRET-active Cy5, the final acceptor in three-color FRET. , the concentration of free Cy5 is not present in Eqs. (15) and (16), and it is impossible to measure the free-state concentration of the reddest fluorophore in a multi-color FRET sample through EEM analysis.
As shown in Eqs. (15) and (16), which consist of four linear equations but have seven unknown concentrations, additional model constraints are needed to solve all unknown concentrations. Because binding between fluorescein-labeled ssDNA, Cy3-labeled ssDNA, and Cy5-labled ssDNA do not interfere with each other, there are
| (17) |
Under such constraints, relative ratios of all seven concentrations become solvable. The no-interference condition may not hold in a specific biological study. Nevertheless the model presented here can still serve as guidance for interpretation of measurement results. For example, regardless whether Eq. (17) applies or not, the percentage of quenched fluorescein or Cy3 will decrease when fluorescein or Cy3 becomes overabundant. Changes in percentages of quenched vs. unquenched states still provide valuable information on the ratiometric relationship between interacting molecules.
The multi-step analysis procedure involves only at most a single unknown lifetime parameter at each step, and is therefore more robust than direct multi-decay analysis. The analysis could suffer from error escalating through multiple steps if the signal-to-noise ratio of the time-resolved EEM data is unsatisfactory. In future imaging application, global lifetime analysis [23] could further boost the accuracy and robustness of the time-resolved EEM analysis in live cell experiments.
3. Materials and methods
3.1 Sample preparation
DNA oligonucleotides with fluorescence labels were purchased from Integrated DNA Technologies, Inc. with HPLC purification. A pair of 20 base-pair complimentary oligonucleotides with fluorescence labels on each 5′ ends was first used to validate the two-color FRET model. The sequences of these 20-mer oligonucleotides were Alexa488-5′-TTGAAAACGAGA-GAGACATA-3′ and Alexa546-5′-TATGTCTCTCTCGTTTTCAA-3′. The oligonucleotides were mixed with 1:1 molar ratio in 10 mM Tris-HCl, 1mM EDTA, pH 8.0 buffer at a concentration of 0.5 μM, heated to 90 °C for 3 minutes, and slowly cooled down to room temperature.
The three-color FRET experiments were performed using oligonucleotides with internal fluorescein, Cy3 and Cy5 labels. The sequences of these oligonucleotides were: 66-mer, 5′- ATACGAGCCCCTACACAGGCAGGTG AGTTCCGCAACTCCGACAGCAGTACCATCTCAATGTGACG −3′; 30-mer, 5′-CGTCACATTGACGATGGTACTGCTGTCGGA −3′; and 36-mer, 5′- GTTGCGGAACTCACCTGCCTGTGTAGGGGCTCGTAT −3′. The italic T base represents the positions where the internal fluorescence labels were attached. A schematic of the oligonucleotide design is shown in Fig. 3 . The 66-mer, 30-mer and 36-mer were labeled with Cy3, fluorescein, and Cy5, respectively. Oligonucleotides of identical sequences but without the internal fluorescence labels were obtained for control experiments. dsDNA were formed by mixing the three complimentary oligonucleotides in 10 mM Tris-HCl, 1 mM EDTA, pH 8.0 buffer solution, heating the solution to 90 °C for 3 minutes and slowly cooling to room temperature. When the oligonucleotides hybridized, they formed a linear structure with fluorescein on one side, Cy3 at the center, and Cy5 on the other side. The distances between two adjacent fluorophores were 15 base-pair (5.1 nm) each. No purification was performed after the DNA hybridization, thus free ssDNA and incomplete dsDNA with only two labels might exist.
Fig. 3.
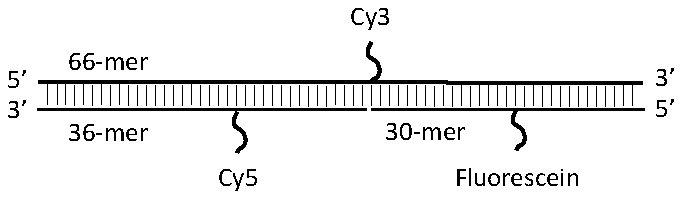
Structure of the triple-labeled dsDNA. The distances between fluorescein and Cy3, or Cy3 and Cy5 were 15 base-pairs or 5.1 nm.
3.2 Experimental setup
The FLEEM spectrometer operates with the Fourier lifetime method we previously reported [19]. A schematic of the FLEEM system is shown in Fig. 4 . The instrument is based on Fourier transform spectroscopy that utilizes a Michelson interferometer with a high-speed optical delay line to modulate a multi-wavelength continuous wave laser source. Laser lines at different wavelengths are modulated at different frequencies inversely proportional to their wavelength ω = 2πv/λ, where v is the optical delay scanning speed and λ is the wavelength of the laser lines. The scan velocity of the optical delay line varies linearly from about −100 m/s to 100 m/s within 45.5 μs, producing a linear round trip modulation frequency scan between 0 and ~150 MHz for visible wavelengths. The modulated output of the interferometer is then used to excite a fluorescent sample. Fluorescence emission associated with a given excitation wavelength is modulated at the same frequency as the excitation laser and can be separated by Fourier analysis. The high modulation frequency allows nanosecond frequency domain lifetime measurements at multiple excitation lines in parallel.
Fig. 4.
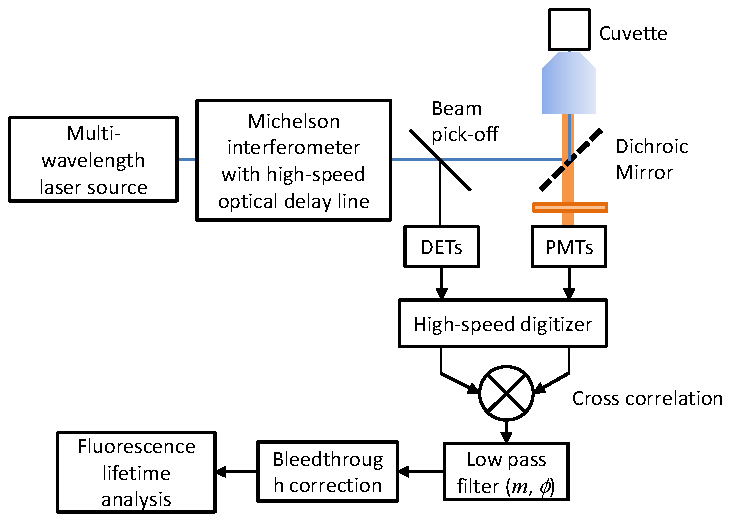
Schematic of the FLEEM system and data processing. A Michelson interferometer is used to modulate a multi-wavelength CW laser source. The modulated output of the interferometer then excites a fluorescent sample. The laser references and fluorescence emission signals at different excitation-emission wavelengths combinantions are digitized and cross-correlated to obtain frequency response of the sample as an EEM.
The fluorescence output of the samples are further split into multiple emission detection channels at different wavelengths by dichroic mirrors and emission filters, and detected with multiple PMTs (Hamamatsu H7420). The excitation laser lines and emission spectral windows are shown in Fig. 1(b). A beam pick-off mirror reflects a portion of the modulated excitation light, which is detected by multiple amplified silicon detectors (Thorlabs PDA10) as references. The fluorescence and reference signals are digitized simultaneously by a high-speed digitizer, and transferred onto a computer.
For each EEM channel, the fluorescence emission and reference laser excitation signals are cross-correlated through digital mixing followed by a low-pass filter and integrated over short time segments (typically 1 μs) [19]. The resulting cross-correlated signal is the frequency response of the fluorescent sample over a continuous frequency range at the excitation-emission wavelength combination, i.e. an EEM channel. Complete EEM of the sample is measured by cross-correlating over all possible excitation-emission wavelength combinations. Spectral bleedthrough is then removed from the measured EEM according to Eq. (3), using bleedthrough matrices calibrated from single labeled samples. The frequency responses of the bleedthrough corrected EEM channels are then analyzed by the models described in Section 2.
4. Results and discussions
4.1 Validation of time-resolved EEM analysis in two-color FRET
We first tested the time-resolved EEM analysis with double-labeled dsDNA after complete hybridization. 20 base-pair long complimentary sequences of oligonucleotides were labeled on each 5′-end with Alexa 488 (donor) and Alexa 546 (acceptor). Pure donor- and acceptor-labeled oligonucleotides were first measured to establish their lifetime baselines and bleedthrough matrices. Lifetimes of pure Alexa 488- and Alexa 546- oligonucleotides were measured to be τAlexa488 = 4.1 ± 0.1 ns and τAlexa546 = 3.4 ± 0.1 ns. The error represents the measurement uncertainty of a single facet scan in 46 μs.
EEM of completely hybridized dsDNA were then measured. Frequency responses of the donor/acceptor EEM channels and the FRET EEM channel were measured simultaneously. Modulation and phase responses of the donor and acceptor EEM channels of fully hybridized DNA are compared to pure donor and acceptor responses in Figs. 5(a) and 5(b), respectively. Single exponential decay model was used to fit both channels. The fluorescence lifetime for the donor decreased from τAlexa488 = 4.1 ± 0.1 ns to τAlexa488-quench = 3.0 ± 0.1 ns in the presence of FRET, while the lifetime for the acceptor remained unchanged at τAlexa546 = 3.4 ± 0.1 ns. Figure 5(c) plots the modulation and phase of the bleedthrough-corrected FRET channel signal. The experimental data are overlaid with the theoretical model on the complex frequency response of FRET signals (Eq. (8)), using τAlexa488-quench = 3.0 ns and τAlexa546 = 3.4 ns. Both the modulation and phase model of the FRET channel agrees well with the experimental measurements. The phase in the FRET channel exceeds π/2, which is a defining signature of FRET. The spectral configuration of the EEM channels is plotted in Fig. 5(d). By comparing the donor lifetime with and without FRET, with an R0 value of 6.4 nm between Alexa 488 and Alexa 546 dyes, the FRET distance was calculated to be 7.7 ± 0.1 nm. The result was in good agreement with the length of 20-base-pair dsDNA plus two 6-carbon chains that linked the two fluorophores to the 5′ ends of each ssDNA.
Fig. 5.
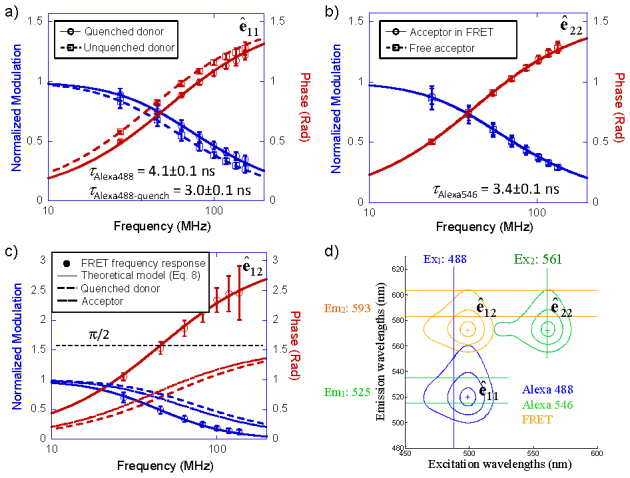
Time-resolved EEM measurements on double-labeled dsDNA. (a) Modulation and phase of quenched Alexa488 (donor EEM channel ê11), in comparison with unquenched Alexa488. The fluorescence lifetime of Alexa488 decreased from 4.1 ± 0.1 ns to 3.0 ± 0.1 ns due to FRET. (b) Modulation and phase of Aelxa546 (acceptor EEM channel ê22). The fluorescence lifetime of Alexa546 was 3.4 ± 0.1 ns, same as pure Alexa546. The acceptor EEM channel is unrelated to FRET, thus the lifetime remains constant. (c) Modulation and phase of the Alexa488-Alexa546 FRET EEM channel ê12. Experimental results in (c) were overlaid with the theoretical model (Eq. (8)). The phase delay in the FRET EEM channel exceeded π/2, which was a signature of FRET. (d) Spectral configuration of the EEM measurement. Error bars in (a~c) represent standard deviations of multiple 46-μs frequency sweeps.
4.2 Two-color FRET in the presence of free donor
We next tested time-resolved EEM analysis of two-color FRET in the presence of free donor molecules. The 66-mer ssDNA with internal Cy3 label was hybridized with either the 30-mer oligonucleotide with internal fluorescein label, or the 36-mer oligonucleotides with internal Cy5 label. Time-resolved EEM measurements of single-labeled samples were first performed to obtain unquenched lifetimes and EEM bleedthrough matrices. The lifetimes of free fluorescein, Cy3 and Cy5 were measured at 3.9 ± 0.1 ns, 1.5 ± 0.1 ns and 1.8 ± 0.1 ns, respectively. The error represents measurement uncertainty with an integration time of 1 ms.
The EEM channel of hybridized mixtures with different donor-to-acceptor ssDNA concentration ratios were then measured simultaneously. Figure 6 shows frequency responses of bleedthrough corrected EEM channels from a hybridized mixture of 2 μM fluorescein-ssDNA (donor) and 0.5 μM Cy3-ssDNA (acceptor). The fluorescein-ssDNA was overabundant.
Fig. 6.
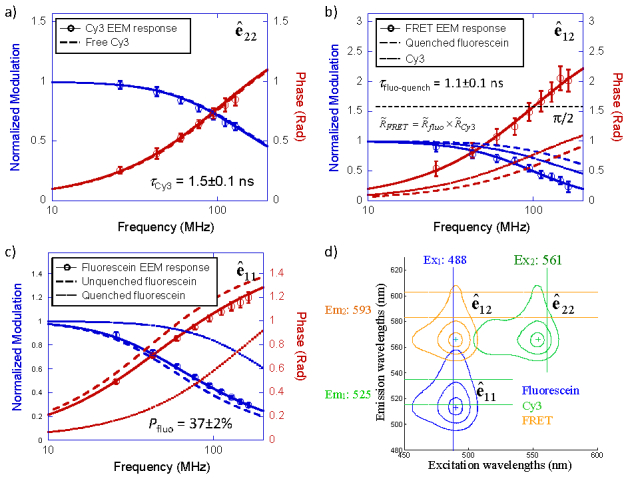
Frequency responses of bleedthrough corrected EEM channels from a mixture of two-color FRET complexes and free donors. (a) Cy3 EEM channel ê22 fitted with single exponential decay. The acceptor lifetime was found to be τCy3 = 1.5 ± 0.1 ns. (b) Fluorescein-Cy3 FRET EEM channel ê12 fitted with the FRET frequency response model (Eq. (8)). The quenched lifetime of fluorescein was calculated as τfluo-quench = 1.0 ± 0.1 ns (c) Fluorescein EEM channel ê11 fitted with a double exponential decay model, in which two lifetime, quenched and unquenched lifetime were fixed at knowing values. The percentage of quenched fluorescein was found to be Pfluo = 37 ± 2%. (d) Spectral configuration of the EEM measurement. Error bars in (a~c) represent standard deviations of multiple measurements with 1 ms integration time. Fitted curves of quenched/unquenched donor and acceptor response are plotted in (a~c) for reference.
The EEM spectra configuration for analyzing the two-color FRET mixture is shown in Fig. 6(d). Time-resolved analysis of EEM starts from the Cy3 EEM channel [, Fig. 6(a)], which only contains acceptor fluorescence decay. This channel was fitted with a single exponential lifetime decay, and the resulting acceptor lifetime remained at τCy3 = 1.5 ± 0.1 ns, same as pure acceptor sample. This step will serve as a reference point for any possible systematic drifts in future time lapse imaging study. Next the bleedthrough corrected fluorescein-Cy3 FRET channel [, Fig. 6(b)] was analyzed. The phase response in this channel exceeds π/2, which is a clear indication of FRET. The FRET EEM channel contained only signals from FRET complex. It was fitted with Eq. (8), in which the acceptor lifetime was fixed at 1.5 ns. The fitting yielded the quenched donor lifetime in FRET complex, τfluo-quench = 1.1 ± 0.1 ns. Fitted curves of quenched donor and acceptor responses were plotted in Fig. 6(b) together with measured and fitted FRET channel response. Figure 6(b) clearly shows that the modulation of the FRET channel response is the product of the quenched donor and acceptor modulation responses, and the phase of the FRET channel response is the sum of the quenched donor and acceptor phase responses. The Förster distance between fluorescein and Cy3 is 5.6 nm [24]. Based on the quenched and unquenched lifetimes of fluorescein, the distance between fluorescein and Cy3 in dsDNA was determined to be 4.8 ± 0.1 nm (equivalent to 14.3 ± 0.3 base-pair distance), consistent with the 15-base-pair sequence between fluorescein and Cy3 labeling sites. Finally, the fluorescein EEM channel (, Fig. 6(c)) contained fluorescence decays from both quenched and unquenched donors. As shown in Fig. 6(c), the measured fluorescein response fell between frequency responses of quenched and unquenched donor. The channel was analyzed with a double exponential decay model, in which quenched and unquenched fluorescein lifetimes were fixed at known values (τfluo-quench = 1.1 ± 0.1 ns and τfluo = 3.9 ± 0.1 ns). The percentage of quenched fluorescein Pfluo was determined as 37 ± 2%. Thus 63 ± 2% of fluorescein were not paired with Cy3 due to concentration imbalance or unlabeled ssDNA.
Same measurements and analysis procedure were performed on hybridized mixtures made at different donor-to-acceptor molar ratios. Table 1 shows the intensity and lifetime fitting results of the time-resolved EEM analysis for the two different FRET constructs, fluorescein-Cy3 and Cy3-Cy5. In both cases, the acceptor ssDNA concentration was fixed at 0.5 μM, while the donor ssDNA concentration varied from 0.25 μM to 2 μM. For both constructs, regardless the varying percentages of quenched donor (Pfluo or PCy3), which was due to donor overabundance and/or incomplete hybridization, the quenched donor lifetime (τfluo-quench or τCy3-quench) obtained from the bleedthrough corrected FRET channel remained constant. Acceptor intensity (ICy3 or ICy5) and lifetime (τCy3 or τCy5) remained constant at all donor concentrations as expected. Increasing donor concentration changed the donor intensity (Ifluo or ICy3) and the apparent average donor lifetime (or ) obtained by single exponential fitting on the donor fluorescence response. When the donor concentrations were overabundant, the intensities of the bleedthrough corrected FRET channel IFRET remained at a constant saturating level, indicating that in these cases the FRET signal was limit by the availability of acceptor. When all acceptor molecules were already bound in FRET complexes, increasing the donor concentration would not generate more FRET complexes. FRET channel intensity was proportional to the concentration of FRET complexes, and therefore remained at a saturated level when the donor ssDNA is overabundant.
Table 1. Intensities, Lifetimes and Percentages of Quenched Donor for Different Donor-acceptor ratio FRET Mixtures.
| Donor Conc. (μM)* | Molar Ratio (D:A) | Cy3 - Cy5 |
||||||
|---|---|---|---|---|---|---|---|---|
| ICy5 (a.u.) | τCy5 (ns) | IFRET (a.u.) | τCy3-quench (ns) | ICy3 (a.u.) | (ns) | P Cy3 | ||
| 0.25 | 0.5:1 | 88 ± 5 | 1.8 ± 0.1 | 4.1 ± 0.5 | 0.87 ± 0.2 | 10 ± 0.6 | 0.9 ± 0.1 | 0.99 ± 0.08 |
| 0.5 | 1:1 | 82 ± 5 | 1.8 ± 0.1 | 12 ± 2 | 0.74 ± 0.1 | 20 ± 1.5 | 0.9 ± 0.1 | 0.84 ± 0.05 |
| 1 | 2:1 | 81 ± 5 | 1.8 ± 0.1 | 15 ± 3 | 0.79 ± 0.1 | 70 ± 5 | 1.2 ± 0.1 | 0.44 ± 0.05 |
| 2 | 4:1 | 89 ± 5 | 1.8 ± 0.1 | 16 ± 3 | 0.81 ± 0.1 | 121 ± 7 | 1.3 ± 0.1 | 0.30 ± 0.03 |
| Fluorescein – Cy3 | ||||||||
| ICy3 (a.u.) | τCy3 (ns) | IFRET (a.u.) | τfluo-quench (ns) | Ifluor (a.u.) | (ns) | P fluo | ||
| 0.25 | 0.5:1 | 80 ± 8 | 1.5 ± 0.1 | 13 ± 2 | 1.0 ± 0.1 | 60 ± 6 | 1.7 ± 0.1 | 0.83 ± 0.04 |
| 0.5 | 1:1 | 82 ± 7 | 1.5 ± 0.1 | 24 ± 4 | 1.1 ± 0.1 | 115 ± 10 | 1.8 ± 0.1 | 0.83 ± 0.03 |
| 1 | 2:1 | 81 ± 8 | 1.5 ± 0.1 | 29 ± 4 | 1.2 ± 0.1 | 252 ± 19 | 2.5 ± 0.1 | 0.60 ± 0.02 |
| 2 | 4:1 | 75 ± 7 | 1.6 ± 0.1 | 23 ± 4 | 1.1 ± 0.1 | 460 ± 34 | 3.1 ± 0.1 | 0.37 ± 0.02 |
*Molar concentrations of ssDNAs may not represent true values of fluorophore concentrations due to incomplete labeling.
With a Förster distance of 5.4 nm between Cy3 and Cy5 [25], the distance between the Cy3 and Cy5 labels was calculated at 5.2 ± 0.1 nm (equivalent to 15.3 ± 0.3 base-pairs). Together with fluorescein-Cy3 distance at 4.8 ± 0.1 nm (equivalent to 14.3 ± 0.3 base-pairs), the measured distances were consistent with the designed 15 base-pair distances between labeling sites.
The accuracy of the time-resolved EEM analysis method in calculating the quenched donor lifetime is far superior to the conventional double-exponential fitting on the donor EEM channel. Table 2 compares fitting results from both methods. In our approach of conventional donor double experiential fitting, the unquenched donor life was fixed to the calibrated value from pure samples, whereas the quenched donor lifetime and the percentages of quenched donor were fitted. Both methods were applied to the same data sets taken from the two FRET constructs. The results of quenched donor lifetime (τfluo-quench or τCy3-quench) and percentages of quenched donors (Pfluo or PCy3) are listed in Table 2. In the case of the fluorescein-Cy3 construct, because of its relatively large separation between quenched and unquenched lifetimes (3.9 ± 0.1 ns to 1.1 ± 0.1 ns, 72% FRET efficiency), at low fluorescein concentrations where most of fluorescein molecules were quenched, both methods obtained similar results with comparable accuracy. When free fluorophore molecules were largely abundant, the double exponential fitting became more inaccurate. The reason is that the donor EEM channel response is an ensemble average between quenched and unquenched fluorophores. When free donors are overabundant, the averaged response is less sensitive to changes in quenched donor lifetime, which only contributes to a small portion of the measured signal. For the Cy3-Cy5 construct, the FRET efficiency was lower, and the decrease in Cy3 lifetime was not as strong (1.5 ± 0.1 ns to 0.8 ± 0.1 ns, 47% FRET efficiency). In such case, the double exponential fitting performed considerably worse at all donor concentrations, and even failed to converge at the highest donor concentration. In contrast, results from the time-resolved EEM analysis were not affected by either the free donor concentration, or the lifetime separation between quenched and unquenched donors. The side-by-side comparison shows that the time-resolved EEM analysis is more robust compared to conventional double-exponential fitting.
Table 2. Comparison between EEM Analysis and Double Exponential Fitting of the Donor EEM Channel.
| Donor Conc. (μM) | Molar ratio (D:A) | Cy3-Cy5 EEM analysis |
Cy3-Cy5 double exponential fitting |
||
|---|---|---|---|---|---|
| τCy3-quench (ns) | PCy3 | τCy3-quench (ns) | PCy3 | ||
| 0.25 | 0.5:1 | 0.87 ± 0.2 | 0.99 ± 0.08 | 1.0 ± 0.4 | 0.95 ± 0.25 |
| 0.5 | 1:1 | 0.74 ± 0.1 | 0.84 ± 0.05 | 0.7 ± 0.4 | 0.75 ± 0.22 |
| 1 | 2:1 | 0.79 ± 0.1 | 0.44 ± 0.05 | 0.3 ± 0.5 | 0.55 ± 0.42 |
| 2 | 4:1 | 0.81 ± 0.1 | 0.30 ± 0.03 | Failed | Failed |
| Fluorescein–Cy3 EEM analysis |
Fluorescein–Cy3 double exponential fitting |
||||
| τfluo-quench (ns) | Pfluo | τfluo-quench (ns) | Pfluo | ||
| 0.25 | 0.5:1 | 1.0 ± 0.1 | 0.83 ± 0.04 | 1.2 ± 0.1 | 0.89 ± 0.04 |
| 0.5 | 1:1 | 1.1 ± 0.1 | 0.83 ± 0.03 | 1.2 ± 0.1 | 0.90 ± 0.03 |
| 1 | 2:1 | 1.2 ± 0.1 | 0.60 ± 0.02 | 1.1 ± 0.2 | 0.61 ± 0.03 |
| 2 | 4:1 | 1.1 ± 0.1 | 0.37 ± 0.02 | 1.3 ± 0.3 | 0.36 ± 0.03 |
The percentages of quenched donor PCy3 and Pfluo reveals incomplete hybridization of ssDNAs. In Cy3-Cy5 hybridization, under 1:1 ssDNA molar ratio, Cy3 in fact is 84% quenched, indicating that 16% of Cy3 is unpaired. In fluorescein-Cy3 hybridization, only 83% of fluorescein is quenched even when Cy3-ssDNA is overabundant, indicating the hybridization is not 100% complete. In the past, purifications were needed to obtain pure FRET complexes before precise structural measurements can be performed [14]. The time-resolved EEM analysis allows robust measurements of the absolute FRET efficiency without the need of purification. The method can be applied to live cell imaging in the future, where purification is not applicable.
4.3 Three-color FRET
We next proceed to analyze three-color FRET in a mixture of triple-, double- and single-labeled DNA. Single-labeled ssDNA were first measured to establish lifetime baselines and spectral bleedthrough correction matrices. Single-labeled 66-mer, 30-mer and 36-mer ssDNA (structure shown in Fig. 3) were then mixed with a 1:1:1 molar ratio at 0.5 μM concentration and hybridized. Time resolved EEM measurements were performed on the mixture after hybridization, without purification.
The spectral configuration of the three-color FRET EEM is depicted in Fig. 1(b). The EEM frequency responses of the hybridized mixture (shown in Fig. 7 were corrected for spectral bleedthrough with Eq. (3). The data was then analyzed following the method discussed in Section 2.4.2.
Fig. 7.
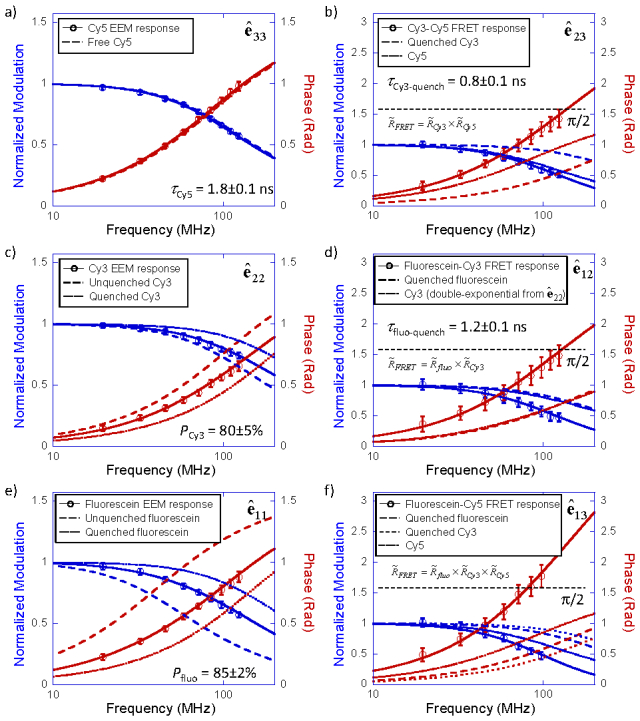
Frequency responses of bleedthrough corrected EEM channels for a three-color FRET mixture with incomplete FRET complexes and free fluorophores. (a) Cy5 EEM channel ê33. The lifetime of the final acceptor Cy5 remained unchanged at τCy5 = 1.8 ± 0.1 ns. (b) Cy3-Cy5 FRET channel ê23. The lifetime of quenched Cy3 was measured as τCy3-quench = 0.8 ± 0.1 ns. (c) Cy3 EEM channel ê22. A double exponential fit found the percentage of quenched Cy3 is PCy3 = 80 ± 5%. (d) Fluorescein-Cy3 FRET channel ê12. The quenched fluorescein lifetime was measured as τfluo-quench = 1.2 ± 0.1 ns. (e) Fluorescein EEM channel ê11. The percentage of quenched fluorescein was measured as Pfluo = 85 ± 2%. (f) Fluorescein-Cy5 FRET channel ê13. This channel contained signal from the two-step FRET (fluorescein-Cy3 then Cy3-Cy5). The measured frequency response was overlaid with the theoretical two-step FRET model, which was a product of individual frequency responses of quenched fluorescein, quenched Cy3 and Cy5. Fitted curves of quenched/unquenched donor and acceptor response were plotted for reference. Error bars represent standard deviations of multiple measurements with 1 ms integration time.
Analysis of the three-color FRET EEM follows the following steps as described in Section 2.4 and depicted in Fig. 2.
-
1)
The analysis started from Cy5 EEM channel [, Fig. 7(a)], the EEM channel of the reddest fluorophore Cy5, which served strictly as acceptor. The decay of Cy5 was fitted with a single-exponential decay mode, and the lifetime was verified to be the same as pure Cy5-ssDNA at τCy5 = 1.8 ± 0.1 ns.
-
2)
Next step of the analysis dealt with the Cy3-Cy5 FRET channel [, Fig. 7(b)] This channel contained signal arising from one-step FRET between Cy3 and Cy5. The phase response of the Cy3-Cy5 FRET channel exceeded π/2, clearly indicating the signal came from FRET. The one-step FRET model (Eq. (8)) was applied to fit channel to obtain the quenched lifetime of Cy3. During the fitting, Cy5 lifetime was fixed at a known 1.8 ns (from step 1, or measured from pure Cy5-ssDNA). The quenched lifetime of Cy3 was determined as τCy3-quench = 0.8 ± 0.1 ns, same as previous results from Cy3-Cy5 two-color FRET. The frequency responses of Cy5 and quenched Cy3 were also plotted in Fig. 7(b), showing that the product of the two complex frequency responses formed the FRET frequency response.
-
3)
The Cy3 EEM channel [, Fig. 7(c)] contained fluorescence decay signals from both quenched and unquenched Cy3. The Cy3 EEM channel frequency response fell between these two single-exponential decays of lifetimes at 0.8 ± 0.1 ns and 1.5 ± 0.1 ns, respectively. The frequency response of channel was fitted with double exponential decay model, in which the quenched and unquenched lifetimes of Cy3 were fixed at known values. The percentage of quenched Cy3, PCy3 was determined as 80 ± 5%. Therefore 20 ± 5% of Cy3 was unpaired with Cy5. The quenched Cy3 percentage obtained here was identical to the percentage from the Cy3-Cy5 two-color FRET experiment in the previous section, which was conducted with identical ssDNAs, except the fluorescein-ssDNA was not hybridized into the sample. Because binding of any two of the three ssDNAs would not interfere with each other, the pairing of Cy3-Cy5 was not affected by the present of fluorescein-ssDNA.
-
4)
The signal in fluorescein-Cy3 FRET channel [, Fig. 7(d)] contained contributions from both three-color complex fluorescein-Cy3-Cy5 and two-color complex fluorescein-Cy3. Based on the result of Step 3, we deduced that the molar ratio between fluorescein-Cy3-Cy5 and fluorescein-Cy3 was 80%:20%. Cy3 lifetime in these two complexes were known (0.8 ± 0.1 ns and 1.5 ± 0.1 ns). Thus when applying Eq. (8) to analyze this channel, the acceptor (Cy3) lifetime response was fixed as a known double exponential decay, identical to the double exponential decay in Cy3 EEM channel [, Fig. 7(c)]. The sole unknown in fitting Eq. (8) was the quenched donor (fluorescein) lifetime response, which was determined as τfluo-quench = 1.2 ± 0.1 ns, identical to the previous result from fluorescein-Cy3 two-color FRET.
-
5)
Fluorescein EEM channel [, Fig. 7(e)] contained fluorescence decay of quenched and unquenched fluorescein. The measured fluorescein frequency response fell between the single exponential decays of quenched and unquenched fluorescein. Channel was fitted with the double exponential decay model, using known quenched and unquenched lifetimes at 1.2 ± 0.1 ns and 3.9 ± 0.1 ns, respectively. The percentage of quenched fluorescein, Pfluo was determined as Pfluo = 85 ± 2%, identical to the result obtained in two-color fluorescein-Cy3 FRET.
-
6)
Fluorescein-Cy5 EEM channel [, Fig. 7(f)] contained signal generated from the two-step FRET (fluorescein-Cy3 then Cy3-Cy5), and possible one-step FRET directly from fluorescein to Cy5. In this final step, as a validation of the multi-color FRET model, the frequency response of EEM channel was overlaid with the two-step FRET model (Eq. (9)), which was a product of complex frequency responses with fluorescence lifetimes of 1.2 ns (quenched fluorescein), 0.8 ns (quenched Cy3) and 1.8 ns (Cy5). The measured frequency responses agreed well with the two-step FRET model. This validated that there was no measurable direct FRET between fluorescein and Cy5.
Combining the information obtained from the time-resolved 3-by-3 EEM, we recovered the molecular structure of the three-color FRET complexes as fluorescein-Cy3 distance at 4.9 ± 0.1 nm (14.4 ± 0.3 base-pair equivalent), and Cy3-Cy5 distance of 5.2 ± 0.1 nm (15.3 ± 0.3 base-pair equivalent). This information was obtained in a background of double- and single-labeled DNA, which caused 15% unpaired fluorescein and 20% unpaired Cy3. The time-resolved EEM analysis allows quantification of three-color FRET in triple-labeled complexes in the presence of incomplete complexes and single-labeled molecules.
Control samples made by hybridizing two labeled ssDNA and one unlabeled ssDNA were measured as further validation of the three-color FRET results. Table 3 shows the comparison of two-color control dsDNA measurements with the three-color dsDNA results, as well as lifetime baselines from single labeled samples. Measurements of the three-color FRET complexes matched well with two-color control samples. The fluorescein-Cy5 complex showed no measurable decrease in the donor lifetime. This validated that FRET between fluorescein and Cy5 was negligible.
Table 3. Comparison of Three-color FRET with Two-color Controls.
| τfluo (ns) | τCy3 (ns) | τCy5 (ns) | Pfluo | P Cy3 | Dfluo-Cy3 (nm) | DCy3-Cy5 (nm) | |
|---|---|---|---|---|---|---|---|
| Three-color | 1.2 ± 0.1 | 0.8 ± 0.1 | 1.8 ± 0.1 | 0.85 ± 0.02 | 0.80 ± 0.04 | 4.9 ± 0.1 | 5.2 ± 0.1 |
| Fluo-Cy3 | 1.1 ± 0.1 | 1.5 ± 0.1 | 0.83 ± 0.02 | 4.8 ± 0.1 | |||
| Cy3-Cy5 | 0.8 ± 0.1 | 1.8 ± 0.1 | 0.84 ± 0.05 | 5.2 ± 0.1 | |||
| Fluo-Cy5 | 3.9 ± 0.1 | 1.8 ± 0.1 | |||||
| Single label | 3.9 ± 0.1 | 1.5 ± 0.1 | 1.8 ± 0.1 |
5. Summary
In this paper we established a multi-color FRET analysis method based on the Fourier lifetime excitation-emission matrix (FLEEM) spectroscopy. The method directly measures the frequency response of the FRET process, which detects only quenched donors but not unquenched donors that are not participating FRET. Time-resolved analysis of EEM was used to quantitatively analyze three-color FRET complexes in the presence of background such as free fluorophores and incomplete FRET complexes, which almost always exist in live cell studies. Future research will apply the method to live cell imaging of multi-color FRET, where structure and ratiometric information captured by the time-resolved EEM analysis could yield deep insight into macromolecule interactions in vivo.
Acknowledgments
This work was supported by NIH grants R00EB008737 and R01EB015481.
References and links
- 1.Forster T., “Zwischenmolekulare energiewanderung und fluoreszenz,” Ann. Phys. (Berlin) 437(1-2), 55–75 (1948). 10.1002/andp.19484370105 [DOI] [Google Scholar]
- 2.Stryer L., “Fluorescence energy transfer as a spectroscopic ruler,” Annu. Rev. Biochem. 47(1), 819–846 (1978). 10.1146/annurev.bi.47.070178.004131 [DOI] [PubMed] [Google Scholar]
- 3.Elangovan M., Day R. N., Periasamy A., “Nanosecond fluorescence resonance energy transfer-fluorescence lifetime imaging microscopy to localize the protein interactions in a single living cell,” J. Microsc. 205(1), 3–14 (2002). 10.1046/j.0022-2720.2001.00984.x [DOI] [PubMed] [Google Scholar]
- 4.Blanchard S. C., Kim H. D., Gonzalez R. L., Jr, Puglisi J. D., Chu S., “tRNA dynamics on the ribosome during translation,” Proc. Natl. Acad. Sci. U.S.A. 101(35), 12893–12898 (2004). 10.1073/pnas.0403884101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar S., Alibhai D., Margineanu A., Laine R., Kennedy G., McGinty J., Warren S., Kelly D., Alexandrov Y., Munro I., Talbot C., Stuckey D. W., Kimberly C., Viellerobe B., Lacombe F., Lam E. W. F., Taylor H., Dallman M. J., Stamp G., Murray E. J., Stuhmeier F., Sardini A., Katan M., Elson D. S., Neil M. A. A., Dunsby C., French P. M. W., “FLIM FRET technology for drug discovery: automated multiwell-plate high-content analysis, multiplexed readouts and application in situ,” ChemPhysChem 12(3), 609–626 (2011). 10.1002/cphc.201000874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jares-Erijman E. A., Jovin T. M., “FRET imaging,” Nat. Biotechnol. 21(11), 1387–1395 (2003). 10.1038/nbt896 [DOI] [PubMed] [Google Scholar]
- 7.Piston D. W., Kremers G. J., “Fluorescent protein FRET: the good, the bad and the ugly,” Trends Biochem. Sci. 32(9), 407–414 (2007). 10.1016/j.tibs.2007.08.003 [DOI] [PubMed] [Google Scholar]
- 8.Lakowicz J. R., Balter A., “Theory of phase-modulation fluorescence spectroscopy for excited-state processes,” Biophys. Chem. 16(2), 99–115 (1982). 10.1016/0301-4622(82)85012-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lakowicz J. R., Balter A., “Analysis of excited-state processes by phase-modulation fluorescence spectroscopy,” Biophys. Chem. 16(2), 117–132 (1982). 10.1016/0301-4622(82)85013-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laptenok S. P., Borst J. W., Mullen K. M., van Stokkum I. H. M., Visser A. J., van Amerongen H., “Global analysis of Forster resonance energy transfer in live cells measured by fluorescence lifetime imaging microscopy exploiting the rise time of acceptor fluorescence,” Phys. Chem. Chem. Phys. 12(27), 7593–7602 (2010). 10.1039/b919700a [DOI] [PubMed] [Google Scholar]
- 11.Borst J. W., Laptenok S. P., Westphal A. H., Kühnemuth R., Hornen H., Visser N. V., Kalinin S., Aker J., van Hoek A., Seidel C. A. M., Visser A. J. W. G., “Structural changes of yellow cameleon domains observed by quantitative FRET analysis and polarized fluorescence correlation spectroscopy,” Biophys. J. 95(11), 5399–5411 (2008). 10.1529/biophysj.107.114587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.J. R. Lakowicz, Principles of fluorescence spectroscopy, third edition (Springer, 2006). [Google Scholar]
- 13.Lakowicz J. R., Laczko G., Cherek H., Gratton E., Limkeman M., “Analysis of fluorescence decay kinetics from variable-frequency phase shift and modulation data,” Biophys. J. 46(4), 463–477 (1984). 10.1016/S0006-3495(84)84043-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watrob H. M., Pan C. P., Barkley M. D., “Two-step FRET as a structural tool,” J. Am. Chem. Soc. 125(24), 7336–7343 (2003). 10.1021/ja034564p [DOI] [PubMed] [Google Scholar]
- 15.Klostermeier D., Sears P., Wong C. H., Millar D. P., Williamson J. R., “A three-fluorophore FRET assay for high-throughput screening of small-molecule inhibitors of ribosome assembly,” Nucleic Acids Res. 32(9), 2707–2715 (2004). 10.1093/nar/gkh588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Y. S., Wallrabe H., Booker C. F., Day R. N., Periasamy A., “Three-color spectral FRET microscopy localizes three interacting proteins in living cells,” Biophys. J. 99(4), 1274–1283 (2010). 10.1016/j.bpj.2010.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galperin E., Verkhusha V. V., Sorkin A., “Three-chromophore FRET microscopy to analyze multiprotein interactions in living cells,” Nat. Methods 1(3), 209–217 (2004). 10.1038/nmeth720 [DOI] [PubMed] [Google Scholar]
- 18.Grant D. M., Zhang W., McGhee E. J., Bunney T. D., Talbot C. B., Kumar S., Munro I., Dunsby C., Neil M. A., Katan M., French P. M., “Multiplexed FRET to image multiple signaling events in live cells,” Biophys. J. 95(10), L69–L71 (2008). 10.1529/biophysj.108.139204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao M., Peng L., “Multiplexed fluorescence lifetime measurements by frequency-sweeping Fourier spectroscopy,” Opt. Lett. 35(17), 2910–2912 (2010). 10.1364/OL.35.002910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee J., Lee S., Ragunathan K., Joo C., Ha T., Hohng S., “Single-molecule four-color FRET,” Angew. Chem. Int. Ed. 49(51), 9922–9925 (2010). 10.1002/anie.201005402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Millican D. W., McGown L. B., “Fluorescence lifetime selectivity in excitation emission matrices for qualitative-analysis of a 2-component system,” Anal. Chem. 61(6), 580–583 (1989). 10.1021/ac00181a017 [DOI] [Google Scholar]
- 22.Millican D. W., McGown L. B., “Fluorescence lifetime resolution of spectra in the frequency-domain using multiway analysis,” Anal. Chem. 62(20), 2242–2247 (1990). 10.1021/ac00219a017 [DOI] [Google Scholar]
- 23.Beechem J. M., “Global analysis of biochemical and biophysical data,” Methods Enzymol. 210, 37–54 (1992). 10.1016/0076-6879(92)10004-W [DOI] [PubMed] [Google Scholar]
- 24.Ha T., Rasnik I., Cheng W., Babcock H. P., Gauss G. H., Lohman T. M., Chu S., “Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase,” Nature 419(6907), 638–641 (2002). 10.1038/nature01083 [DOI] [PubMed] [Google Scholar]
- 25.Norman D. G., Grainger R. J., Uhrín D., Lilley D. M. J., “Location of cyanine-3 on double-stranded DNA: Importance for fluorescence resonance energy transfer studies,” Biochemistry 39(21), 6317–6324 (2000). 10.1021/bi992944a [DOI] [PubMed] [Google Scholar]