

Abstract
The marine toxin bistratene A (BisA) potently induces cytostasis and differentiation in a variety of systems. Evidence that BisA is a selective activator of protein kinase C (PKC) δ implicates PKC δ signaling in the negative growth-regulatory effects of this agent. The current study further investigates the signaling pathways activated by BisA by comparing its effects with those of the PKC agonist phorbol 12-myristate 13-acetate (PMA) in the IEC-18 intestinal crypt cell line. Both BisA and PMA induced cell cycle arrest in these cells, albeit with different kinetics. While BisA produced sustained cell cycle arrest in G0/G1 and G2/M, the effects of PMA were transient and involved mainly a G0/G1 blockade. BisA also produced apoptosis in a proportion of the population, an effect not seen with PMA. Both agents induced membrane translocation/activation of PKC, with BisA translocating only PKC δ and PMA translocating PKC α, δ, and ε in these cells. Notably, while depletion of PKC α, δ, and ε abrogated the cell cycle-specific effects of PMA in IEC-18 cells, the absence of these PKC isozymes failed to inhibit BisA-induced G0/G1 and G2/M arrest or apoptosis. The cell cycle inhibitory and apoptotic effects of BisA, therefore, appear to be PKC-independent in IEC-18 cells. On the other hand, BisA and PMA both promoted PKC-dependent activation of Erk 1 and 2 in this system. Thus, intestinal epithelial cells respond to BisA through activation of at least two signaling pathways: a PKC δ-dependent pathway, which leads to activation of mitogen-activated protein kinase and possibly cytostasis in the appropriate context, and a PKC-independent pathway, which induces both cell cycle arrest in G0/G1 and G2/M and apoptosis through as yet unknown mechanisms.
Keywords: Bistratene A, Protein kinase C, Cell cycle, Signal transduction, Intestinal epithelial cells, Mitogen-activated protein kinase
1. Introduction
The cyclic polyether toxin BisA, isolated from the marine ascidian Lissoclinum bistratum, induces cell cycle arrest and differentiation in several cell types. Treatment with this agent leads to growth inhibition of A549 lung carcinoma cells and MCF-7 breast adenocarcinoma cells [1], and G2/M arrest of HL-60 promyeloid leukemia cells [2] and MM96E melanoma cells [3]. BisA also induces partial differentiation of HL-60 cells along the monocyte/macrophage pathway [4] and stimulates melanogenesis in MM96E human melanoma cells [3]. Other reported effects of this agent include alterations in cell morphology [3,5], changes in cytoskeletal organization [5], increased protein phosphorylation [6], and apoptosis [7,8].
It has been proposed that the effects of BisA on cell growth and differentiation are mediated through selective activation of PKC δ [2,3]. The PKC family consists of at least ten distinct serine/threonine kinases that play central roles in cell signaling, cell cycle control, differentiation, and other fundamental cellular functions [9–12]. PKC molecules are divided into three subfamilies, based on differential activator/cofactor requirements; PKC δ belongs to the “novel” PKC subfamily of Ca2+-independent, phorbol ester-responsive isozymes. Increasing evidence from studies in several systems supports a role for PKC δ in inhibition of cell growth/cell cycle progression and induction of differentiation [13–17]. In addition, recent findings point to the involvement of this isozyme in the execution of the apoptotic program induced by chemicals and irradiation [18–20]. In cultured cells, BisA induces the translocation/activation of PKC δ, but not other PKC isozymes, at concentrations similar to those required to produce growth inhibitory [2, 21], differentiation-promoting [3], or apoptotic [8] events.
The present study further investigates the signaling pathways involved in BisA-induced cell cycle regulation. Cellular effects of BisA were examined in the IEC-18 rat intestinal crypt cell line, which has been shown to undergo cell cycle arrest in response to a variety of PKC agonists [22, 23]. Treatment of IEC-18 cells with phorbol esters (e.g. PMA) or DAG analogs activates PKC α, δ, and ε, leading to cell cycle arrest in G0/G1 phase and delayed progression through G2/M. Both induction and maintenance of these effects are dependent upon the presence of membrane-associated/active PKC [22]. The current study compares the cell cycle-specific effects of BisA with those of PMA in IEC-18 cells, and directly examines the role of PKC δ in cellular response(s) to BisA in this system.
Both BisA and PMA produced cell cycle arrest in IEC-18 cells, albeit with different kinetics. Notably, although BisA treatment resulted in translocation of PKC δ to the membrane fraction of IEC-18 cells, the cell cycle blockade induced by this agent was not abrogated by depletion of PKC α, δ, and ε, and thus appeared to be PKC-independent. BisA also induced PKC-independent apoptosis in a proportion of the cells. On the other hand, both BisA and PMA elicited a PKC-dependent activation of the MAPK signaling pathway, confirming that BisA-induced signaling through PKC δ has downstream consequences in IEC-18 cells. Thus, BisA appears to stimulate at least two distinct pathways in intestinal epithelial cells. Activation of PKC δ leads to phosphorylation/activation of Erk 1 and 2 and may contribute to the observed BisA-induced cell cycle effects, but is not required for BisA-mediated arrest in G0/G1 and G2/M or apoptosis in these cells. An additional, as yet unidentified, target(s) appears to be a potent mediator(s) of BisA-induced cell cycle inhibition and apoptosis in intestinal epithelial cells.
2. Materials and methods
2.1. Cell culture and PKC activation protocols
The IEC-18 cell line (CRL-1589; American Type Culture Collection) is an immature, non-transformed cell line derived from rat ileal epithelium, which maintains many characteristics of proliferating intestinal crypt cells [24]. Cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 4 mM glutamine, 10 μg/mL of insulin, and 5% fetal bovine serum. IEC-18 cells express the phorbol ester-responsive PKC isozymes α, δ, and ε and the atypical isozymes ζ and ι. PKC α, δ, and ε were activated in these cells by treatment with 100 nM PMA (Sigma Chemical Co.). PKC δ alone was activated with 100 nM BisA [2]. Depletion of PKC α, δ, and ε was accomplished by overnight exposure to 1 μM PDBu followed by return to complete medium, as previously described [22].
2.2. Antibodies
Monoclonal PKC α -specific antibody was purchased from Upstate Biotechnology. Polyclonal rabbit anti-PKC δ, -PKC ε, and -PKC ζ antibodies were obtained from Santa Cruz Biotechnology. Polyclonal rabbit anti-MAPK antibody recognizing only the fully-phosphorylated, active forms of Erk 1 and 2 was obtained from New England Biolabs. HRP-conjugated goat anti-rabbit IgG was obtained from Boehringer-Mannheim, and HRP-conjugated rat anti-mouse IgG was purchased from Jackson ImmunoResearch Laboratories. Primary antibody dilutions used were as follows: 1:1000 for MAPK and PKC ζ, 1:2000 for PKC δ and ε, and 1:3000 for PKC α. HRP-conjugated secondary antibodies were used at 1:2000.
2.3. Flow cytometric analysis of IEC-18 cell cycle distribution
Cells were harvested by trypsinization, fixed overnight in 70% EtOH, and resuspended in 20 mM Tris (pH 7.5), containing 250 mM sucrose, 5 mM MgCl2, 0.37% NP-40 (Sigma), and 0.04 mg/mL of RNase (Sigma). Cellular DNA was stained with 25 μg/mL of PI (Sigma) in 0.05% sodium citrate and quantified by flow cytometry. Cell cycle analysis was performed using Winlist (Verity Software House).
2.4. Flow cytometric analysis of apoptosis in IEC-18 cells
Apoptosis was measured using an Annexin V-FITC apoptosis detection kit (R&D Systems). Briefly, cells were harvested by trypsinization, washed twice in cold PBS, stained for 15 min with 5 μg/mL of PI and 0.25 μg/mL of Annexin V-FITC in binding buffer [10 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, and 1.8 mM CaCl2], and then diluted 1:5 in binding buffer. PI and Annexin V-FITC binding were quantified by flow cytometry; data analysis was performed using Winlist.
2.5. Whole cell lysates and subcellular fractionation
Whole cell lysates were prepared by solubilizing subconfluent IEC-18 cell populations in boiling SDS lysis buffer [10 mM Tris (pH 7.4), 1% SDS]. Samples were boiled for 5 min, cellular DNA was sheared by passing extracts through a 27-gauge needle, and lysates were cleared by centrifugation (10 min at 10,000 g). Cytosolic and membrane subcellular fractions of IEC-18 cells were prepared essentially as previously described [22,25]. Briefly, cells were collected on ice in an extraction buffer containing 20 mM Tris (pH 7.5), 2 mM EGTA, 2 mM EDTA, 0.5 mg/mL of digitonin, 1 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 25 μg/mL of leupeptin, and 25 μg/mL of aprotinin (digitonin buffer). Digitonin-soluble (cytosolic) and -insoluble (particulate) fractions were separated by ultracentrifugation at 100,000 g for 30 min at 4°. The cytosolic protein in the supernatant was precipitated by incubation with 10% trichloroacetic acid for 10 min on ice, pelleted by centrifugation (10,000 g for 30 min at 4°), washed in acetone, resolubilized in 100 mM NaOH, and neutralized with 100 mM HCl. The membrane fraction was extracted from the particulate pellet (4° for 30 min) in digitonin buffer containing 1% Triton X-100, and cleared by centrifugation (10,000 g for 30 min at 4°). All cellular extracts were boiled in Laemmli sample buffer [26] and subjected to SDS–PAGE and western blot analysis.
2.6. Western blot analysis
Whole cell lysates (30 μg) or subcellular fractions (20 μg soluble fraction and an equal amount of the respective membrane fraction) were separated by SDS–PAGE, using 10% polyacrylamide gels (76:1 acrylamide to bis-acrylamide ratio). Protein was electrophoretically transferred to nitrocellulose membrane (Bio-Rad Laboratories), and membranes were blocked in Tris-buffered saline (TBS; 20 mM Tris–HCl, 137 mM NaCl, pH 7.6), containing 5% non-fat dried milk and 0.1% Tween-20 (TBST/milk), for 30 min at 37°. Membranes were incubated for 2 hr at room temperature or overnight at 4° with primary antibody in TBST/milk, followed by six 5-min washes in TBST/milk. Blots were then incubated for 1 hr at room temperature in secondary HRP-conjugated antibody in TBST/milk. After six 5-min washes in TBST/milk and three 5-min washes in TBS, bound HRP was detected using the SuperSignal ECL system (Pierce Chemical Co.).
3. Results
3.1. Induction of cell cycle arrest by both BisA and PMA in IEC-18 cells
To compare the cell cycle-specific effects of PMA and BisA in IEC-18 cells, cells were treated with either agent (100 nM) for 6, 12, and 24 hr, and cell cycle distribution was determined by flow cytometry. As shown in Fig. 1, a decrease in the number of cells in S phase was observed by 6 hr following treatment with either agent, indicating an inhibition of G1→S progression. DNA synthesis apparently was not affected, as cells in S phase at the beginning of drug exposure progressed into G2/M. By 12 hr, PMA-treated cells had accumulated almost exclusively in G0/G1 (following a variable delay in G2/M progression as described previously [22]), and the cell cycle arrest induced by this agent reversed between 12 and 16 hr of treatment, coincident with the down-regulation of PMA-responsive PKC isozymes (Fig. 1 and [22]). BisA-treated cells, on the other hand, underwent a sustained cell cycle arrest in both G0/G1 and G2/M lasting for at least 24 hr; reversal of this effect was not seen until 48 hr (data not shown). Furthermore, BisA induced apoptosis in a proportion of the cells (~14%) by 24 hr (see Table 1), a response not seen following PMA treatment. Thus, the pattern of cell cycle-specific effects produced by BisA in IEC-18 cells differs from that induced by other PKC agonists in this system.
Fig. 1.

IEC-18 cell cycle arrest following exposure to PMA or BisA. Asynchronously growing IEC-18 cells were treated with either 100 nM PMA or 100 nM BisA for the indicated times (C, vehicle control). Cell cycle distribution was determined by flow cytometric analysis. Data shown are representative of three independent experiments.
Table 1.
PKC-independent induction of apoptosis by BisA
| Condition | % Apoptosis |
|---|---|
| Vehicle control | 0.4 ± 0.1 |
| PMA, 100 nM | 0.8 ± 0.2 |
| PKC-depleted | 0.9 ± 0.3 |
| BisA, 100 nM | 14.1 ± 3.8 |
| PKC-depleted/BisA, 100 nM | 18.4 ± 1.7 |
IEC-18 cells were treated for 24 hr with vehicle, PMA, or BisA, with or without preincubation with 1 μM PDBu to deplete cells of PKC α, δ, and ε. Cells were harvested and stained with Annexin V and PI as described in “Materials and methods,” and apoptosis was assayed by flow cytometry. Percentages reflect the number of cells in the Annexin V-positive, PI-negative population. Results are averages (±SD) of duplicate samples and are representative of two independent experiments.
3.2. Membrane translocation of PKC δ but not PKC α or ε by BisA in IEC-18 cells
A possible explanation for the differences in the effects produced by BisA and other PKC agonists in IEC-18 cells is that BisA is a selective activator of PKC δ, while phorbol esters and DAG analogs stimulate PKC α, δ, and ε in this system. Evidence for the specificity of BisA for PKC δ includes the observations that BisA (a) stimulates PKC activity only in the PKC δ-containing fraction of HL-60 extracts separated on hydroxyapatite columns [2], and (b) induces selective translocation of PKC δ to the membrane subcellular fraction (a potential indicator of PKC activation; [27]) in HL-60 cells [2, 28], fibroblasts [5], melanocytes [3], and colon carcinoma cells [8]. In addition, BisA has been shown to stimulate the activity of recombinant PKC δ against a PKC ε pseudosubstrate peptide in in vitro enzyme assays.2 To compare the effects of BisA and PMA on PKC signaling in intestinal epithelial cells, membrane translocation of PKC isozymes was determined in IEC-18 cells treated with either agent for 10 min and for 1 hr. As shown in Fig. 2, western blot analysis of soluble and membrane fractions using PKC isozyme-specific antibodies demonstrated that PMA treatment results in complete translocation of PKC α, δ, and ε to cellular membranes. In contrast, BisA treatment stimulated increased, albeit incomplete, accumulation only of PKC δ in the membrane fraction. Neither agent had any effect on the subcellular distribution of the phorbol ester-insensitive PKC ζ. Thus, consistent with previous findings, BisA treatment results in apparent activation of only one PKC isozyme, PKC δ, in IEC-18 cells.
Fig. 2.
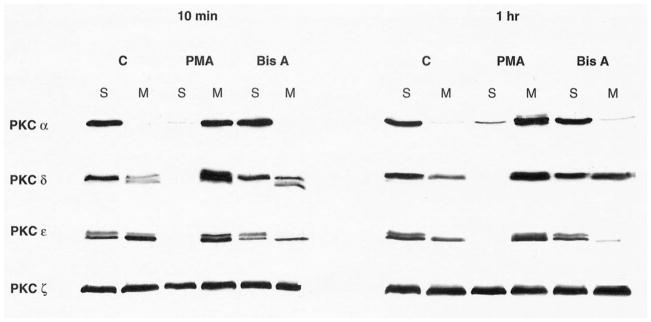
Selective translocation of PKC δ to the membrane fraction of intestinal epithelial cells by BisA. Asynchronously growing IEC-18 cell populations were treated with PMA or BisA for the indicated times (C, control). Cytosolic/soluble (S) and membrane (M) fractions were prepared and subjected to western blot analysis, using antibodies specific for individual PKC isozymes. The presence of more than one immunoreactive band in some of the samples likely reflects differential phosphorylation of these molecules [19, 29]. Data shown are representative of three independent experiments.
3.3. PKC-independent induction of intestinal epithelial cell cycle arrest and apoptosis by BisA
The specific involvement of PKC in mediating an observed response to PKC agonists can be confirmed by loss of the response in PKC-depleted cells. To evaluate whether activation of PKC δ is required for BisA-induced cell cycle arrest and apoptosis in IEC-18 cells, cultures were exposed to 1 μM PDBu for 16 hr to down-regulate all phorbol ester-sensitive isozymes (i.e. α, δ, and ε; Fig. 3a). Depletion of PKC α, δ, and ε completely abrogated the ability of PMA to induce cell cycle arrest in this system (Fig. 3b and [22]). Surprisingly, however, neither BisA-induced arrest in G0/G1 and G2/M nor apoptosis was affected by PKC depletion (Fig. 3b, Table 1). Thus, the cell cycle-specific and apoptotic effects of BisA in IEC-18 cells appear to be PKC-independent, and likely involve modulation of the activity of an additional, as yet unknown, cellular target(s) other than PKC δ.
Fig. 3.

Induction of PKC-independent intestinal epithelial cell cycle arrest by BisA. IEC-18 cells were pretreated with 1 μM PDBu for 16 hr to down-regulate PKC α, δ, and ε. (a) Whole cell lysates of untreated (U) or PDBu-pretreated (D) IEC-18 cells were examined by western blot analysis for PKC isozyme expression. (b) Untreated (U) or PKC-depleted (D) cells were exposed to 100 nM PMA or 100 nM BisA for 12 hr, and cell cycle distribution was determined by flow cytometric analysis. C, control. Data shown are representative of three independent experiments.
3.4. PKC-dependent activation of MAPK by PMA and BisA in IEC-18 cells
To investigate further the signaling pathways activated by BisA in IEC-18 cells, whole cell lysates from control and BisA-treated populations were subjected to western blot analysis using an antibody specific for the phosphorylated, active forms of the MAPK molecules Erk 1 and 2. Samples of PMA-treated IEC-18 cells were included for comparison. As shown in Fig. 4, both BisA and PMA induced Erk 1 and 2 phosphorylation by 10 min of treatment. PMA-induced Erk 1/2 activation was longer-lived than that produced by BisA, with increased levels of phospho-Erk sustained for at least 4 hr. In contrast, BisA-induced MAPK activation was reversed by 1 hr post-treatment. The kinetics of PMA-induced Erk 1/2 activation are consistent with a role for the MAPK pathway in PKC-mediated cell cycle arrest, since several reports have linked sustained activation of the MAPK pathway to cell cycle exit in other cell types [30, 31]. Notably, Erk 1/2 activation by either agent was abolished by depletion of PKC α, δ, and ε (Fig. 5). Thus, unlike the PKC-independent effects on cell cycle distribution/apoptosis induced by BisA in these cells, MAPK activation by either BisA or PMA is dependent upon the presence of phorbol ester-responsive PKC. The transient nature of Erk activation by BisA may reflect either the limited amount of PKC δ translocated/activated by this agent or a requirement for PKC α and/or ε for maintenance of the effect.
Fig. 4.
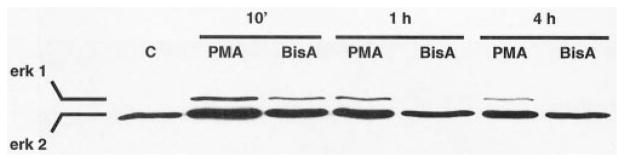
Activation of Erk 1 and 2 by PMA and BisA in intestinal epithelial cells. Proliferating IEC-18 cell populations were treated with 100 nM PMA, 100 nM BisA, or vehicle (C) for the indicated times; PMA, BisA, or vehicle was added directly to the plating medium. Whole cell lysates were prepared and subjected to western blot analysis, using an antibody specific for the dually phosphorylated, active forms of Erk 1 and 2. A single representative vehicle control is shown, since changes in Erk phosphorylation were not observed in control cells over the 4-hr experimental period. Data shown are representative of three independent experiments.
Fig. 5.

PKC-dependent activation of MAPK by PMA and BisA in IEC-18 cells. Untreated IEC-18 cells or cells pretreated with 1 μM PDBu to deplete PKC α, δ, and ε were exposed to 100 nM PMA or 100 nM BisA for 30 min. Whole cell lysates were prepared and subjected to western blot analysis using an antibody specific for the dually phosphorylated, active forms of Erk 1 and 2. U, untreated; U+, no depletion, treated with PMA or BisA for 30 min; D, PKC-depleted; D+, PKC-depleted, retreated with PMA or BisA for 30 min. Data shown are representative of three independent experiments.
Together, the data presented in this report demonstrate that, while BisA treatment results in cell cycle arrest and apoptosis in intestinal epithelial cells, these effects are largely PKC-independent and likely involve interaction of the drug with an additional cellular target(s) other than PKC δ. On the other hand, both BisA and PMA induce MAPK activation in this system in a PKC-dependent fashion, indicating that BisA is capable of stimulating, through selective PKC δ activation, at least some components of the pathways associated with PKC-dependent growth arrest induced by other PKC agonists.
4. Discussion
Previous studies in a variety of systems have clearly demonstrated the potent cytostatic effects of BisA [1–4, 32], and studies in Burkitt’s lymphoma [7] and colon carcinoma cells [8] have pointed to the ability of this agent to induce apoptosis under certain conditions. Here, we show that IEC-18 cells are sensitive to the negative growth-regulatory and apoptotic effects of this agent, arresting in both G0/G1 and G2/M phases by 6 hr of treatment and undergoing limited apoptosis by 24 hr. BisA does not directly affect DNA synthesis in this system, consistent with findings in other cell types [33]. The cell cycle-specific effects of BisA in IEC-18 cells parallel those seen in other systems; accumulation of cells in G0/G1 has been noted following BisA treatment of non-small-cell lung carcinoma cells [32], while G2/M arrest has been described in HL-60 promyeloid leukemia cells [33] and melanoma cells [3]. The G2/M effects of BisA observed in previous studies appear to be the result of inhibition of cytokinesis [32–34], possibly through control of the cytoplasmic protein stathmin. This important regulator of microtubule stability during mitosis [35, 36] has been shown to be rapidly phosphorylated in response to BisA treatment [3, 21]. Evidence indicates that stathmin phosphorylation abrogates its microtubule-destabilizing activity, preventing formation of a functional mitotic spindle and causing cell cycle blockade in M phase [34]. Whether similar mechanisms are involved in BisA-induced G2/M arrest in IEC-18 cells remains to be determined. Furthermore, since stathmin has not been reported to regulate G1→S progression, some other mechanism(s) must underlie BisA-mediated G0/G1 arrest in this system.
BisA has been shown to selectively induce the translocation/activation of PKC δ [2] and is currently the only known specific activator of this PKC isozyme. Interestingly, several of the cellular effects of BisA parallel reported effects of PKC δ overexpression/activation (for a review, see Ref. 12). For example, overexpression of PKC δ in NIH3T3 cells results in decreased cellular adherence and inhibition of cell growth [13], and PMA-induced myeloid differentiation involves signaling through PKC δ [14]. Furthermore, activation of PKC δ in keratinocytes or vascular smooth muscle cells leads to cell cycle arrest in the G1 phase [15, 16], and phorbol ester treatment of CHO cells overexpressing PKC δ results in G2 arrest and the appearance of multinucleate cells as a result of inhibition of cytokinesis [17]. PKC δ has also been implicated in apoptosis; activation of this isozyme by caspases has been linked to stimulation of the apoptotic program [37], overexpression of an active PKC δ catalytic fragment induces apoptosis in HeLa cells [38], and PKC δ has been proposed to be an apoptotic lamin kinase [28]. Together, these findings point to PKC δ as a major mediator of BisA-induced cellular responses. Surprisingly, however, although BisA treatment of IEC-18 cells resulted in rapid translocation of PKC δ to the membrane, depletion of PKC α, δ, and ε from IEC-18 cells did not abrogate either the G0/G1 and G2/M blockade or the apoptotic effects of this agent. These data provide the first demonstration of the ability of BisA to induce cell cycle arrest and apoptosis in the absence of PKC δ (and other phorbol ester-responsive PKC isozymes), and strongly point to the presence of an additional cellular target(s) for this agent in intestinal epithelial cells. In this regard, it should be noted that a role for the atypical PKC isozymes ζ and ι in the cell cycle-specific effects of BisA has not been formally ruled out, although to date BisA-induced changes in their subcellular localization have not been observed in any system examined [2, 3, 5].
This study also demonstrates for the first time that, like PMA ( [39] and Fig. 5), BisA can activate Erk 1 and 2, and that activation of these kinases is PKC-dependent. Since BisA appears to be a specific activator of PKC δ, this finding links PKC δ in particular to activation of the MAPK pathway (also see [40]). Taken together with the demonstration that BisA induces the accumulation and phosphorylation of JNK in HL-60 cells [20], these data suggest that activation of MAPK signaling is an important component of cellular responses to this agent. The functional consequences of BisA-induced Erk 1 and 2 activation in IEC-18 cells remain to be determined in future studies.
The findings presented in this report provide strong evidence for the ability of BisA to activate at least two signaling pathways in intestinal epithelial cells: one mediated by PKC δ, with currently undefined downstream effects, and at least one PKC-independent pathway leading to cell cycle arrest in G0/G1 and G2/M as well as limited apoptosis. Although PKC-independent pathways seem to predominate in mediating the cell cycle-specific and apoptotic effects of BisA in IEC-18 cells, evidence that PKC δ can produce similar responses in other cell types suggests that PKC-dependent signaling may also be important in the effects of this agent. Whether PKC-dependent or -independent pathways predominate in a particular system may thus be determined by cellular context. In IEC-18 cells, any PKC δ -dependent responses appear to be masked by the dramatic PKC-independent effects induced by BisA in this system.
In summary, the cellular effects of BisA appear to be highly complex. More than one target is likely to be involved, and at least two growth inhibitory signaling pathways can be activated by this agent in mammalian cells. Furthermore, the growth-regulatory effects of BisA seem to be highly system-dependent. Despite this complexity, consistent evidence for its ability to activate PKC δ and to produce cell cycle arrest suggests that a common mechanism(s) underlies the responses to this agent in different systems. The finding that BisA targets more than one signaling pathway to regulate cell growth suggests that it may be particularly useful as a base compound for anticancer therapy.
Acknowledgments
The authors gratefully thank Dr. Adrian Black for critical reading of the manuscript and helpful discussion, and Paula Diegelman for technical assistance with the apoptosis assays. This work was supported by NIH Grants DK54909 and CA16056.
Abbreviations
- BisA
bistratene A
- DAG
diacylglycerol
- HRP
horse-radish peroxidase
- MAPK
mitogen-activated protein kinase
- PDBu
phorbol 12,13-dibutyrate
- PI
propidium iodide
- PKC
protein kinase C
- and PMA
phorbol 12-myristate 13-acetate
References
- 1.Stanwell C, Gescher A, Watters D. Cytostatic and cytotoxic properties of the marine product bistratene A and analysis of the role of protein kinase C in its mode of action. Biochem Pharmacol. 1993;45:1753–61. doi: 10.1016/0006-2952(93)90430-5. [DOI] [PubMed] [Google Scholar]
- 2.Griffiths G, Garrone B, Deacon E, Owen P, Pongracz J, Mead G, Bradwell A, Watters D, Lord J. The polyether bistratene A activates protein kinase C-δ and induces growth arrest in HL60 cells. Biochem Biophys Res Commun. 1996;222:802–8. doi: 10.1006/bbrc.1996.0830. [DOI] [PubMed] [Google Scholar]
- 3.Watters D, Garrone B, Coomer J, Johnson WE, Brown G, Parsons P. Stimulation of melanogenesis in a human melanoma cell line by bistratene A. Biochem Pharmacol. 1998;55:1691–9. doi: 10.1016/s0006-2952(97)00680-1. [DOI] [PubMed] [Google Scholar]
- 4.Watters D, Marshall K, Hamilton S, Michael J, McArthur M, Seymour G, Hawkins C, Gardiner R, Lavin M. The bistratenes: new cytotoxic marine macrolides which induce some properties indicative of differentiation in HL-60 cells. Biochem Pharmacol. 1990;39:1609–14. doi: 10.1016/0006-2952(90)90528-s. [DOI] [PubMed] [Google Scholar]
- 5.Watters D, Garrone B, Gobert G, Williams S, Gardiner R, Lavin M. Bistratene A causes phosphorylation of talin and redistribution of actin microfilaments in fibroblasts: possible role for PKC-δ. Exp Cell Res. 1996;229:327–35. doi: 10.1006/excr.1996.0378. [DOI] [PubMed] [Google Scholar]
- 6.Watters DJ, Michael J, Hemphill JE, Hamilton SE, Lavin MF, Pettit GR. Bistratene A: a novel compound causing changes in protein phosphorylation patterns in human leukemia cells. J Cell Biochem. 1992;49:417–24. doi: 10.1002/jcb.240490412. [DOI] [PubMed] [Google Scholar]
- 7.Song Q, Baxter GD, Kovacs EM, Findik D, Lavin MF. Inhibition of apoptosis in human tumour cells by okadaic acid. J Cell Physiol. 1992;153:550–6. doi: 10.1002/jcp.1041530316. [DOI] [PubMed] [Google Scholar]
- 8.Weller SG, Klein IK, Penington RC, Karnes WE., Jr Distinct protein kinase C isozymes signal mitogenesis and apoptosis in human colon cancer cells. Gastroenterology. 1999;117:848–57. doi: 10.1016/s0016-5085(99)70343-4. [DOI] [PubMed] [Google Scholar]
- 9.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–14. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 10.Clemens MJ, Trayner I, Menaya J. The role of protein kinase C isoenzymes in the regulation of cell proliferation and differentiation. J Cell Sci. 1992;103:881–7. doi: 10.1242/jcs.103.4.881. [DOI] [PubMed] [Google Scholar]
- 11.Dekker LV, Parker PJ. Protein kinase C—a question of specificity. Trends Biochem Sci. 1994;19:73–7. doi: 10.1016/0968-0004(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 12.Black JD. Protein kinase C-mediated regulation of the cell cycle. Front Biosci. 2000;5:D406–23. doi: 10.2741/black. [DOI] [PubMed] [Google Scholar]
- 13.Mischak H, Goodnight J, Kolch W, Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH, Mushinski JF. Overexpression of protein kinase C-δ and -ε in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268:6090–6. [PubMed] [Google Scholar]
- 14.Mischak H, Pierce JH, Goodnight J, Kazanietz MG, Blumberg PM, Mushinski JF. Phorbol ester-induced myeloid differentiation is mediated by protein kinase C-α and -δ and not by protein kinase C-β II, -ε, -ζ, and -η. J Biol Chem. 1993;268:20110–15. [PubMed] [Google Scholar]
- 15.Ishino K, Ohba M, Kashiwagi M, Kawabe S, Chida K, Kuroki T. Phorbol ester-induced G1 arrest in BALB/MK-2 mouse keratinocytes is mediated by δ and η isoforms of protein kinase C. Jpn J Cancer Res. 1998;89:1126–33. doi: 10.1111/j.1349-7006.1998.tb00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukumoto S, Nishizawa Y, Hosoi M, Koyama H, Yamakawa K, Ohno S, Morii H. Protein kinase C δ inhibits the proliferation of vascular smooth muscle cells by suppressing G1 cyclin expression. J Biol Chem. 1997;272:13816–22. doi: 10.1074/jbc.272.21.13816. [DOI] [PubMed] [Google Scholar]
- 17.Watanabe T, Ono Y, Taniyama Y, Hazama K, Igarashi K, Ogita K, Kikkawa U, Nishizuka Y. Cell division arrest induced by phorbol ester in CHO cells overexpressing protein kinase C-δ subspecies. Proc Natl Acad Sci USA. 1992;89:10159–63. doi: 10.1073/pnas.89.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deacon EM, Pongracz J, Griffiths G, Lord JM. Isoenzymes of protein kinase C: differential involvement in apoptosis and pathogenesis. Mol Pathol. 1997;50:124–31. doi: 10.1136/mp.50.3.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, Messing RO. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol. 2000;279:L429–38. doi: 10.1152/ajplung.2000.279.3.L429. [DOI] [PubMed] [Google Scholar]
- 20.Watters DJ, Parsons PG. Critical targets of protein kinase C in differentiation of tumour cells. Biochem Pharmacol. 1999;58:383–8. doi: 10.1016/s0006-2952(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 21.Garrone B, Kedar P, Elarova I, Lavin M, Watters D. Approaches to determine the specific role of the delta isoform of protein kinase C. J Biochem Biophys Methods. 1997;36:51–61. doi: 10.1016/s0165-022x(97)00041-9. [DOI] [PubMed] [Google Scholar]
- 22.Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21waf1/cip1 and p27kip1 and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. J Biol Chem. 1997;272:9424–35. doi: 10.1074/jbc.272.14.9424. [DOI] [PubMed] [Google Scholar]
- 23.Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol. 2000;151:763–77. doi: 10.1083/jcb.151.4.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quaroni A, May RJ. Establishment and characterizaton of intestinal epithelial cell cultures. Methods Cell Biol. 1980;21B:403–27. [PubMed] [Google Scholar]
- 25.Saxon ML, Zhao X, Black JD. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J Cell Biol. 1994;126:747–63. doi: 10.1083/jcb.126.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Kraft AS, Anderson WB. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 1983;301:621–3. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]
- 28.Cross T, Griffiths G, Deacon E, Sallis R, Gough M, Watters D, Lord JM. PKC-δ is an apoptotic lamin kinase. Oncogene. 2000;19:2331–7. doi: 10.1038/sj.onc.1203555. [DOI] [PubMed] [Google Scholar]
- 29.Newton AC. Regulation of protein kinase C. Curr Opin Cell Biol. 1997;9:161–7. doi: 10.1016/s0955-0674(97)80058-0. [DOI] [PubMed] [Google Scholar]
- 30.Traverse S, Gomez N, Paterson H, Marshall C, Cohen P. Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J. 1992;288:351–5. doi: 10.1042/bj2880351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woods D, Parry D, Cherwinski H, Bosch E, Lees E, McMahon M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5598–611. doi: 10.1128/mcb.17.9.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roussakis C, Robillard N, Riou D, Biard JF, Pradal G, Piloquet P, Debitus C, Verbist JF. Effects of bistramide A on a non-small-cell bronchial carcinoma line. Cancer Chemother Pharmacol. 1991;28:283–92. doi: 10.1007/BF00685536. [DOI] [PubMed] [Google Scholar]
- 33.Watters DJ, Beamish HJ, Marshall KA, Gardiner RA, Seymour GJ, Lavin MF. Accumulation of HL-60 leukemia cells in G2/M and inhibition of cytokinesis caused by two marine compounds, bistratene A and cycloxazoline. Cancer Chemother Pharmacol. 1994;33:399–409. doi: 10.1007/BF00686269. [DOI] [PubMed] [Google Scholar]
- 34.Johnson WE, Watters DJ, Suniara RK, Brown G, Bunce CM. Bistratene A induces a microtubule-dependent block in cytokinesis and altered stathmin expression in HL60 cells. Biochem Biophys Res Commun. 1999;260:80–8. doi: 10.1006/bbrc.1999.0854. [DOI] [PubMed] [Google Scholar]
- 35.Marklund U, Larsson N, Gradin HM, Brattsand G, Gullberg M. Oncoprotein 18 is a phosphorylation-responsive regulator of micro-tubule dynamics. EMBO J. 1996;15:5290–8. [PMC free article] [PubMed] [Google Scholar]
- 36.Belmont LD, Mitchison TJ. Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell. 1996;84:623–31. doi: 10.1016/s0092-8674(00)81037-5. [DOI] [PubMed] [Google Scholar]
- 37.Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, Ghayur T, Wong WW, Kamen R, Weichselbaum R, Kufe D. Proteolytic activation of protein kinase C δ by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–56. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, Pandey P, Datta R, Huang Y, Kharbanda S, Allen H, Kamen R, Wong W, Kufe D. Proteolytic activation of protein kinase C δ by an ICE/CED 3-like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das D, Pintucci G, Stern A. MAPK-dependent expression of p21WAF and p27kip1 in PMA-induced differentiation of HL60 cells. FEBS Lett. 2000;472:50–2. doi: 10.1016/s0014-5793(00)01416-2. [DOI] [PubMed] [Google Scholar]
- 40.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol. 1998;18:790–8. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]