

Abstract
Background. The compilation of previous genomewide association studies of AIDS shows a major polymorphism in the HCP5 gene associated with both control of the viral load and long-term nonprogression (LTNP) to AIDS.
Methods. To look for genetic variants that affect LTNP without necessary control of the viral load, we reanalyzed the genomewide data of the unique LTNP Genomics of Resistance to Immunodeficiency Virus (GRIV) cohort by excluding “elite controller” patients, who were controlling the viral load at very low levels (<100 copies/mL).
Results. The rs2234358 polymorphism in the CXCR6 gene was the strongest signal (P = 2.5 × 10−7; odds ratio, 1.85) obtained for the genomewide association study comparing the 186 GRIV LTNPs who were not elite controllers with 697 uninfected control subjects. This association was replicated in 3 additional independent European studies, reaching genomewide significance of Pcombined = 9.7 × 10−10. This association with LTNP is independent of the combined CCR2-CCR5 locus and the HCP5 polymorphisms.
Conclusion. The statistical significance, the replication, and the magnitude of the association demonstrate that CXCR6 is likely involved in the molecular etiology of AIDS and, in particular, in LTNP, emphasizing the power of extreme-phenotype cohorts. CXCR6 is a chemokine receptor that is known as a minor coreceptor in human immunodeficiency virus type 1 infection but could participate in disease progression through its role as a mediator of inflammation.
Previous genomewide association studies (GWASs) of AIDS have revealed a major association involving a genetic polymorphism within the human leukocyte antigen region, the rs2395029 HCP5 single-nucleotide polymorphism (SNP), which tracks HLA-*5701. This SNP was associated with viral load control through analysis of human immunodeficiency virus type 1 (HIV-1) seroconverters [1, 2] and by the comparison of patients with long-term nonprogression (LTNPs) with uninfected control subjects as well [3]. LTNPs are a small percentage (1%–5%) of HIV-1 seroconverters [4–6] and thus constitute a powerful contrasting tool to unravel new genetic factors associated with AIDS progression. Of the LTNPs in the Genomics of Resistance to Immunodeficiency Virus (GRIV) cohort, patients carrying the HCP5 rs2395029-G allele exhibited a significantly lower viral load than the rest of the cohort [3]. Only a minority (ie, 25%) of the GRIV LTNPs exhibited effective viral load control (ie, a very low viral load of <100 copies/mL). Because viral load is known to account for only 34% of the variability in the time to a CD4 T cell decrease of <200 cells/µL [7], we decided to perform a new analysis of the genomewide data for GRIV LTNPs by excluding these “elite controller” subjects (subjects who had a viral load of <100 copies/mL). The aim of the current study was thus to focus on genetic variations affecting LTNP without necessarily controlling the viral load at a very low level. The result is that we have indeed identified a new specific signal in the CXCR6 gene and have replicated this finding in 3 additional independent cohorts of European descent.
Methods
The GRIV Study: Participants, Genotyping, and Analysis
The GRIV cohort. The GRIV study cohort and methods were described in detail in previously published work on the genomewide association study of LTNPs [3]. The GRIV cohort was established in France in 1995 to generate a large collection of DNAs for genetic studies seeking to identify host genes associated with rapid and LTNP to AIDS [8–11]. Only white people who were of European descent and were living in France were eligible for enrollment, to reduce confounding by population substructure. The LTNPs were seroprevalent subjects who were included on the basis of their main clinical outcomes, CD4 T cell count, and time to disease progression: asymptomatic HIV-1 infection for >8 years, no receipt of antiretroviral treatment, and a CD4 T cell count consistently >500 cells/mm3.
Among those in the LTNP group (n = 275), viral load (ie, the plasma HIV-1 RNA concentration) at the time of inclusion could be assessed for 248 individuals. Of these 248 individuals, 186 had a viral load >100 copies/mL. All subjects provided written, informed consent before their enrollment in the GRIV genetic association study.
The control group. The Data from an Epidemiological Study on Insulin Resistance Syndrome (DESIR) program was a 9-year follow-up study designed to clarify the development of the insulin resistance syndrome. During 1994–1996, subjects were recruited from volunteers insured by the French social security system, which offers periodic health examinations free of charge [12]. This control group was comprised of 697 non-obese and normoglycemic individuals, and all were French, of European descent, and HIV-1 seronegative.
Genotyping method and quality control. The GRIV cohort and the control group were genotyped using the Illumina Infinium II HumanHap300 BeadChips (Illumina). Genotyping quality was assessed using BeadStudio software (version 3.1; Illumina). Missing data (>2%), low minor allele frequency (<1%), and deviations from Hardy-Weinberg equilibrium in the control group (P < 1.0 × 10−3) were excluded from analysis during these quality control steps. Moreover, identification of potential population stratification was identified using Structure software (version 2.2) [13], by producing a quantile-quantile plot (see Figure A1A in the Appendix, which appears only in the electronic version of the Journal) and by computing the genomic inflation factor λ. Overall, little effect of stratification was observed, and 283,637 SNPs could be tested statistically for association with LTNP.
Figure 1.

Effect of rs2234358 in the Genomics of Resistance to Immunodeficiency Virus (GRIV), Amsterdam Cohort Study (ACS), Multicenter AIDS Cohort Study (MACS), and USA HIV-1 study groups. A, Allelic frequency of rs2234358-T in the GRIV long-term nonprogressor (LTNP) population (n = 186) and the control group (CTR) (n = 697). Frequencies are also given for the 31 ACS subjects with LTNP (ACS LTNPs), for the remaining 285 ACS participants (ACS*), for the 59 MACS subjects with LTNP (MACS LTNPs), and for the remaining 97 MACS participants (MACS*). B, Kaplan-Meier survival curve derived from the ACS cohort for the time to AIDS-related death. Genotypes GG (green) (n = 76), GT (blue) (n = 171), and TT (black) (n = 69). C, Kaplan-Meier survival curve derived from the MACS cohort for time to clinical AIDS. Genotypes GG (green) (n = 45), GT (blue) (n = 72), and TT (black) (n = 39). D, Kaplan-Meier survival curve derived from the USA HIV-1 cohort for time to AIDS-related death. Genotypes GG (green) (n = 140), GT (blue) (n = 297), and TT (black) (n = 119).
Statistical analysis. For each SNP, we performed a standard case-control analysis, using Fisher's exact tests (with Plink software [14]) to compare allelic distributions between LTNPs and the control subjects. Bonferroni corrections were made to account for multiple comparisons.
SNP imputation. Untyped SNPs present in the HapMap database of chromosome 3 were imputed for all GRIV patients and control subjects, by use of Impute software (version 2.1) [15] and the HapMap release 21 phased data for the white population (CEU) as the reference panel [16].
CXCR6 genotyping by PCR sequencing. Primers and conditions used for polymerase chain reaction (PCR) amplifications were standard. Sequencing reactions were performed according to the Dye Terminator method by use of an ABI Prism 3730XL DNA Analyzer (Applied Biosystems). Alignment, SNP discovery, and genotyping were performed using the software Genalys, which was developed by the Commissariat à l'Énergie Atomique/Centre National de Génotypage [17].
Haplotype inference. Haplotype inference was obtained using the rapid and accurate Shape-IT algorithm [18].
Bioinformatics exploration. To further explore the associations observed, we tried to identify modifications in messenger RNA (mRNA) expression (Genevar [19] and Dixon [20] databases), splicing (NetGene2 [21]), polyadenylation (polyAH [22] and polyApred [23]), and transcription factor-binding sites (SignalScan [24], TESS [25], and TFSearch [26], derived from the TRANSFAC database).
Replication in the Amsterdam Cohort Study: Participants, Genotyping, and Analysis
The Amsterdam Cohort Study (ACS) participants andmethods were described in detail elsewhere [27]. In the present study, 335 HIV-1-infected homosexual men from ACS were analyzed for the course of HIV-1 infection using AIDS-related death as an end point. AIDS-related death is defined as death with AIDSrelated malignancy, death with AIDS opportunistic infections, or death with an AIDS-related cause not specified by the treating physician.
The ACS rs2234358 genotyping data were obtained using Illumina Infinium II HumanHap300 BeadChips (Illumina). Quality control filters were applied to ensure reliable genotyping data. Potential population stratification was also analyzed using Structure software (version 2.2) [13], and 19 participants were thus excluded from further analyses (n = 316) because they differed significantly from the HapMap white population.
Statistical analysis was performed by Kaplan-Meier survival analysis and determination of the log rank P value under the genotypic model, by use of SPSS software (version 16.0; SPSS) and the R package [28].
Because the viral load (ie, the plasma HIV-1 RNA concentration) and the CD4 T cell count were assessed during routine clinical follow-up, we could identify the ACS LTNPs who matched the GRIV definition and exhibited a viral load of >100 copies/mL (n = 31). LTNP status was easily determined for seroconverters because the date of seroconversion was known, and this was also the case for seroprevalent subjects, because the time of seropositivity was imputed (on average, at 18 months before enrollment).
Replication in the Multicenter AIDS Cohort Study: Participants, Genotyping, and Analysis
Multicenter AIDS Cohort Study (MACS) participants and methods previously have been described in detail elsewhere [29]. GWAS data were collected from 156 HIV-1-infected white homosexual men, with time to clinical AIDS used as an end point. This panel was chosen to be enriched with extreme AIDS progression phenotypes.
The MACS rs2234358 genotyping data were obtained using the Affymetrix GeneChip Human Mapping 500K Array (Affymetrix), in which the rs2234358 SNP is tagged by rs4682799 (r2 = 1). Quality control filters were applied to ensure reliable genotyping data, and population stratification was also checked.
Statistical analysis was performed by Kaplan-Meier survival analysis and Cox proportional regression determination of the P value under the genotypic model using the R package.
As with the ACS, viral load (the plasma HIV-1 RNA concentration) and CD4 T cell count were assessed during routine clinical follow-up. We could extract 59 MACS LTNPs, selected from among seroconverters and seroprevalent subjects, who matched the GRIV definition and exhibited a viral load of >100 copies/mL.
Replication in the USA HIV-1 Cohort: Participants, Genotyping, and Analysis
USA HIV-1 cohort patients and methods previously were described in detail elsewhere [30]. For this study, 556 HIV-1 seroconverters of European ancestry were collected from 4 USA-based natural history HIV/AIDS longitudinal cohorts (MACS, San Francisco City Clinic Cohort, Multicenter Hemophilia Cohort Study, and Hemophilia Growth and Development Study), with AIDS-related death used as an end point. Of importance, the 556 USA HIV-1-infected participants did not include subjects overlapping with the 156 MACS participants.
The USA HIV-1 cohort rs2234358 genotyping data were obtained using commercial TaqMan genotype assays (with assay ID C_1929536_1; Applied Biosystems). Conformity to the genotype frequencies expected under Hardy-Weinberg equilibrium was checked.
Statistical analysis was performed by Kaplan-Meier survival analysis and Cox proportional regression for determination of the P value under the genotypic model using the statistical SAS package (version 9.13; SAS Institute).
Independence from the CCR2-CCR5 Locus and HCP5 Polymorphisms
The genotypic data were available for the known CCR2-CCR5 locus and HCP5 polymorphisms in the GRIV, MACS, and USA HIV-1 cohorts, and it was thus possible to assess the independence of the rs2234358 SNP from these polymorphisms.
For the GRIV cohort, multivariate logistic regression analysis was used to adjust effects of covariates CCR2-64I, CCR5-Δ32, CCR5-P1, and HCP5 rs2395029. The same approach was done for the MACS and USA HIV-1 cohorts but by fitting to the data a linear model instead of a logistic model. The independent effect of the rs2234358 SNP on disease phenotype was confirmed by adjusting the model with these covariates: the P values that were obtained were similar with and without the covariate analysis.
Results
After quality control tests, a case-control analysis using Fisher's exact tests was performed to compare allelic distributions of the 283,637 SNPs between the GRIV LTNPs exhibiting a detectable viral load (>100 copies/mL) (n = 186) and uninfected controls (n = 697) (see Methods). The strongest association was found for rs2234358, with a P value close to the 1.7 × 10−7 Bonferroni threshold for genomewide significance (see Figure A1B in the Appendix, which appears only in the electronic version of the Journal): P = 2.5 × 10−7 (odds ratio [OR], 1.85 [95% confidence interval {CI}, 1.46–2.36]). The rs2234358-T allele was associated with not being an LTNP (36.83% in LTNPs vs. 51.94% in controls) (Figure 1A). This allele was not associated with acquisition of HIV-1 infection, because its frequency was similar in seropositive and control groups: 51.16% among GRIV rapid progressors [31], 48.59% in the ACS, 51.92% in the MACS, 48% in the USA HIV-1 cohort, 54.9% in the Dutch control population (CTRACS), 48.3% in the Illumina controls (CTRIllumina), and 50.9% in HapMap CEU (see Figure A2 in the Appendix, which appears only in the electronic version of the Journal).
Figure 2.
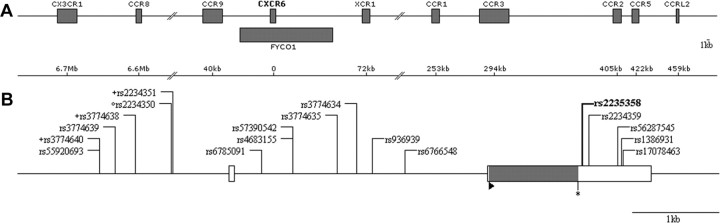
A, Genetic map of the CXCR6 gene region. CXCR6 is localized within the 14th intron of a predicted gene, FYCO1. B, Genetic map of the CXCR6 gene. Exons and untranslated regions are symbolized by full and empty rectangles, respectively. The positions of the ATG and STOP codons are indicated by a triangle (▸) and an asterisk (*), respectively. All single-nucleotide polymorphisms (SNPs) covered by the polymerase chain reaction sequencing study are represented, and the rs2234358 SNP of interest is shown in boldface type. The 3 promoter haplotypes in high linkage disequilibrium (LD) with rs2234358 (r2 = 0.97) correspond to 2-SNP haplotypes composed of the rs2234350 SNP (°), with either one of the SNPs denoted by the + symbol (these 3 latter SNPs exhibit r2 = 0.99).
The rs2234358 SNP lies within the CXCR6 gene in a region of chromosome 3 that is rich in genes encoding chemokine receptors, and it is notably positioned 422 kb from the well-known CCR5 gene [32] (Figure 2A). To eliminate possible tracking effects, we evaluated a potential association between the CXCR6 signal and the CCR2-CCR5 haplotypes (Δ32, P1, and 64I) previously associated with the course of HIV-1 disease [9, 32–33]. First, the rs2234358 SNP had no linkage disequilibrium (LD, r2 < 0.1) with any of the CCR5-Δ32, CCR5-P1, or CCR2-64I haplotypes. Second, we could not find any epistatic effects between rs2234358 and any of these haplotypes, by use of either Plink software [14] or logistic regression using CCR2-CCR5 haplotypes as covariates (version 2.1) (see Methods). Third, the HapMap LD for whites did not reveal any SNP with a high LD (r2 > 0.9) beyond the CXCR6 locus. Of note, we also did not observe an epistatic effect between rs2234358 and the chromosome 6 rs2395029 HCP5/HLA-B*5701 signal. This CXCR6 signal thus represents a new association with LTNP, independent from the well-known CCR2-CCR5 and HCP5/HLA-B*5701 associations. Of interest, we inferred the SNP distribution over the entire chromosome 3, using Impute software (see Methods). Instead of the 20,000 genotyped SNPs present in the Illumina HumanHap300 BeadChip in chromosome 3, a total of 176,000 SNPs could be imputed for which we could not identify a P value better than the one exhibited for rs2234358. The attributable risk for rs2234358-T variant is very strong, because it explains 12% of the prevention of LTNP. For comparison, the attributable risk for CCR5-Δ32 is 5.1% in the GRIV LTNP cohort.
The rs2234358 signal was replicated in 3 independent additional cohort studies of white people of European descent that also evaluated for AIDS progression phenotype (after removal of stratification outliers; see Methods): (1) the European ACS cohort (n = 316) (P = 2.3 × 10−2) (Figure 1B), (2) a European descent subgroup of the American MACS cohort enriched in extreme phenotypes (n = 156) (P = 4.2 × 10−3) (Figure 1C), and (3) a pool of European American HIV-1 cohorts (n = 556) (P = 8.6 × 10−3) (Figure 1D and Table A1, the latter of which may be found in the Appendix, which appears only in the electronic version of the Journal). As shown in Figure 1, the rs2234358-T allele favored progression in all of these cohorts, which is in agreement with a prevention of LTNP. Overall, the combined P value computed by the Fisher method between the 4 cohorts (GRIV, ACS, MACS, and USA HIV-1) passed the Bonferroni genomewide significance threshold: Pcombined = 9.7 × 10−10.
It was surprising to observe significant but rather weak P values in all cohorts except the GRIV cohort, so we assessed whether the effect was specifically amplified in the LTNP subpopulation. We identified 31 and 59 LTNPs fulfilling the GRIV definition and with a detectable viral load (>100 copies/mL) in the ACS and MACS cohorts, respectively. In these groups, the rs2234358-T allele frequency was ∼40%, which is similar to that found among the GRIV LTNPs (Figure 1A). Because no LTNP from these 3 cohorts differed significantly from the HapMap white population, according to the Structure analysis [13] (see Figure A3B in the Appendix, which appears only in the electronic version of the Journal), the ACS and MACS LTNPs were added to those in the GRIV cohort, and we computed a P value comparing this extended LTNP case group (n = 276) with the control group (n = 697). The P value again reached genomewide significance: P = 2.1 × 10−8 (OR, 1.77 [95% CI, 1.44–2.18]), confirming the association of rs2234358-T with prevention of LTNP (Table A1, which may be found in the Appendix, which appears only in the electronic version of the Journal). Of importance, several additional control groups were tested and exhibited a similar allele frequency for rs2234358-T (see Figure A2 in the Appendix, which appears only in the electronic version of the Journal).
To further explore this association, we resequenced the entire CXCR6 gene to detect additional variants (Figure 2B and Table A2, the latter of which may be found in the Appendix, which appears only in the electronic version of the Journal): rs2234358 remained the SNP exhibiting the strongest association. Interestingly, using Shape-IT software (version 2.0) to compute haplotypes [18], we found several haplotypes comprising CXCR6 promoter SNPs in high LD (r2 = 0.97) with rs2234358 (Figure 2B).
The rs2234358 SNP is located in the 3' untranslated region of CXCR6, located 42 bp downstream from the termination codon (Figure 2B), and could thus influence gene expression, mRNA stability, mRNA regulation, or mRNA splicing. According to the Dixon or Genevar mRNA expression databases, none of the genotyped SNPs are predicted to affect CXCR6 or any other chromosome 3 gene expression, and bioinformatics methods failed to predict a modification of splicing or polyadenylation sites (see Methods). Nevertheless, we identified sev eral putative transcription factor-binding sites containing the SNPs included in promoter haplotypes in high LD with rs2234358 (see Methods). Further experiments are required to determine the causative genetic variants and the biological mechanisms at stake.
Discussion
Because the major signal identified in previous AIDS GWASs was associated with control of viral replication, we reanalyzed the genomewide data of the French GRIV LTNP cohort by excluding elite controller patients (ie, patients with a viral load of <100 copies/mL). The comparison of 186 LTNPs exhibiting a viral load of >100 copies/mL with 697 uninfected controls highlighted a strong association for the CXCR6 rs2234358 (P = 2.5 × 10−7). This new signal was replicated by a candidate SNP approach in 3 additional independent European descent cohorts (including 316, 156, and 556 subjects), and the combined P value of the 4 cohorts reached the genomewide significance threshold: Pcombined = 9.7 × 10−10. This chromosome 3 combined association is independent from the well-known neighboring CCR2-CCR5 locus, is not linked with the control of viral load (the GRIV LTNP groups carrying the various rs2234358 genotypes exhibit a similar mean viral load) (P = .72, data not shown), and exhibits a high attributable risk of LTNP of 12%.
This study presents the first non-HLA-replicated association obtained through a GWAS approach. The P value for rs2234358 is very strong in the GRIV cohort, and this signal was confirmed in 3 independent cohorts but with weaker P values. The specific design of the LTNP phenotype can explain this discrepancy. Indeed, the extraction of LTNPs with a viral load >100 copies/mL from the ACS and MACS cohorts confirmed the strength of this common SNP association with LTNP: ∼40% versus ∼50% in several uninfected control groups (see Figure 1A and Figure A2 in the Appendix, the latter of which appears only in the electronic version of the Journal). It emphasizes the critical importance of cohort design and the particular power of extreme phenotypes [5, 34–35], particularly in light of a recent powerful GWAS involving >2500 patients, which solely identified chromosome 6-related signals [36].
The finding of a new chemokine receptor genetic variant contributing to a differential progression to AIDS is not so much of a surprise, because the chemokine system is a major weapon of the early host defense system against infectious diseases and comprises >100 members. An exonic CXCR6 variant present in African Americans (but absent in Europeans) was previously associated with Pneumocystis carinii pneumonia-mediated progression to AIDS [37]. Our CXCR6 genetic variant is not exonic, and its biological effect should rather be a modulation of CXCR6 expression. CXCR6, known as a minor HIV-1 coreceptor [38] and mediator of inflammation [39, 40], is notably expressed in organs (thymus, gut, and bone marrow) and in immune cells [41], which are important for HIV-1 infection. It is involved in the trafficking of effector T cells mediating type 1 inflammation [39] and in the activation and homeostasis of natural killer T cells [42], known to be an important bridge between innate and adaptive immune responses. Interestingly, in simian immunodeficiency virus (SIV) infection, it has been proposed that interleukin-17-secreting natural killer T cells could compensate for the Th17 defect in the gut, because they are essential for controlling mucosal barrier integrity and microbial translocation [43, 44]. These hypotheses are compatible with a major role of CXCR6 as an inflammation mediator in AIDS [39, 40], but they deserve further functional/biological research to enhance our understanding of the molecular pathways to HIV-1 LTNP.
At a time when HIV-1 entry inhibitors such as CCR5 and CXCR4 antagonists are being developed, the identification of a molecular mechanism of AIDS pathogenesis involving a new chemokine receptor is of particular interest and opens new insights for therapeutic drug targets and prediction of HIV-1 progression.
Acknowledgments
We thank all the patients and medical staff who have kindly collaborated with the Genomics of Resistance to Immunodeficiency Virus project. S.L. benefits from a fellowship from the French Ministry of Education, Technology and Research, and S.L.C. benefits from a fellowship of Agence Nationale de Recherches sur le SIDA et les Hépatites Virales Genomic Group.
Financial support. Agence Nationale de Recherche sur le SIDA, Sidaction, Innovation 2007 program of Conservatoire National des Arts et Métiers, AIDS Cancer Vaccine Development Foundation, Neovacs SA, Vax-consulting, and R37 (grant AI047734) from the US National Institutes of Health. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health (NIH; contract HHSN261200800001E) and in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The authors acknowledge funding from the Netherlands Organization for Scientific Research (TOP; registration number 9120.6046). The Hemophilia Growth and Development Study is funded by the National Institutes of Health, National Institute of Child Health and Human Development, 1 R01 HD41224. The Amsterdam Cohort Studies on HIV infection and AIDS, a collaboration between the Amsterdam Health Service, the AcademicMedical Center of the University of Amsterdam, Sanquin Research, and the University Medical Center Utrecht, are part of the Netherlands HIV Monitoring Foundation and are financially supported by the Netherlands National Institute for Public Health and the Environment.
Footnotes
Potential conflicts of interest: none reported.
Financial support: see the Acknowledgments section.
References
- 1.Fellay J, Shianna KV, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalmasso C, Carpentier W, Meyer L, et al. Distinct genetic loci control plasma HIV-RNA and cellular HIV-DNA levels in HIV-1 infection: the ANRS Genome Wide Association 01 study. PLoS ONE. 2008;3:e3907. doi: 10.1371/journal.pone.0003907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Limou S, Le Clerc S, Coulonges C, et al. Genomewide association study of an AIDS-nonprogression cohort emphasizes the role played by HLA genes (ANRS Genomewide Association Study 02) J Infect Dis. 2009;199:419–426. doi: 10.1086/596067. [DOI] [PubMed] [Google Scholar]
- 4.Grabar S, Selinger-Leneman H, Abgrall S, Pialoux G, Weiss L, Costagliola D. Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French Hospital Database on HIV. AIDS. 2009;23:1163–1169. doi: 10.1097/QAD.0b013e32832b44c8. [DOI] [PubMed] [Google Scholar]
- 5.Huber C, Pons O, Hendel H, et al. Genomic studies in AIDS: problems and answers. Development of a statistical model integrating both longitudinal cohort studies and transversal observations of extreme cases. Biomed Pharmacother. 2003;57:25–33. doi: 10.1016/s0753-3322(02)00335-9. [DOI] [PubMed] [Google Scholar]
- 6.Petrucci A, Dorrucci M, Alliegro MB, et al. How many HIV-infected individuals may be defined as long-term nonprogressors? A report from the Italian Seroconversion Study? Italian Seroconversion Study Group (ISS) J Acquir Immune Defic Syndr Hum Retrovirol. 1997;14:243–8. doi: 10.1097/00042560-199703010-00008. [DOI] [PubMed] [Google Scholar]
- 7.Mellors JW, Margolick JB, Phair JP, et al. Prognostic value of HIV-1 RNA, CD4 cell count, and CD4 cell count slope for progression to AIDS and death in untreated HIV-1 infection. JAMA. 2007;297:2349–2350. doi: 10.1001/jama.297.21.2349. [DOI] [PubMed] [Google Scholar]
- 8.Rappaport J, Cho YY, Hendel H, Schwartz EJ, Schachter F, Zagury JF. 32 bp CCR-5 gene deletion and resistance to fast progression in HIV-1 infected heterozygotes. Lancet. 1997;349:922–923. doi: 10.1016/S0140-6736(05)62697-9. [DOI] [PubMed] [Google Scholar]
- 9.Winkler CA, Hendel H, Carrington M, et al. Dominant effects of CCR2-CCR5 haplotypes in HIV-1 disease progression. J Acquir Immune Defic Syndr. 2004;37:1534–1538. doi: 10.1097/01.qai.0000127353.01578.63. [DOI] [PubMed] [Google Scholar]
- 10.Hendel H, Caillat-Zucman S, Lebuanec H, et al. New class I and II HLA alleles strongly associated with opposite patterns of progression to AIDS. J Immunol. 1999;162:6942–6946. [PubMed] [Google Scholar]
- 11.Flores-Villanueva PO, Hendel H, Caillat-Zucman S, et al. Associations of MHC ancestral haplotypes with resistance/susceptibility to AIDS disease development. J Immunol. 2003;170:1925–1929. doi: 10.4049/jimmunol.170.4.1925. [DOI] [PubMed] [Google Scholar]
- 12.Balkau B. An epidemiologic survey from a network of French Health Examination Centres, (D.E.S.I.R.): epidemiologic data on the insulin resistance syndrome. Rev Epidemiol Sante Publique. 1996;44:373–375. [in French] [PubMed] [Google Scholar]
- 13.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 16.International HapMap Project http://www.hapmap.org Accessed 22 July 2010.
- 17.Takahashi M, Matsuda F, Margetic N, Lathrop M. Automated identification of single nucleotide polymorphisms from sequencing data. J Bioinform Comput Biol. 2003;1:253–265. doi: 10.1142/s021972000300006x. [DOI] [PubMed] [Google Scholar]
- 18.Delaneau O, Coulonges C, Zagury JF. Shape-IT: new rapid and accurate algorithm for haplotype inference. BMC Bioinformatics. 2008;9:540. doi: 10.1186/1471-2105-9-540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge D, Zhang K, Need AC, et al. WGAViewer: software for genomic annotation of whole genome association studies. Genome Res. 2008;18:640–643. doi: 10.1101/gr.071571.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon AL, Liang L, Moffatt MF, et al. A genome-wide association study of global gene expression. Nat Genet. 2007;39:1202–1207. doi: 10.1038/ng2109. [DOI] [PubMed] [Google Scholar]
- 21. http://www.cbs.dtu.dk/services/NetGene2/ NetGene2 Server. Accessed 22 July 2010.
- 22. http://linux1.softberry.com/berry.phtml?topic=polyah&group=programs&subgroup=promoterSoftBerry.com Accessed 30 July 2010.
- 23. http://www.imtech.res.in/raghava/polyapred/submission.html Accessed 22 July 2010.
- 24. http://www-bimas.cit.nih.gov/molbio/signal/.WWWSignalScan Accessed 22 July 2010.
- 25. http://www.cbil.upenn.edu/cgi-bin/tess/tess?RQ=WELCOMETranscriptionElementSearchSystem Accessed 22 July 2010.
- 26. http://www.cbrc.jp/research/db/TFSEARCH.html TFSearch: Searching transcription factor binding sites (ver 1.3) Accessed 22 July 2010.
- 27.van Manen D, Kootstra NA, Boeser-Nunnink B, Handulle MA, van't Wout AB, Schuitemaker H. Association of HLA-C and HCP5 gene regions with the clinical course of HIV-1 infection. AIDS. 2009;23:19–28. doi: 10.1097/QAD.0b013e32831db247. [DOI] [PubMed] [Google Scholar]
- 28. http://www.r-project.org R Project Accessed 22 July 2010.
- 29.Herbeck JT, Gottlieb GS, Winkler CA, et al. Multi-stage genome-wide association study identifies a locus at 1q41 associated with rate of HIV-1 disease progression to clinical AIDS. J Infect Dis. 2010;201:618–626. doi: 10.1086/649842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.An P, Duggal P,Wang LH, et al. Polymorphisms of CUL5 are associated with CD4+ T cell loss in HIV-1 infected individuals. PLoS Genet. 2007;3:e19. doi: 10.1371/journal.pgen.0030019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Le Clerc S, Limou S, Coulonges C, et al. Genomewide association study of a rapid progression cohort identifies new susceptibility alleles for AIDS (ANRS Genomewide Association Study 03) J Infect Dis. 2009;200:1194–1201. doi: 10.1086/605892. [DOI] [PubMed] [Google Scholar]
- 32.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 33.Martin MP, Dean M, Smith MW, et al. Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science. 1998;282:1907–1911. doi: 10.1126/science.282.5395.1907. [DOI] [PubMed] [Google Scholar]
- 34.Froguel P, Blakemore AI. The power of the extreme in elucidating obesity. N Engl J Med. 2008;359:891–893. doi: 10.1056/NEJMp0805396. [DOI] [PubMed] [Google Scholar]
- 35.Zhang G, Nebert DW, Chakraborty R, Jin L. Statistical power of association using the extreme discordant phenotype design. Pharmacogenet Genomics. 2006;16:401–413. doi: 10.1097/01.fpc.0000204995.99429.0f. [DOI] [PubMed] [Google Scholar]
- 36.Fellay J, Ge D, Shianna KV, et al. Common genetic variation and the control of HIV-1 in humans. PLoS Genet. 2009;5:e1000791. doi: 10.1371/journal.pgen.1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duggal P, An P, Beaty TH, et al. Genetic influence of CXCR6 chemokine receptor alleles on PCP-mediated AIDS progression among African Americans. Genes Immun. 2003;4:245–250. doi: 10.1038/sj.gene.6363950. [DOI] [PubMed] [Google Scholar]
- 38.Deng HK, Unutmaz D, KewalRamani VN, Littman DR. Expression cloning of new receptors used by simian and human immunodeficiency viruses. Nature. 1997;388:296–300. doi: 10.1038/40894. [DOI] [PubMed] [Google Scholar]
- 39.Kim CH, Kunkel EJ, Boisvert J, et al. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J Clin Investig. 2001;107:595–601. doi: 10.1172/JCI11902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landrø L, Damås JK, Halvorsen B, et al. CXCL16 in HIV infection—a link between inflammation and viral replication. Eur J Clin Invest. 2009;39:1017–1024. doi: 10.1111/j.1365-2362.2009.02207.x. [DOI] [PubMed] [Google Scholar]
- 41.Koprak S, Matheravidathu S, Springer M, Gould S, Dumont FJ. Down-regulation of cell surface CXCR6 expression during T cell activation is predominantly mediated by calcineurin. Cell Immunol. 2003;223:1–12. doi: 10.1016/s0008-8749(03)00130-8. [DOI] [PubMed] [Google Scholar]
- 42.Germanov E, Veinotte L, Cullen R, Chamberlain E, Butcher EC, Johnston B. Critical role for the chemokine receptor CXCR6 in homeostasis and activation of CD1d-restricted NKT cells. J Immunol. 2008;181:81–91. doi: 10.4049/jimmunol.181.1.81. [DOI] [PubMed] [Google Scholar]
- 43.Campillo-Gimenez L, Cumont MC, Fay M, et al. AIDS progression is associated with the emergence of IL-17-producing cells early after simian immunodeficiency virus infection. J Immunol. 2010;184:984–992. doi: 10.4049/jimmunol.0902316. [DOI] [PubMed] [Google Scholar]
- 44.Raffatellu M, Santos RL, Verhoeven DE, et al. Simian immunodeficiency virus-induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]