

Abstract
We have developed a cylindrical insert that can be inserted in the fornix for extended release of glaucoma drug timolol. The insert is prepared by incorporating timolol-loaded nanoparticles into a poly hydroxyl ethyl methacrylate (p-HEMA) matrix. A 1-mm diameter, 7.5-mm long insert with 25% (w/w) particles can release timolol for about 10 days at an average rate of about 15 μg/day, which may be therapeutically effective. The increase in particle fraction increases drug loading, but also increases the release duration. The net effect of increasing the particle fraction is a significant increase in release duration, but a decrease in daily drug release rates, in the first few weeks. The release duration increases to about 1 and 3 months on increasing the particle fraction to 50% and 75%, respectively. The average daily release rates in the first 3 weeks are 15, 9, and 3 μg/day for the inserts with 50%, 75%, and 100% (w/w) particles, respectively. The mechanism of release is hydrolysis of the ester bond that links timolol to the propoxylated glyceryl triacrylate matrix, and thus the release profiles fit a first order reaction model. The water content of the inserts decreases from 31% to almost zero on increasing the particle loading from 25% to 100%. The rate constant for the hydrolysis decreases with an increase in particle loading in the insert most likely due to the reduction in the water content. The inserts can be packaged in wet conditions and stored in a refrigerator, but the inserts will exhibit a burst release caused by release of the drug from the particles into the p-HEMA matrix during the shelf life. Also, the magnitude of drug release after the initial burst is reduced due to the storage. The burst effect could potentially be avoided by packaging the inserts in a dry state, with hydration before insertion.
Introduction
Glaucoma is the second largest cause of blindness in the world (largest among African-Americans) and affects about 66.8 million people, leaving 6.7 million with bilateral blindness.1,2 The World Health Organization estimates that by 2020, the number of cases for blindness due to glaucoma will double to 12 million.2 In the U.S., approximately 120,000 are blind from glaucoma, accounting for 9% to 12% of all cases of blindness.
Glaucoma can be managed effectively through several intraocular pressure (IOP)-lowering medications, including prostaglandins, beta-blockers, and alpha-adrenergic agonists. In spite of the availability of effective medications, glaucoma continues to progress in several patients eventually leading to partial or total vision loss. Managing glaucoma effectively requires patient-compliance, which is frequently low for eye drop-based glaucoma therapies.3,4 The compliance is less than 50%5 for glaucoma eye drops, and even lower for multiple drops and/or multiple drug therapies.6 In addition to the lack of compliance, low bioavailability of glaucoma drugs delivered through eye drops is a major problem, which can lead to mild to severe side effects. The corneal bioavailability of glaucoma drugs ranges in 1%–5% because of the rapid tear turn over accompanied by drug transport into the conjunctiva, which has a larger area and permeability compared to the cornea.7–10
The deficiencies of the eye drop formulations for delivering ophthalmic drugs have resulted in research and development for improved formulations and novel devices for sustained release, including fornix inserts.11–13 There are also a few commercial successes, including Ocusert® (Alza), a highly successful pilocarpine releasing ocular insert that was placed in the cul de sac of the eye. Ocusert was shown in clinical studies to produce a constant reduction in IOP for over 7 days.14 The total dosage of pilocarpine administered by one Ocusert system over 7 days was about one-eighth of the amount provided by the 28 applications (4 each day) of the 2% eyedrops.15 The simplified regimen and reduced side effects from Ocusert encouraged patient compliance.16 Unfortunately, the manufacturer of Ocusert has discontinued making them likely due to some drawbacks, including retention problems, twisting to change shape, sight impediment due to dislocation of the insert in front of the pupil, but most important a sudden burst release in 0.4% of cases that could cause toxicity.17 Additionally, pilocarpine has been replaced by several newer drugs, such as prostaglandins, sometimes accompanied by beta-blockers, as the first line of defense against glaucoma.
Our goal in this article is to develop an ophthalmic insert that releases a glaucoma drug for an extended period of time of about 1 week, and can be safely and easily inserted in the fornix. Timolol, a beta-adrenergic receptor antagonist, is used as the drug because it has become the gold standard drug for IOP reduction since its approval by FDA in 197918 and also because of the potential of significant cardiac side effects from systemic exposure to timolol.19 Recently, Jung and Chauhan developed a nanoparticle poly hydroxylethyl methacrylate (p-HEMA) contact lens in which the drug molecules were crosslinked with the propoxylated glyceryl triacrylate (PGT) nanoparticles through an ester bond.20 The particle-loaded gels released timolol for about a month at room temperature due to slow hydrolysis of the ester bond.20 Contact lenses have also been designed for extended delivery of several other drugs, including dry eye drug cyclosporine, anti-inflammatory dexamethasone, anesthetic lidocaine, antibiotics, antivirals, and antifungals.21–25 The drug-eluding contact lenses are potentially very useful for ophthalmic drug delivery, but may not be used by patients that do not require vision correction. A drug-eluding fornix insert could be useful for a wider patient base and so, here we focus on developing inserts for extended delivery of timolol by incorporating drug-loaded nanoparticles into p–HEMA inserts. Also, inserts are not required to be transparent and so particle size and loadings could be higher than those in contact lenses, affording the possibility of longer release durations with larger daily release amounts. The increase in particle loadings can have a significant effect on the release profiles, as discussed later in this article.
Several researchers have developed fornix inserts11–13,16 and Ocusert and Lacrisert® have been commercialized to treat glaucoma and dry eyes, respectively. Lacrisert is a cylindrical insert 3.5 mm long and 1.27 mm in diameter made of hydroxypropyl cellulose.26 The insert dissolves over a period of a day after insertion leading to increased tear viscosity and lubrication. The inserts proposed here were chosen to be geometrically similar to Lacrisert and, thus, have a length of about 7 mm and a diameter of about 1 mm. While retention of the fornix inserts is typically a concern, cylindrical-shaped inserts are considered best for retention in the conjunctival sac.27,28 The materials for designing the inserts were chosen to be HEMA and PGT, which are similar to the materials commonly used in ocular applications, such as contact lenses. Also, the insert is nondegradable and so there is no concern from toxicity from the degradation products.
Methods
Materials
HEMA monomer, timololmaleate, Azobisisobutylonitrile (AIBN), and Dulbecco's phosphate-buffered saline (PBS) were purchased from Aldrich Chemicals (St Louis, MO). PGT was kindly provided by Sartomer (Exton, PA); Benzoyl peroxide (BP) (97%) was purchased from Aldrich Chemicals (Milwaukee, WI).
Preparation of highly crosslinked PGT nanoparticles
The drug-loaded nanoparticles were prepared by thermal polymerization of a mixture of the timolol base and the PGT. The details of the particle preparation process and characterization are available elsewhere.20 Briefly, timolol maleate was converted to the oily base form by increasing the pH of the aqueous solution. The timolol base (240 mg) was added to 1 g of the crosslinker (PGT) and 7.5 mg of the initiator BP. The ratio of the timolol base and PGT was varied to prepare particles with various drug loadings. The mixture was then added to 5 mL of deionized (DI) water, and then 1.65 mL of 2.08 M NaOH was added to the mixture. The mixture was purged with nitrogen for 15 min, and then heated in an 80°C hot water bath under stirring at 1100 rpm for 8 h. The thermal polymerization results in the formation of drug-loaded nanoparticles, which are about 4 nm in size.20 The particles are separated from the suspension by centrifugation for 15 min during which the particles aggregate and separate into an oily phase that contains particles along with the unreacted PGT.
Preparation of nanoparticle-laden inserts
The timolol-PGT particle mixture was added to the HEMA monomer in various ratios ranging from 25:75 (w/w) to 100:0 to create the polymerization mixture for fabricating the inserts. It is noted that the nanoparticle preparation step does not consume the entire amount of crosslinker PGT. In fact, the solution at the end of the particle preparation step is designed to be liquid for ease of separation and to avoid aggregation of particles into larger aggregates. The unreacted fraction of the PGT reacts in the next step of insert fabrication. In the inserts with 100% particles, both particles and the matrix are composed of PGT. The mixture was purged with nitrogen for 15 min to reduce the amount of dissolved oxygen. Next, 30 mg of a thermal initiator AIBN was added to the mixture, and stirred for 15 min. The mixture was then poured into a 1.02-mm inner diameter Silastic® tubing, which served as molds for the polymerization. The silicone molds were sealed at both ends, and then submerged into a water bath at 80°C for 40 min for polymerization. After overnight drying, the cylindrical inserts were gently pulled out of the silicone molds. The cured inserts were 1 mm in diameter and were cut into 7.5-mm-long sections. Control p-HEMA inserts were also prepared by following the same procedure as described above except that the polymerization mixture contained a mixture of HEMA monomer with timolol base.
Drug release experiments
The 7.5-mm long 1-mm diameter inserts were first submerged in 30 mL DI water under minimal stirring (140 rpm) and at room temperature for 24 h to extract the unreacted monomer. Next, the inserts were transferred to 3 mL of fresh PBS for the drug release experiments. During the release experiments, the drug concentration was measured every 24 h, with 2 additional measurements at 1 and 4 h after the soaking in PBS. The time-dependent concentrations of timolol in PBS were determined by measuring the absorbance as a function of time by UV-Vis spectrophotometer in the 252–312-nm wavelength range. The concentration of timolol was determined by fitting the spectra from the various solutions in the range 270–305 nm to the reference timolol spectra measured at various concentrations using a linear interpolation and a least-square fit. By utilizing the spectra over a range of wavelength, we can clearly determine whether release of some other component from the insert is contributing to the absorption. The absorption spectra of timolol at several concentrations are shown in Fig. 1A and the fits between the measured and the fitted spectra are shown in Fig. 1B. The fitting procedure was robust and the fits were good with typical root mean square errors of less than 0.5%.
FIG. 1.

(A) Absorption spectra of timolol at several concentrations. (B) Least square fitting of the spectra in the drug release experiments to the timolol spectra to determine the timolol concentration.
Packaging tests
The inserts could be packaged either in a dry or in a hydrated state. Hydrated inserts would likely be more comfortable compared to the dry inserts, but packaging in hydrated state could lead to loss of drugs during packaging. To explore the impact of wet-packaging, the 7.5-mm long, 1-mm diameter inserts were packaged in 1 mL of PBS in a refrigerator at 4°C for 3 months. The inserts were subjected to the initial extraction in 30 mL DI water for 24 h before the packaging. After 3 months of storage in the refrigerator, the inserts were submerged in 3 mL of fresh PBS for the drug release experiments.
Results
A photograph of the dried nanoparticle-loaded insert is shown in Fig. 2. The dried inserts are rigid and clear, but become soft and lose transparency on hydration. The drug release profiles from the p-HEMA insert without nanoparticles are shown in Fig. 3 and the profiles for the inserts with various loadings of nanoparticles are shown in Fig. 4A–C. The cumulative mass of drug release is plotted as a function of time in Fig. 4A, while the percentage release is plotted in Fig. 4B. The plot in Fig. 4C shows the cumulative percentage release profiles as a function of the square root of time. The effect of increase in drug loadings in the particles, while keeping the particle fraction in the inserts fixed at 25%, is shown in Fig. 5. The cumulative drug release profiles from nanoparticle-loaded inserts with 25% particle loading after 3 month packaging in 1 mL PBS at 4°C is shown in Fig. 6.
FIG. 2.

Photograph of the nanoparticle-laden fornix insert.
FIG. 3.

Cumulative drug release profiles from control poly hydroxyl ethyl methacrylate (without nanoparticles) inserts of 1 mm diameter and 7.5 mm length. Data are shown as mean±std (n=3).
FIG. 4.

Cumulative drug release (A) and percentage drug release (B) profiles from nanoparticle-loaded inserts. The 4 curves correspond to different particle loadings. The cumulative percentage release is plotted as a function of square root of time in (C) to determine the transport mechanism. Data are shown as mean±std (n=3). Error bars are not included in B and C for clarity of presentation.
FIG. 5.

Effect of timolol:propoxylated glyceryl triacrylate (PGT) ratio in nanoparticles on cumulative drug release profiles from inserts. The nanoparticle loading was 25% for both inserts, but the ratio of timolol:PGT was 0.24 and 0.36 for the curves with circles and diamond markers, respectively. Data are shown as mean±std (n=3).
FIG. 6.

Cumulative drug release profiles from nanoparticle-loaded inserts with 25% particle loading after 3-month packaging at 4°C. The cumulative release before packaging is also included for comparison. Data are shown as mean±std (n=3).
Discussion
Drug release from control p-HEMA inserts
Figure 3 shows the drug release profiles from the control p-HEMA insert in which timolol was added directly to the polymerization mixture before thermal curing. The drug diffuses out in about 0.25 days, which is inadequate for the desired goal of about 1 week of extended release.
Effect of timolol-PGT particle loading in inserts on drug release
Figure 4A shows the cumulative amount of drug released as a function of time from the particle-loaded inserts. The 4 curves correspond to the various particle loadings, which are indicated in the legend. The solid lines are fits of a model described below to the experimental data. The cumulative release profiles for the inserts with 25% and 50% particles reached a plateau in 2 and 3 months, respectively. The inserts with 75% and 100% particles did not reach a plateau during the 100 days of measurements showing that the release duration is longer than 100 days for these inserts. The maximum mass of drug released from the inserts was determined by measuring the mass of drug released after soaking the inserts in 3-mL PBS at 95°C for 2 days. The mass of drug released at high temperatures is listed in Table 1 for the 4 different particle loadings. The mass of drug released at high temperatures is comparable to the total mass of drug released at room temperature for the insert with 25% and 50% particles (Table 1). The total mass of drug released at room temperature was obtained by extrapolating the measured data to infinite time (M∞) using a model described below in Equation 1. The values of M∞ in Table 1 match the drug-released amounts at high temperature for all particle loadings. The ratio of the cumulative release and the total mass released at high temperature is considered to be the cumulative percentage release and is plotted in Fig. 4B. The data in Fig. 4A and B clearly show that there is an extended drug release from the inserts that lasts for 1 month or longer, which is significantly longer than the 0.25-day drug release duration from the p-HEMA inserts without particles (Fig. 3). The increase in particle loading increases the total amount of drug release, but it is accompanied by an increase in the release duration as well. The increase in total amount of drug released is expected because an increased particle loading results in an increased total drug loading in the insert. The increase in the drug release duration is interesting; particularly, since the drug release rates are controlled by release from the particles and so an increase in particle loading is not expected to impact release durations. The increased duration could potentially be caused by an increased crosslinking in the gel due to the presence of the particles and also due to the PGT fraction that remained unreacted during the particle preparation step. To explore whether diffusion limitations due to increased crosslinking are leading to the increased release duration, the cumulative percentage release profiles are plotted as a function of t1/2 in Fig. 4C. If diffusion is the rate limiting step, the cumulative drug release should scale as t1/2 at very short times and, thus, the plot of mass release as a function of t1/2 should be linear for about 15%–20% of the cumulative release from a cylindrical device.29 The curves in Fig. 4C are clearly not linear for the first 15%–20% release showing that diffusion through the gel is not rate limiting for any of the 4 cases shown in Fig. 4. The likely rate controlling mechanism and the potential reasons for an increase in release duration with an increase in particle loadings are described below.
Table 1.
Summary of Drug Release Parameters from Nanoparticle-Loaded Inserts
| Particle:HEMA | Total Release at 25°C(μg) | Total Release at 95°C(μg) | M∞(μg) | K(1/day) | Total release in initial extraction(μg) |
|---|---|---|---|---|---|
| 25:75 | 273.70±34.05 | 283.08±4.48 | 273.88±38.69 | 0.1159±0.018 | 31.67±3.07 |
| 50:50 | 431.13±60.00 | 485.88±6.51 | 494.08±20.41 | 0.0360±0.007 | 33.12±5.17 |
| 75:25 | - | 639.81±38.87 | 682.82±13.62 | 0.0184±0.004 | 5.68±0.79 |
| 100:0 | - | 743.27±25.40 | 762.20±59.81 | 0.0062±0.0005 | 4.32±0.41 |
Comparison of release rates from inserts to therapeutic doses
The usual dose of timolol is one drop of 0.25% timolol maleate in the affected eye(s) twice per a day. Assuming a volume of 25 μL for each drop, the daily dosage of timolol is 125 μg each day. The bioavailability of timolol delivered through eye drops is only about 1%–2%, which implies that the therapeutic requirement of timolol is about 2.5 μg/day. Unfortunately, there is no data on therapeutic release rates from fornix inserts and comparing the data from inserts with that from drops is not appropriate because of differences in release profiles and location of release, which could contribute to differences in bioavailability and spatial differences in concentrations. Since there is no data on bioavailability of timolol released from fornix inserts, we cannot conclusively determine the therapeutic requirement for timolol. To obtain an initial estimate of the therapeutic rates, we use the data from pilocarpine releasing inset Ocusert. The total dosage of pilocarpine administered by one Ocusert insert over 7 days was about one-eighth of the amount provided by the 28 applications (4 each day) of the 2% eyedrops. Assuming that the increase in bioavailability will be comparable for timolol and pilocarpine, a therapeutically desired release rate for the timolol insert could be about 15 μg/day. The insert with 25% particles releases about 15 μg/day for the first 10 days and, thus, it might be suitable for use as a 1-week release system. After the 1-week period, the insert will need to be removed and replaced by a fresh insert. The inserts with higher particle loadings release timolol over longer durations and also at rates closer to the zero-order. However, the release rates decrease with increased particle loadings, and the insert with 100% particles release only about 3 μg/day. Although the release rates could potentially be increased by either increasing the length of the insert or by increasing drug loadings in the particles, wear duration of longer than a week may be difficult to achieve due to retention issues. It is noted that the in vitro release profiles reported were measured under perfect sink conditions, but the conditions in the lower fornix may not correspond to perfect sink. Also, the tear fluid in the eyes may not be at the same concentration as that in the lower fornix due to mixing limitations, and this may also impact the drug delivery rates to the cornea.
Effect of timolol loading in the particles on drug release
To explore whether the release rates from the inserts could be increased by increasing the ratio of timolol to PGT in the particles, we prepared nanoparticles by following the same procedure as described in the Methods section except that 360 mg of timolol base was added to 1 g of PGT. The drug release profiles from the 7.5-mm insert with 25% particle loadings and increased drug percentage in the particles are shown in Fig. 5, along with the profiles for the similar insert with the lower drug fraction in the particles. The data show that increased drug loading in the particles leads to an increase in the release rates without impacting the total release duration. However, the increase in the release rates for the 1st week is only about 33% even though the drug loadings in the particles increased by about 50%. Most likely, this discrepancy is due to drug loss during the insert preparation steps.
Mechanism of release
The mechanism of extended drug release from p-HEMA contact lenses loaded with the timolol-PGT particles was shown previously to be hydrolysis of the ester bond between timolol and the particle network.30 The mechanism for the ester bond formation during the particle preparation step is also described in Ref. 21. Accordingly, the drug release data should fit the first order reaction model, that is,
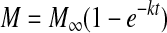 |
(1) |
where M is the amount of drug released at a time t and M∞ is the amount of drug released after infinite time, and k is the rate constant. In the previous studies with contact lenses, the above model fitted the release data, but the particle loadings were substantially smaller compared to the loadings in the inserts. To test whether the same model can describe the drug release profiles even at very high particle loadings in the inserts, the release data in Fig. 4A was fitted to Eq. 1. The best fit curves are included as the solid lines in the figure and the parameters k and M∞ for the fits are included in Table 1. The model fits the data well proving that the release is controlled by the hydrolysis reaction. The rate constant k decreases with an increase in the particle loading most likely because the water content of the inserts (Table 2) decrease with an increase in particle loading. The hydrolysis reaction could be rate limited by the availability of water, thus leading to an increase in release durations with an increase in particle loadings. The decrease in k leads to an increase in the total release duration. The value of M∞ increases linearly with increasing particle loadings, but then levels off due to an increase in an irreversibly trapped drug fraction or an increased loss of the particles to the silicone tubing mold during the insert fabrication. It is typical for drug delivery devices of various types, including transdermal patches to retain a small fraction of drug even after a long time due to irreversible entrapment. The values of M∞ also match the mass of drug released at the elevated temperature (Table 1), further supporting the validity of the first-order reaction model. A fraction of the drug loaded in the inserts is also extracted during the initial 24 h soaking in DI water for extraction of the unencapsulated drug and unreacted monomer. The mass of drug released during the initial extraction is also included in Table 1. The ratio of drug released during the initial extraction and the total release decrease significantly with an increase in particle loading, likely because the degree of crosslinking in the insert matrix increases with increasing particle loading due to the unreacted PGT fraction during particle preparation. The PGT that was unreacted during the particle preparation eventually reacts during the insert fabrication, potentially encapsulating the drug that remained unreacted after the particle preparation.
Table 2.
Water Content in Nanoparticle-Loaded Inserts
| Particle:HEMA | EWC (%) |
|---|---|
| 25:75 | 31.57 |
| 50:50 | 5.34 |
| 75:25 | 2.06 |
| 100:0 | 0.66 |
HEMA, hydroxyl ethyl methacrylate; EWC, equilibrium water content.
Effect of packaging
To explore the effect of packaging, the particle-laden inserts were soaked in 1 mL of packaging solution (PBS) for a period of 3 months in a refrigerator, and then subjected to drug release studies with protocols described in the Methods section. The results of these studies are presented in Fig. 6. The data for drug release before packaging are also included for ease of comparison. The release profiles after packaging exhibit a slight burst release, followed by an extended release for about 20 days, but at a much reduced rate compared to the inserts that were not packaged. The burst release is due to diffusion of the drug that was released from the particles into the gel due to hydrolysis during the packaging duration. Although hydrolysis is slowed down in refrigerator, the burst release after packaging is undesirable, and thus it would be preferable to package the inserts in the dry state and hydrate them just before insertion. Dry packaging could also minimize the potential introduction of bacteria into the eyes due to bacterial growth in the packaging liquid during the long shelf life.
Conclusions
Glaucoma therapy through eye drops has several drawbacks, including low bioavailability and low compliance. Extended release of glaucoma drugs through ophthalmic inserts could potentially increase both bioavailability and compliance. We have developed a particle-loaded insert for extended delivery of beta blocker timolol. The release is controlled by hydrolysis of the ester bond that links timolol to the particle matrix. The cumulative release profiles can be fitted to a first order reaction model, and thus, the daily release rates decrease exponentially with time. The release durations could be adjusted from about 10 days to a month by changing the fraction of particles in the insert. The release rates could also be adjusted by the drug loading in the particles. Based on an approximate estimation, a 7.5-mm and 1-mm diameter insert with 25% particles could be suitable for extended delivery for about a week. The inserts should be stored in dry state and hydrated before insertion to minimize the loss of drug to the packaging liquid during the long shelf life. The approach of incorporation of timolol encapsulated nanoparticles into the conjunctival inserts to prolong the drug release could potentially find application in several other drug delivery applications, such as puncta plugs, ophtha coils, retinal implants, transdermal patches, and wound-healing patches. While the results of this in vitro design study are encouraging, these need to be supplemented with animal studies to explore retention and therapeutic efficacy.
Acknowledgments
This research was partially supported by a research grant from the National Science Foundation (CBET CMMI Grant 1129932).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Quigley H.A. Number of people with glaucoma worldwide. Br. J. Ophthalmol. 1996;80:389–393. doi: 10.1136/bjo.80.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Resnikoff S. Pascolini D. Etya'ale D. Kocur I. Pararajasegaram R. Pokharel G.P. Mariotti S.P. Global data on visual impairment in the year 2002. Bull. World Health Organ. 2004;82:844–851. [PMC free article] [PubMed] [Google Scholar]
- 3.Kholdebarin R. Campbell R.J. Jin Y.P. Buys Y.M. Canadian Compliance Study G. Multicenter study of compliance and drop administration in glaucoma. Can. J. Ophthalmol. 2008;43:454–461. doi: 10.1139/i08-076. [DOI] [PubMed] [Google Scholar]
- 4.Mackean J.M. Elkington A.R. Compliance with treatment of patients with chronic open-angle glaucoma. Br. J. Ophthalmol. 1983;67:46–49. doi: 10.1136/bjo.67.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman D.S. Nordstrom B. Mozaffari E. Quigley H.A. Glaucoma management among individuals enrolled in a single comprehensive insurance plan. Ophthalmology. 2005;112:1500–1504. doi: 10.1016/j.ophtha.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 6.Bron A.M. Hermann M.M. Creuzot-Garcher C. Diestelhorst M. Monitoring individual compliance in glaucoma patients used to topical therapy. Program and abstracts of the Association for Research in Vision and Ophthalmology 2007 Annual Meeting; May 6–10;2007 ; Fort Lauderdale, Florida. Abstract 5580. [Google Scholar]
- 7.Zhu H. Chauhan A. A mathematical model for ocular tear and solute balance. Cur. Eye. Res. 2005;30:841–854. doi: 10.1080/02713680591004077. [DOI] [PubMed] [Google Scholar]
- 8.Zhu H. Chauhan A. Tear dynamics model. Cur. Eye. Res. 2007;32:177–197. doi: 10.1080/02713680601186706. [DOI] [PubMed] [Google Scholar]
- 9.Edwards A. Prausnitz M.R. Predicted permeability of the cornea to topical drugs. Pharm. Res. 2001;18:1497–1508. doi: 10.1023/a:1013061926851. [DOI] [PubMed] [Google Scholar]
- 10.Prausnitz M.R. Noonan J.S. Permeability of cornea, sclera, and conjunctiva: A literature analysis for drug delivery to the eye. J. Pharm. Sci. 1998;87:1479–1488. doi: 10.1021/js9802594. [DOI] [PubMed] [Google Scholar]
- 11.Colo G.D. Burgalassi S. Chetoni P. Fiaschi M.P. Zambito Y. Saettone M.F. Gel forming erodible inserts for ocular controlled delivery of ofloxacin. Int. J. Pharm. 2001;215:101–111. doi: 10.1016/s0378-5173(00)00671-2. [DOI] [PubMed] [Google Scholar]
- 12.Colo G.D. Zambito Y. Burgalassi S. Serafini A. Saettone M.F. Effect of chitosan on in vitro release and ocular delivery of ofloxacin from erodible inserts based on poly(ethylene oxide) Int. J. Pharm. 2002;248:115–122. doi: 10.1016/s0378-5173(02)00421-0. [DOI] [PubMed] [Google Scholar]
- 13.Sultana Y. Aqil M. Ali A. Ocular inserts for controlled delivery of pefloxacin mesylate: preparation and evaluation. Acta Pharm. 2005;55:305–314. [PubMed] [Google Scholar]
- 14.Akerblom T. Aurell E. Cristiansson J. Kriisakunnos V. Wiebert O. A multicenter study of the effect and tolerance of OCUSERT-P-40. Acta Ophthalmol. 1980;58:617–623. doi: 10.1111/j.1755-3768.1980.tb08303.x. [DOI] [PubMed] [Google Scholar]
- 15.Zaffaroni A. Systems for controlled drug delivery. Med. Res. Rev. 1981;1:373–386. doi: 10.1002/med.2610010404. [DOI] [PubMed] [Google Scholar]
- 16.Saettone MF. Salminen L. Ocular inserts for topical delivery. Adv. Drug. Deliv. Rev. 1995;16:95–106. [Google Scholar]
- 17.Leydhecker W. OCUSERT - summary of panel discussion. Klin. Monatsbl. Augenheilkd. 1975;167:917–918. [PubMed] [Google Scholar]
- 18.Marquis R.E. Whitson J.T. Management of glaucoma: focus on pharmacological therapy. Drugs Aging. 2005;22:1–21. doi: 10.2165/00002512-200522010-00001. [DOI] [PubMed] [Google Scholar]
- 19.Fraunfelder F.T. Meyer S.M. Systemic side-effects from ophthalmic timolol and their prevention. J. Ocul. Pharmacol. 1987;3:177–184. doi: 10.1089/jop.1987.3.177. [DOI] [PubMed] [Google Scholar]
- 20.Jung H.J. Chauhan A. Temperature sensitive contact lenses for triggered ophthalmic drug delivery. Biomaterials. 2012;33:2289–2300. doi: 10.1016/j.biomaterials.2011.10.076. [DOI] [PubMed] [Google Scholar]
- 21.Hiratani H. Alvarez-Lorenzo C. Timolol uptake and release by imprinted soft contact lenses made of N,N-diethylacrylamide and methacrylic acid. J. Control. Rel. 2002;83:223–230. doi: 10.1016/s0168-3659(02)00213-4. [DOI] [PubMed] [Google Scholar]
- 22.Peng C.C. Kim J. Chauhan A. Extended delivery of hydrophilic drugs from silicone-hydrogel contact lenses containing Vitamin E diffusion barriers. Biomaterials. 2010;31:4032–4047. doi: 10.1016/j.biomaterials.2010.01.113. [DOI] [PubMed] [Google Scholar]
- 23.Ciolino J.B. Hoare T.R. Iwata N.G. Behlau I. Dohlman C.H. Langer R. Kohane D.S. A drug-eluting contact lens. Invest. Ophthalmol. Vis. Sci. 2009;50:3346–3352. doi: 10.1167/iovs.08-2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alvarez-Lorenzo C. Hiratani H. Concheiro A. Contact lenses for drug delivery: achieving sustained release with novel systems. Am. J. Drug Deliv. 2006;4:131–151. [Google Scholar]
- 25.Peng C.C. Chauhan A. Extended cyclosporine delivery by silicone contact lenses. J. Control. Rel. 2011;154:267–274. doi: 10.1016/j.jconrel.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 26.Lacrisert [package insert] Aton Pharma, Inc.; Lawrenceville, NJ: 2007. [Google Scholar]
- 27.Katz I.M. Blackman W.M. A soluble sustained-release ophthalmic delivery unit. Am. J. Ophthalmol. 1977;83:728–734. doi: 10.1016/0002-9394(77)90141-6. [DOI] [PubMed] [Google Scholar]
- 28.Lamberts D.W. Pavan-Langston D. Chu W. A clinical study of slow-releasing artificial tears. Ophthalmology. 1978;85:794–800. doi: 10.1016/s0161-6420(78)35610-4. [DOI] [PubMed] [Google Scholar]
- 29.Ritger P.L. Peppas N.A. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J. Control. Rel. 1987;5:23–36. [PubMed] [Google Scholar]