

Abstract
EpCAM [epithelial cell adhesion molecule; CD326 (cluster of differentiation 326)] is highly expressed on epithelium-derived tumours and can play a role in cell proliferation. Recently, RIP (regulated intramembrane proteolysis) has been implicated as the trigger for EpCAM-mediated proliferative signalling. However, RIP does not explain all EpCAM-derived protein fragments. To shed light on how proteolytic cleavage is involved in EpCAM signalling, we characterized the protein biochemically using antibodies binding to three different EpCAM domains. Using a newly generated anti-EpCAM antibody, we find that EpCAM can be cleaved at multiple positions within its ectodomain in addition to described peptides, revealing that EpCAM is processed via distinct proteolytic pathways. Here, we report on four new peptides, but also discuss the previously described cleavage products to provide a comprehensive picture of EpCAM cleavage at multiple positions. The complex regulation of EpCAM might not only result in the absence of full-length EpCAM, but the newly formed EpCAM-derived proteins may have their own signalling properties.
Keywords: cell–cell contact, EpCAM, notch-like signalling, regulated intramembrane proteolysis, polypeptides, shedding
Abbreviations: ADAM, a disintegrin and metalloprotease; APP, amyloid precursor protein; CAM, cell adhesion molecule; CD326, cluster of differentiation 326; CTE, congenital tufting enteropathy; CTF, C-terminal fragment; DAPT, N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester; DMEM, Dulbecco’s modified Eagle’s medium; ECD, extracellular domain; EGFP, enhanced green fluorescent protein; EpCAM, epithelial cell adhesion molecule; EpEX, EpCAM cleaved extracellular domain; EpICD, EpCAM cleaved intracellular domain; ER, endoplasmic reticulum; FCS, foetal calf serum; GFP, green fluorescent protein; HEK-293T cells, human embryonic kidney cells; ICD, intracellular domain; MW, molecular mass; NTF, N-terminal fragment; PNGase F, peptide N-glycosidase F; RIP, regulated intramembrane proteolysis; TEM, tetraspanin-enriched microdomain; TNFα, tumour necrosis factor α; TY, thyroglobulin
INTRODUCTION
EpCAM [epithelial cell adhesion molecule; CD326 (cluster of differentiation 326); Figure 1] is a transmembrane glycoprotein that plays a role in balancing cell proliferation and differentiation. In healthy tissue, high EpCAM levels are associated with proliferation during morphogenesis [1], tissue regeneration [2,3] and stem cell maintenance [4]. High EpCAM expression has been found to promote tumour progression in epithelial cancer, and often corresponds to a poor prognosis [2,5–7]. In contrast with high EpCAM levels, absence of EpCAM because of gene mutations results in CTE (congenital tufting enteropathy), a rare disease causing severe diarrhea in newborns due to abnormal development of the intestinal epithelium [8]. How EpCAM is functionally involved in development and tumour biology is still not completely understood. Because of its tumour-specific overexpression, EpCAM has been explored as a prognostic/diagnostic marker and anti-cancer target since the 1970s [5]. However, parts of the protein might be absent during diagnosis, since EpCAM is subject to limited proteolysis by a variety of enzymes.
Figure 1. EpCAM amino acid sequence and post-translational modification.

Arrow 1: signal peptide cleavage site; arrow 2: N-terminal cleavage site between Arg-80/Arg-81. EpCAM motif 1 (green): six-cysteine motif with a unique disulfide linkage pattern [11]; TY type-1 repeat (motif 2, purple): second cysteine-rich domain, displaying sequence homology to a TY type-1A repeat, a domain exhibiting protease inhibitor activity. Approximate regions of the epitopes recognized by monoclonal antibodies MOC31 and 311-1K2 and the polyclonal rabbit antibody P6052, recognizing EpCAM's ICD, are indicated. See Table 1 for the defined epitope location.
During the synthesis, EpCAM's signal peptide for proper ER (endoplasmic reticulum) targeting is removed [9–11] (Figure 1, arrow 1). Furthermore, EpCAM can be cleaved between two arginine residues within its ectodomain [10,12,13] (Figure 1, arrow 2). Although this cleavage site was detected soon after EpCAM's discovery, the functional consequence is still unknown. More recently, EpCAM-mediated proliferative signalling has been proposed to be activated by RIP (regulated intramembrane proteolysis) [14], an evolutionarily conserved mechanism combining regulated ectodomain shedding with the consecutive release of the ICD (intracellular domain) from transmembrane proteins [15]. Generally, RIP can (i) activate signalling events triggered by the shed ectodomain (e.g. as soluble growth factors) and/or the ICD (e.g. as cytosolic signalling molecules or nuclear transcription cofactors); and (ii) lead to degradation, thus functional inactivation, of transmembrane proteins (Lichtenthaler et al. [16] and references therein). The best-studied substrates for RIP are Notch, EGF (epidermal growth factor), TNFα (tumour necrosis factor α) and APP (amyloid precursor protein) (reviewed in [17]). In addition, several CAMs (cell adhesion molecules) have been identified as substrates, e.g. epithelial E-cadherin, neuronal N-cadherin, CD44 and L1-CAM (reviewed in [18]). ADAM17 [a disintegrin and metalloprotease 17; also known as TACE (TNFα converting enzyme)] is the metalloprotease that has been reported to initiate EpCAM cleavage at the extracellular site, resulting in shedding of its ectodomain EpEX (EpCAM cleaved extracellular domain). Subsequently, an additional cleavage by a γ-secretase with PS-2 (presenilin-2) as the active subunit causes the release of EpICD (EpCAM cleaved ICD). EpICD may interact with (co)transcription factors in the nucleus, altering gene transcription [14].
Here, we address where and when EpCAM is proteolytically cleaved, and discuss how the different proteolytic steps may be implicated in EpCAM's function. Owing to EpCAM's discovery in a screen for tumour-specific cell surface proteins, the majority of available antibodies are directed against EpCAM's N-terminal extracellular domain. To distinguish between this domain and the short cytoplasmic tail, we first made an antibody directed against EpCAM's C-terminal domain. While characterizing EpCAM biochemically using three different antibodies recognizing distinct domains, we found four novel C-terminal proteolytic fragments. We conclude that EpCAM is proteolytically processed by an additional pathway, thus highlighting the importance of proteolysis in the regulation of EpCAM signalling. Whether the novel cleavage sites only lead to functional down-regulation of EpCAM or may serve additional functions remains to be established.
EXPERIMENTAL
Cell culture and transfection
HEK-293T cells (human embryonic kidney cells; ATCC) were cultured in DMEM (Dulbecco's modified Eagle's medium; 1 g/l glucose) supplemented with 5% (v/v) FCS (foetal calf serum) and penicillin (100 units/ml)/streptomycin (1 mg/ml). Human epithelial colon carcinoma cell lines HT29, CaCo2 and HCT116, human keratinocyte cell line HaCaT and epidermoid carcinoma cell line A431, human hepatoma cell line HepG2, human breast cancer cell line MCF7, human ovarian cancer cell line OVCAR-3 (all ATCC), and head and neck carcinoma cell lines FaDu, SCC23 and SCC32 (kind gift from E. Schuuring, UMCG) were cultured in DMEM (4.5 g/l glucose) supplemented with 10% (v/v) FCS, 1× MEM non-essential amino acids (Invitrogen) and penicillin (100 units/ml)/streptomycin (1 mg/ml). Cells were cultured at 37°C in a humidified incubator in the presence of 5% (v/v) CO2. Media and supplementation were obtained from PAA. HEK-293T cells were transfected with FuGene6 (Roche) according to the manufacturer's protocol.
Plasmids
Human EpCAM cDNA (a gift from V. Cirulli, Department of Medicine, University of Washington, Seattle, WA, U.S.A.) [19] was subcloned into pcDNA3.1. Using this as template, the C66Y mutation was introduced (G>A substitution) [19a] by QuikChange® Mutagenesis (Stratagene). Plasmids were verified by sequencing. Either the non-tagged versions were used, or the C-terminal EGFP [enhanced GFP (green fluorescent protein)]-tagged versions with the linker ‘RSAAAT’. The ER-marker ER-superluc-EGFP (calnexin pre-sequence) has been described [20].
Antibodies and reagents
For detection of EpCAM, we used mouse monoclonal antibodies MOC31, binding EpCAM motif 1, and 311-1K2, binding within the cysteine-free motif (hybridomas kindly provided by L.F.M.H. de Leij, UMCG) [21]. EpCAM CTE-mutant W143_T164del [8] is not recognized by antibody 311-1K2, revealing its binding site (see Table 1). Furthermore, rabbit polyclonal antibody P6052, raised against EpCAM's intracellular domain (immunizing peptide: CEIKEMGEMHRELNA) was designed in our laboratory and generated by BioGenes (Germany). Beta-Tubulin antibody (B512), DAPT {N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester} and PNGase F (peptide N-glycosidase F) were obtained from Sigma-Aldrich.
Table 1. Recognition of EpCAM domains by the antibodies used in the present study.
| Antibody | Domain/motif | Region of binding (amino acids, aa) |
|---|---|---|
| MOC31 | Extracellular domain/EpCAM motif 1 | 27–59 [22] |
| 311-1K2 | Extracellular domain/cysteine-free region | 143–164 |
| P6052 | Intracellular domain | 301–314 |
Western blot
SDS/PAGE and Western blot procedures were conducted according to the standard protocols (equipment from BioRad; Millipore), using 10 or 15% gels. Cells were plated in 6-well plates and grown until confluency. If indicated, cells were incubated with DMSO [0.1% (v/v)] or 10 μM DAPT for 24 h. Cells were lysed using either reducing [1% (v/v) β-mercaptoethanol] or non-reducing Laemmli sample buffer (62.5 mmol/l Tris/HCl; 20 g/l SDS; 100 ml/l glycerol; bromophenol blue; pH 6.8). Following incubation with the primary and an HRP (horseradish peroxidase)-conjugated secondary antibody, proteins were detected by enhanced chemiluminescence (antibodies, ECL detection kit, film: GE Healthcare).
Medium concentration
Confluent cells were incubated for 24 h in medium with 0.1% (v/v) FCS to enable Western blot analysis after concentration of the supernatant. First, the medium was centrifuged for 15 min at room temperature (20°C) and 4500 g to pellet dead cells and cell debris. The filtered medium (0.2 μm; Whatman) was concentrated using Amicon Ultra-4 centrifugal concentration filter tubes (Millipore) with a 10 kDa MW (molecular mass) cut-off. The final concentrate was diluted with 5× Laemmli sample buffer (non-reducing), denatured at 95°C for 5–10 min, and analysed by Western blot. To separate microvesicles and soluble proteins, the medium was cleared by centrifugation and filtering and subjected to ultracentrifugation at 100000 g for 1 h at 4°C. The resulting pellet was dissolved in 1.2× non-reducing Laemmli sample buffer and the supernatant was concentrated as described above.
Immunofluorescent staining
HEK-293T cells, co-transfected with ER-GFP (green fluorescent protein) and either wtEpCAM or EpCAM-C66Y for at least 24 h, were fixed with 10% (v/v) formalin [equals 4% (v/v) formaldehyde; Sigma-Aldrich] for 30 min and permeabilized with 0.1% (v/v) Triton X-100 (Merck)/1% (w/v) BSA/PBS for 15 min. Following blocking with 1% (w/v) BSA/PBS for 15 min, primary antibody MOC31 and Alexa Fluor® 568-conjugated secondary antibody (Invitrogen, The Netherlands), diluted in 1% (w/v) BSA/PBS, were applied for 1 h each and cells were mounted with Vectashield (Vector). All steps of the immunostaining procedure were conducted at room temperature. Fluorescent images were acquired using a Leica SP2 AOBS confocal microscope (Leica Microsystems).
RESULTS
In addition to newly discovered polypeptides, we provide a complete overview of all EpCAM fragments, including the NTFs (N-terminal fragments) that have been reported previously.
Signal peptide
We did not detect EpCAM with the signal peptide (cleavage at aa 23), which will be removed during EpCAM translation by a signal peptidase in the ER lumen [23], and therefore will never be part of full-length EpCAM after translation is completed.
N-terminal cleavage
Another cleavage site at the N-terminus of EpCAM is between Arg-80/Arg-81, originally identified by Thampoe and Ng [12]; and Schön et al. [10,12,13]. Following cleavage, the domains predictably will stay bound together by the disulphide bridge in EpCAM's TY (thyroglobulin)-like motif (Figures 1 and 2), which will be broken under reducing conditions. When EpCAM is subjected to reduction, Arg-80/Arg-81 cleavage is detected in EpCAM-expressing HEK-293T cells using antibody 311-1K2 (Figure 2A). Similarly, the cleavage occurs in numerous cancer cell lines expressing EpCAM endogenously (Figure 2B). The cleaved NTF has a predicted MW of 6 kDa (non-glycosylated). Based on the size difference between non-cleaved EpCAM and the remaining 32 kDa part on Western blots, the glycosylated cleaved fragment has a size of 10 kDa (Figure 3). Notably, only a fraction of total EpCAM is cleaved, and the ratio of cleaved to non-cleaved protein varies between cell lines (Figure 2) as well as between experiments (results not shown).
Figure 2. EpCAM is cleaved in the N-terminal region.

Lysates of HEK-293T cells transfected with EpCAM (A) and various cancer cell lines expressing EpCAM endogenously (B) were analysed by Western blot using an antibody binding to EpCAM's ectodomain (cysteine-free region; 311-1K2). Arg-80/Arg-81 cleavage (red arrow) is apparent under reducing (R) but not under non-reducing conditions (NR). Lysate of non-transfected HEK-293T cells serves as an antibody specificity control.
Figure 3. Sizes of EpCAM-derived fragments.

MW of EpCAM and its fragments deduced from Western blots (using ImageJ) or predicted with and without N-glycosylation (2 kDa per glycosylation site). aa, amino acids. *Without signal peptide; †calculated by the difference between full-length EpCAM and EpCAMΔ6kD; ‡complete extracellular domain; ∥if cleaved after aa 152; ¶if cleaved after aa 198.
EpCAM ectodomain shedding
To determine whether EpCAM undergoes RIP (Maetzel et al. [14]), resulting in shedding of its ectodomain (EpEX) and release of the cytoplasmic peptide (EpICD) [14], we analysed cell lysates and cell-free medium. Using antibodies directed against EpCAM's ECD (extracellular domain; antibody 311-1K2) or ICD (antibody P6052), the full-length protein is detected in cell lysates of the colon carcinoma cell line HT29 (Figure 4A, LYS). In the concentrated cell-free medium, only the antibody binding to EpCAM's ectodomain, but not the one directed against the ICD, detects cleaved EpEX (Figure 4A, MED), demonstrating that the intracellular domain is absent. Based on analysis of Western blots, EpEX has a MW of 35 kDa (Figure 3). Notably, full-length EpCAM is detectable in the cell-free medium, possibly as part of microvesicles, which are secreted by cells and only will be pelleted after ultracentrifugation [24] (Figure 4B). The levels of EpCAM on microvesicles or the amount of vesicles, respectively, as well as the amount of EpEX in the medium depends on the cell line (Figure 4C). Whereas all cell lines release EpCAM-containing microvesicles, the level of EpEX is near the detection limit in most cell lines.
Figure 4. EpCAM undergoes RIP.

(A–D) Size determination of EpCAM by Western blot. (A) Lysates (LYS) and concentrated medium (MED) from HT29 cells were analysed with antibodies against EpCAM's ECD or ICD as indicated. (B) HT29 lysate, concentrated medium before and after ultracentrifugation (100000 g for 1 h), and the pelleted fraction resulting from ultracentrifugation were immunoblotted against EpCAM's ectodomain (311-1K2). (C) Distinct ratios of EpEX and EpCAM on microvesicles in different cell lines as indicated. (D) HEK-293T cells expressing EpCAM–EGFP were incubated with DMSO (0.1%) or 10 μM DAPT for 24 h, lysed, and immunoblotted against EpCAM's ICD (P6052). TMD, transmembrane domain.
EpICD cleavage
Following ectodomain shedding, EpCAM may be cleaved within the transmembrane domain, releasing a soluble cytoplasmic peptide (EpICD). Detection of EpICD is impeded by its predicted size of only 2–3 kDa. To visualize EpICD, we used an EpCAM–EGFP fusion protein to increase the MW by 26 kDa. Indeed, in cells expressing EpCAM–EGFP, several protein fragments are recognized by the antibody binding EpCAM's ICD (P6052; Figure 4D). The lowest three bands with masses of 27–29 kDa correspond to EpICD–EGFP; the 32 kDa fragment to the intermediate cleavage product, consisting of EpCAM's transmembrane domain and ICD only. To address which protease is responsible for the intramembranous cleavage, cells were treated with γ-secretase inhibitor DAPT. Treatment resulted in an increase of the intermediate cleavage product and decrease of the 29 kDa EpICD–EGFP band (Figure 4D, DAPT versus DMSO), pointing to γ-secretase involvement in the second cleavage step of RIP.
Novel detected proteolytic peptides
Our new antibody directed against EpCAM's ICD detected several novel proteolytic fragments: (i) a 20 kDa fragment [CTF20 (C-terminal fragment of 20 kDa); Figure 5A], and (ii) fragments ranging from 12 to 15 kDa [CTFs12–15: C-terminal fragments of 12–15 kDa; Figure 5A and Figure 3; see Supplementary Figure S1 for more cell lines (http://www.bioscirep.org/bsr/033/bsr033e030add.htm)]. These CTFs are not recognized by the antibody against EpCAM's ECD (311-1K2; Figure 5A, anti-ECD), but can also be detected with the anti-ICD antibody E144 [commercially available; Supplementary Figure S2 (http://www.bioscirep.org/bsr/033/bsr033e030add.htm)]. To rule out that the CTFs did not arise due to trypsinization when passaging the cells, we detached cells using EDTA for several weeks, showing that the CTFs are not a result of trypsinization (Figure 5B). As the size differs from the fragments discussed before, CTFs are distinct from those generated by RIP (Figure 3). The low levels of CTF20 in comparison with CTFs12–15 in all cell lines might indicate that the two cleavage events occur sequentially (Figure 5D). Incubation of cells with DAPT leads to increased CTFs12–15 levels (Figure 5C), most prominently seen in HEK-293T cells, and to a lesser extent in HT29 cells, suggesting that these fragments can be further processed by a γ-secretase (Figure 5D).
Figure 5. EpCAM is processed via an alternative proteolytic pathway.

Blots were probed against EpCAM's cytoplasmic domain (ICD; antibody P6052), unless noted otherwise. (A) Lysates of HT29 cells or HEK-293T cells transfected with empty vector (EV) or EpCAM. Note that the protein fragments are not recognized by the antibody against the ECD (first lane, MOC31). (B) HT29 cells, passaged with trypsin versus EDTA for several weeks and analysed under reducing conditions. (C) HEK-293T cells were transfected as indicated and HT29 cells were treated with DMSO (0.1%) or 10 μM DAPT for 24 h. Lysate of non-transfected HEK-293T cells serves as antibody-specificity control (lane was spiked with MW marker). (D) Schematic representation of EpCAM's cleavage. Orange arrow, EpICD cleavage. Antibody-binding sites are indicated.
The trigger of proteolysis remains unclear for most of the fragments. For activation of EpCAM signalling by RIP, cell–cell contact, or binding of EpEX as a ligand, has been suggested to be the initial trigger [25]. We addressed whether the CTFs arise upon cell–cell contact formation during cell growth (Figure 6A). One day after plating, single cells or small cell clusters were present, whereas at day 5 cells were grown to confluency (Figure 6B). In all cell lines tested, the levels of CTFs did not change in a cell-density-dependent manner (Figure 6A). To test whether EpCAM needs to be present at the cell surface to allow CTF generation, we used an EpCAM point mutant (C66T) that causes CTE [8]. EpCAM-C66Y (Figure 6C) does not reach the cell surface because it is retained in the ER [19a]. Interestingly, ER retention leads to a reduction, but not a complete absence of CTFs (Figure 6D), indicating that cleavage of the new CTFs occurs during synthesis in the Golgi apparatus or in the ER lumen.
Figure 6. Alternative cleavage is independent from cell–cell contacts and EpCAM's subcellular localization.

(A) HT29, HaCaT and CaCo2 cells were lysed after 1, 3 or 5 days of culture and analysed by Western blot, using P6052 antibody against EpCAM's ICD. n-fold values represent intensities of the 12 kDa EpCAM band normalized against tubulin (Tub). (B) Brightfield images of cells used in (A). (C) HEK-293T cells co-transfected with ER-EGFP and wtEpCAM or EpCAM-C66Y immunostained with MOC31 and fluorescently conjugated secondary antibody. Confocal images are shown. (D) Cells expressing wtEpCAM or EpCAM-C66Y analysed by Western blotting using P6052. (E) Cell lysates treated with a deglycosylase (5 units of PNGase F for 18 h), and immunoblotted with P6052.
The exact cleavage positions would help to reveal responsible proteases and the functional consequences of EpCAM proteolysis. However, MS did not reveal conclusive results because the purity of the isolated protein was below the required threshold (results not shown). An alternative approach to narrow down the estimated cleavage sites giving rise to CTFs was performed by deglycosylation using PNGase F (Figure 6E). Since EpCAM contains three N-glycosylation sites (Figure 1), PNGase F treatment resulted in a shift of the full-length EpCAM from 39 to 35 kDa (Figure 6E, upper arrow; Figure 3). Moreover, a shift of CTF20 down to 18 kDa is visible (Figure 6E, lower arrow). Deglycosylation did not affect any of the CTFs12–15 bands. Thus, CTFs12–15 are generated C-terminally of EpCAM's third glycosylation site at aa 198 (Figures 1 and 7B).
Figure 7. EpCAM is cleaved in multiple distinct ways.
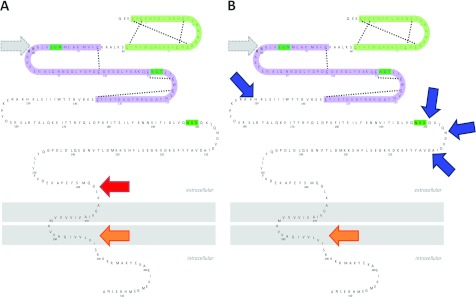
(A) Cleavage by RIP comprises ectodomain shedding (1st step, red arrow) and intramembranous cleavage (2nd step, orange arrow). (B) Alternative proteolysis presented here includes cleavage within EpCAM's ectodomain (blue arrows), followed by intramembrane cleavage (3rd step, orange arrow). In both (A) and (B), intramembrane cleavage involves a γ-secretase. Additionally, EpCAM may be cleaved at position Arg-80/Arg-81 in both pathways (grey dotted arrows).
DISCUSSION
Originally discovered as a protein overexpressed in epithelial cancers, and therefore used as a surface tumour marker, we show that EpCAM can have different faces at the extracellular site. The different cleavage events in EpCAM's ectodomain may lead to loss of its detection in EpCAM-based diagnostic tests. EpCAM's diverse C-terminal fragments, which may have their own signalling properties, may therefore often remain undetected. Below, we discuss how proteolysis of EpCAM, including the newly identified cleavage sites presented in this study, might affect EpCAM's function.
N-terminal cleavage at position Arg-80/Arg-81
Soon after EpCAM's discovery, the cleavage at position Arg-80/Arg-81 was identified [10,12,13]. In all cell-lines tested we find that this cleavage partially occurs, resulting in a 6 kDa NTF that remains bound to the protein backbone by the first disulfide bond within the TY-like domain. Most likely, the cleavage will lead to structural changes in the ectodomain, which may influence the homophilic binding properties or recognition of EpCAM by other proteins, e.g. proteases. Alternatively, EpCAM might be synthesized in a zymogen form (inactive precursor) [26], which is converted to its active form by this proteolytic action [12,13]. Notably, only a certain percentage of EpCAM undergoes cleavage, which could be due to the subcellular localization of responsible proteases. The presence of predominantly non-cleaved EpCAM in all cell lines tested might reflect that EpCAM needs to be exposed at the cell surface to be available for proteases.
Both serine and cysteine proteases are able to conduct the Arg-80/Arg-81 cleavage [13]. It has been suggested that EpCAM might be able to affect certain cysteine proteases, the cathepsins, via its conserved TY domain [5]. TY type-1A domains have been shown to inhibit certain cathepsins, which can be localized in lysosomes and in the extracellular space [27], by binding their active site cleft [28]. Cathepsins can promote migration and invasion of tumour cells by enhancing extracellular matrix degradation [27,29]. EpCAM has been proposed to function as a membrane-bound protease inhibitor, possibly protecting tumour cells from their own secreted cathepsins during tumour progression [5]. Cells could thus benefit from EpCAM overexpression either because of enhanced inhibition of cathepsins via the TY domain, but also because EpCAM may serve as a ‘decoy’ cathepsin substrate (Arg-80/Arg-81). Notably, cleavage within EpCAM's ectodomain by RIP or the newly identified proteolysis events results in truncation of the TY-domain, which would impair a protective role of EpCAM. Whether non-cleaved EpCAM is able to bind or inhibit cathepsins or serves as a substrate remains to be established.
EpCAM is subject to RIP
RIP of EpCAM has been reported to induce downstream signalling [14]. Since we find EpCAM's ectodomain EpEX derived from various cancer cell lines, RIP seemingly is a general phenomenon in vitro. By measuring the size, we predict that the cleavage site generating EpEX is close to the plasma membrane. Our results are concordant with the findings of Maetzel et al. [14] and confirm that a γ-secretase is involved in the second step of EpCAM RIP. Notably, in our Western blots we detect multiple intracellular peptides generated by intramembrane cleavage. γ-secretases are able to cleave the same substrate at multiple sites [30]; however, only the 29 kDa fragment decreased in response to inhibition with DAPT (Figure 4D), suggesting that other intramembranously cleaving proteases [30] might be involved.
Ex vivo, EpEX has been reported to be present in the serum of cancer patients [31–33]; however, the detection methods did not distinguish between full-length EpCAM and cleaved EpEX. Since EpCAM is present on microvesicles, conclusions about the in vivo occurrence and relevance of EpEX shedding cannot be made. As yet, only a few studies have used antibodies against EpCAM's intracellular domain in cancer tissue, showing that nuclear or cytoplasmic staining is exclusively detectable in tumours, but not in normal tissue [14,34,35]. In thyroid cancer, abundant nuclear and cytoplasmic staining has been represented as a prognostic marker for poor prognosis and tumour aggressiveness [34]. However, based on our own immunostaining data, we cannot exclude artifacts due to lack of antibody specificity in cancer tissues (results not shown). Using fluorescent proteins as well as immunostaining in vitro, we could not detect EpCAM's intracellular domain in the nuclei of cancer cell lines [36].
EpCAM is cleaved via an additional pathway
We generated an antibody directed against EpCAM's cytoplasmic tail that, for the first time, identified novel CTFs in multiple cell lines. Commonly, only antibodies recognizing EpCAM's ectodomain have been used, including to visualize EpCAM fragments [12,13]. Hence, CTFs lacking extracellular epitopes due to cleavage would have been missed. The corresponding NTFs cannot be detected in cell lysates, indicating that these fragments are either degraded or secreted into the medium.
The processing of EpCAM by RIP seems to occur exclusively at the plasma membrane. Homophilic binding, either due to cell–cell contact or EpEX as a ligand for non-cleaved EpCAM, has been shown to be the initial trigger for RIP [14,25]. In contrast, cleavage of the new CTFs seems to be independent from cell–cell contact and to occur both at the plasma membrane and during protein synthesis: retention of a point mutant of EpCAM in the ER, as determined by microscopy and FACS analysis [19a] decreases cleavage, but does not prevent it. Alternatively, traces of EpCAM that cannot be detected by microscopy or FACS analysis may reach the surface. Moreover, treatment with DAPT led to an increase of CTFs12–15, indicating that these fragments are intermediate cleavage products that are further processed by intramembrane proteolysis. Therefore we hypothesize that EpCAM can be processed in two distinct ways (see Figure 7): (1) RIP, resulting in shedding of EpEX and the release of EpICD (in agreement with [14]); and (2) consecutive cleavage at two sites within the cysteine-poor motif in EpCAM's ectodomain, followed by intramembrane proteolysis, releasing EpICD.
Cross-talk between the two proteolytic pathways
EpCAM-induced proliferation has been shown to be dependent on the cleaved intracellular domain EpICD, which may translocate to the nucleus, inducing target genes [14]. Our experiments show that the alternative proteolytic pathway is sensitive to inhibition of γ-secretases, pointing to generation of EpICD. However, in contrast with the alternative (CTF) cleavage, RIP also generates EpEX, which might function as a ligand for non-cleaved EpCAM [14]. This homophilic binding may induce RIP and thereby a positive feedback-loop, contributing to loss of proliferation control. In contrast, the CTF proteolytic pathway seems to be constitutive and ligand-independent. Since RIP has been found only to occur in tumour tissue [14,34,35], in which ADAMs are up-regulated [37], the alternative proteolysis could be involved in the regulation of EpCAM signalling in healthy tissue, i.e. during development and maintenance. Notably, although generating distinct extracellular fragments, both proteolytic pathways will lead to loss of EpCAM-mediated cell–cell adhesion. Ectodomain shedding of CAMs may represent an important regulatory mechanism for cell adhesion and contact inhibition [38].
Is EpCAM proteolysis regulated by its localization?
The best-studied protein processed via two competing RIP pathways is the APP [16]. Processing of APP by a β-secretase and γ-secretase generates the Alzheimer's disease-causing amyloid β peptide (Aβ), whereas cleavage by α-secretase ADAM10 prevents its generation [16,39]. While processing of APP via the β-secretase occurs in lipid rafts [40], ADAM10 cleaves APP in non-raft regions [41,42]. In addition to ADAM17, ADAM10 represents a candidate for EpCAM cleavage [14]. In fact, both EpCAM and ADAM10 have been found in TEMs (tetraspanin-enriched microdomains), membrane microdomains distinct from lipid rafts [43,44], which have been implicated in the regulation of ADAMs [45–48]. The localization of EpCAM and its interaction with certain binding partners in TEMs have been found to promote tumour progression in colon carcinoma cells [49]. Hence, localization of EpCAM and relevant proteases in distinct membrane compartments may play a role in the regulation of different cleavage pathways.
Additionally, tetraspanins have been identified as the major components of exosomes, microvesicles released by cells into the extracellular environment that can be taken up by other cells via different mechanisms [50]. Exosomes can function as intercellular communicators by transfering mRNA and microRNA [50], and might influence the tumour microenvironment, promoting tumour progression and metastasis [24,50,51]. Non-cleaved EpCAM on microvesicles, likely exosomes, is present in the medium of all cancer cell lines we tested. In ovarian carcinoma patients, EpCAM has been found on exosomes derived from malignant ascites [24]. Shedding of EpCAM [51] as well as L1-CAM and CD44 has been shown to occur on exosomes, conducted by ADAM10 in the latter cases [52]. Hence exosomes may enable long-distance transport of non-cleaved proteins which can further be cleaved or act as ligands, thereby inducing downstream signalling events or interfering with cell–cell adhesion [18]. In general, EpCAM's spatiotemporal localization within cells, on the cell surface or on exosomes, may be decisive for the distinct proteolytic cleavage events.
Concluding remarks
EpCAM-mediated proliferative signalling has been linked to proteolytic cleavage. In this study, we uncovered novel EpCAM-derived polypeptides, and provide a confirmation of previously reported EpCAM fragments. EpCAM proteolysis might be crucial in the regulation of EpCAM's multiple functions, including cell–cell adhesion and cell proliferation. Additionally, cleavage of EpCAM via two distinct pathways may account for the loss of proliferation control in cancer in contrast with normal tissue. Unraveling the identity of responsible proteases and how the different cleavage events and derived peptides are involved in EpCAM signalling will aid in the understanding of EpCAM's role in health and disease.
AUTHOR CONTRIBUTION
All authors contributed to the study concept and design, data interpretation and analysis. Acquisition of data was carried out by Ulrike Schnell and Jeroen Kuipers. Cloning of constructs was carried out by Ben N.G. Giepmans. Drafting of the manuscript was carried out by Ulrike Schnell and Ben N.G. Giepmans. Study supervision was carried out by Ben N.G. Giepmans. Funding was obtained by Urike Schnell and Ben N.G. Giepmans.
ACKNOWLEDGEMENTS
We thank N. Govorukhina and R. Bischoff (University of Groningen) for the mass spectrometry effort, L. de Leij (University Medical Center Groningen) for providing antibodies, H. Kampinga (University Medical Center Groningen) for helpful discussions, and V. Cirulli (University of Washington) for EpCAM cDNA.
FUNDING
Part of this work was supported by the Jan Kornelis de Cock Stichting, the Groningen University Graduate School of Medical Sciences, and a Marie Curie International Reintegration Grant within the 7th European Community Framework Programme to B.N.G.G. Experiments were partially performed at the University Medical Center Groningen Microscopy and Imaging Center, which is sponsored by Netherlands Organization for Scientific Research [grant numbers 40-00506-98-9021, 175-010-2009-023].
References
- 1.Cirulli V., Crisa L., Beattie G. M., Mally M. I., Lopez A. D., Fannon A., Ptasznik A., Inverardi L., Ricordi C., Deerinck T., et al. KSA antigen Ep-CAM mediates cell–cell adhesion of pancreatic epithelial cells: morphoregulatory roles in pancreatic islet development. J. Cell Biol. 1998;140:1519–1534. doi: 10.1083/jcb.140.6.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trzpis M., McLaughlin P. M. J., de Leij L. M. F. H., Harmsen M. C. Epithelial cell adhesion molecule: more than a carcinoma marker and adhesion molecule. Am. J. Pathol. 2007;171:386–395. doi: 10.2353/ajpath.2007.070152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Boer C. J., van Krieken J. H., Janssen-van Rhijn C. M., Litvinov S. V. Expression of Ep-CAM in normal, regenerating, metaplastic, and neoplastic liver. J. Pathol. 1999;188:201–206. doi: 10.1002/(SICI)1096-9896(199906)188:2<201::AID-PATH339>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Sundberg M., Jansson L., Ketolainen J., Pihlajamäki H., Suuronen R., Skottman H., Inzunza J., Hovatta O., Narkilahti S. CD marker expression profiles of human embryonic stem cells and their neural derivatives, determined using flow-cytometric analysis, reveal a novel CD marker for exclusion of pluripotent stem cells. Stem Cell Res. 2009;2:113–124. doi: 10.1016/j.scr.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle P. A., Gires O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer. 2007;96:417–423. doi: 10.1038/sj.bjc.6603494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Gun B. T. F., Melchers L. J., Ruiters M. H. J., de Leij L. F. M. H., McLaughlin P. M. J., Rots M. G. EpCAM in carcinogenesis: the good, the bad or the ugly. Carcinogenesis. 2010;31:1913–1921. doi: 10.1093/carcin/bgq187. [DOI] [PubMed] [Google Scholar]
- 7.Patriarca C., Macchi R. M., Marschner A. K., Mellstedt H. Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat. Rev. 2012;38:68–75. doi: 10.1016/j.ctrv.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Sivagnanam M., Mueller J. L., Lee H., Chen Z., Nelson S. F., Turner D., Zlotkin S. H., Pencharz P. B., Ngan B.-Y., Libiger O., et al. Identification of EpCAM as the gene for congenital tufting enteropathy. Gastroenterology. 2008;135:429–437. doi: 10.1053/j.gastro.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strnad J., Hamilton A. E., Beavers L. S., Gamboa G. C., Apelgren L. D., Taber L. D., Sportsman J. R., Bumol T. F., Sharp J. D., Gadski R. A. Molecular cloning and characterization of a human adenocarcinoma/epithelial cell surface antigen complementary DNA. Cancer Res. 1989:314–317. [PubMed] [Google Scholar]
- 10.Szala S., Froehlich M., Scollon M., Kasai Y., Steplewski Z., Koprowski H., Linnenbach A. J. Molecular cloning of cDNA for the carcinoma-associated antigen GA733-2. Proc. Natl. Acad. Sci. U.S.A. 1990;87:3542–3546. doi: 10.1073/pnas.87.9.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong J. M., Speicher D. W. Determination of disulfide bond assignments and N-glycosylation sites of the human gastrointestinal carcinoma antigen GA733-2 (CO17-1A, EGP, KS1-4, KSA, and Ep-CAM) J. Biol. Chem. 2001;276:5804–5813. doi: 10.1074/jbc.M008839200. [DOI] [PubMed] [Google Scholar]
- 12.Thampoe I., Ng J. Biochemical analysis of a human epithelial surface antigen: differential cell expression and processing. Arch. Biochem. Biophys. 1988;267:342–352. doi: 10.1016/0003-9861(88)90040-9. [DOI] [PubMed] [Google Scholar]
- 13.Schön M. P., Schön M., Mattes M. J., Stein R., Weber L., Alberti S., Klein C. E. Biochemical and immunological characterization of the human carcinoma-associated antigen MH 99/KS 1/4. Int. J. Cancer. 1993;55:988–995. doi: 10.1002/ijc.2910550619. [DOI] [PubMed] [Google Scholar]
- 14.Maetzel D., Denzel S., Mack B., Canis M., Went P., Benk M., Kieu C., Papior P., Baeuerle P. A., Münz M., et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat. Cell Biol. 2009;11:162–171. doi: 10.1038/ncb1824. [DOI] [PubMed] [Google Scholar]
- 15.Brown M. S., Ye J., Rawson R. B., Goldstein J. L. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 16.Lichtenthaler S. F., Haass C., Steiner H. Regulated intramembrane proteolysis–lessons from amyloid precursor protein processing. J. Neurochem. 2011;117:779–796. doi: 10.1111/j.1471-4159.2011.07248.x. [DOI] [PubMed] [Google Scholar]
- 17.Arribas J., Borroto A. Protein ectodomain shedding. Chem. Rev. 2002;102:4627–4638. doi: 10.1021/cr010202t. [DOI] [PubMed] [Google Scholar]
- 18.van Kilsdonk J. W. J., van Kempen L. C. L. T., van Muijen G. N. P., Ruiter D. J., Swart G. W. M. Soluble adhesion molecules in human cancers: sources and fates. Eur. J. Cell Biol. 2010;89:415–427. doi: 10.1016/j.ejcb.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 19.Winter M. J., Cirulli V., Briaire-de Bruijn I. H., Litvinov S. V. Cadherins are regulated by Ep-CAM via phosphaditylinositol-3 kinase. Mol. Cell. Biochem. 2007;302:19–26. doi: 10.1007/s11010-007-9420-y. [DOI] [PubMed] [Google Scholar]
- 19a.Schnell U., Kuipers J., Mueller J. L., Veenstra-Algra A., Sivagnanam M., Giepmans B. N. G. Absence of cell surface EpCAM in congenital tufting enteropathy. Hum. Mol. Genet. 2013:doi: 10.1093/hmg/ddt105. doi: 10.1093/hmg/ddt105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hageman J., Vos M. J., van Waarde M. A, Kampinga H. H. Comparison of intra-organellar chaperone capacity for dealing with stress-induced protein unfolding. J. Biol. Chem. 2007;282:34334–34345. doi: 10.1074/jbc.M703876200. [DOI] [PubMed] [Google Scholar]
- 21.De Leij L., Helfrich W., Stein R., Mattes M. J. SCLC-cluster-2 antibodies detect the pancarcinoma/epithelial glycoprotein EGP-2. Int. J. Cancer. 1994;8:60–63. doi: 10.1002/ijc.2910570713. [DOI] [PubMed] [Google Scholar]
- 22.Balzar M., Winter M. J., de Boer C. J., Litvinov S. V. The biology of the 17-1A antigen (Ep-CAM) J. Mol. Med. 1999;77:699–712. doi: 10.1007/s001099900038. [DOI] [PubMed] [Google Scholar]
- 23.Tuteja R. Type I signal peptidase: an overview. Arch. Biochem. Biophys. 2005;441:107–111. doi: 10.1016/j.abb.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Runz S., Keller S., Rupp C., Stoeck A., Issa Y., Koensgen D., Mustea A., Sehouli J., Kristiansen G., Altevogt P. Malignant ascites-derived exosomes of ovarian carcinoma patients contain CD24 and EpCAM. Gynecol. Oncol. 2007;107:563–571. doi: 10.1016/j.ygyno.2007.08.064. [DOI] [PubMed] [Google Scholar]
- 25.Denzel S., Maetzel D., Mack B., Eggert C., Bärr G., Gires O. Initial activation of EpCAM cleavage via cell-to-cell contact. BMC Cancer. 2009;9:402. doi: 10.1186/1471-2407-9-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neurath H., Walsh K. A. Role of proteolytic enzymes in biological regulation (a review) Proc. Natl. Acad. Sci. U.S.A. 1976;73:3825–3832. doi: 10.1073/pnas.73.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turk V., Stoka V., Vasiljeva O., Renko M., Sun T., Turk B., Turk D. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochim. Biophys. Acta. 2012;1824:68–88. doi: 10.1016/j.bbapap.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mihelic M., Turk D. Two decades of thyroglobulin type-1 domain research. Biol. Chem. 2007;388:1123–1130. doi: 10.1515/BC.2007.155. [DOI] [PubMed] [Google Scholar]
- 29.Nomura T., Katunuma N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J. Med. Invest. 2005;52:1–9. doi: 10.2152/jmi.52.1. [DOI] [PubMed] [Google Scholar]
- 30.Beel A. J., Sanders C. R. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 2008;65:1311–1334. doi: 10.1007/s00018-008-7462-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe H., Kuroki M., Imakiire T., Yamauchi Y., Yamada H., Arakawa F., Kuroki M. Preparation of recombinant MK-1/Ep-CAM and establishment of an ELISA system for determining soluble MK-1/Ep-CAM levels in sera of cancer patients. J. Immunol. Methods. 2002;270:227–233. doi: 10.1016/s0022-1759(02)00332-0. [DOI] [PubMed] [Google Scholar]
- 32.Kimura H., Kato H., Faried A., Sohda M., Nakajima M., Fukai Y., Miyazaki T., Masuda N., Fukuchi M., Kuwano H. Prognostic significance of EpCAM expression in human esophageal cancer. Int. J. Oncol. 2007;30:171–179. [PubMed] [Google Scholar]
- 33.Petsch S., Gires O., Rüttinger D., Denzel S., Lippold S., Baeuerle P. A., Wolf A. Concentrations of EpCAM ectodomain as found in sera of cancer patients do not significantly impact redirected lysis and T-cell activation by EpCAM/CD3-bispecific BiTE antibody MT110. MAbs. 2011;3:31–37. doi: 10.4161/mabs.3.1.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ralhan R., Cao J., Lim T., Macmillan C., Freeman J. L., Walfish P. G. EpCAM nuclear localization identifies aggressive thyroid cancer and is a marker for poor prognosis. BMC Cancer. 2010;10:331. doi: 10.1186/1471-2407-10-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ralhan R., He H. C.-H., So A. K.-C., Tripathi S. C., Kumar M., Hasan M. R., Kaur J., Kashat L., MacMillan C., Chauhan S. S., et al. Nuclear and cytoplasmic accumulation of Ep-ICD is frequently detected in human epithelial cancers. PloS ONE. 2010;5:e14130. doi: 10.1371/journal.pone.0014130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schnell U., Dijk F., Sjollema K. A., Giepmans B. N. G. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods. 2012;9:152–158. doi: 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- 37.Edwards D. R., Handsley M. M., Pennington C. J. The ADAM metalloproteinases. Mol. Aspects Med. 2008;29:258–289. doi: 10.1016/j.mam.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Craig S. E. L., Brady-Kalnay S. M. Cancer cells cut homophilic cell adhesion molecules and run. Cancer Res. 2011;71:303–309. doi: 10.1158/0008-5472.CAN-10-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhn P.-H., Wang H., Dislich B., Colombo A., Zeitschel U., Ellwart J. W., Kremmer E., Rossner S., Lichtenthaler S. F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cordy J. M., Hussain I., Dingwall C., Hooper N. M., Turner A. J. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11735–11740. doi: 10.1073/pnas.1635130100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris B., Pereira I., Parkin E. Targeting ADAM10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 2009;1296:203–215. doi: 10.1016/j.brainres.2009.07.105. [DOI] [PubMed] [Google Scholar]
- 42.Ehehalt R., Keller P., Haass C., Thiele C., Simons K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le Naour F., André M., Greco C., Billard M., Sordat B., Emile J.-F., Lanza F., Boucheix C., Rubinstein E. Profiling of the tetraspanin web of human colon cancer cells. Mol. Cell. Proteomics. 2006;5:845–857. doi: 10.1074/mcp.M500330-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Le Naour F., André M., Boucheix C., Rubinstein E. Membrane microdomains and proteomics: lessons from tetraspanin microdomains and comparison with lipid rafts. Proteomics. 2006;6:6447–6454. doi: 10.1002/pmic.200600282. [DOI] [PubMed] [Google Scholar]
- 45.Xu D., Sharma C., Hemler M. E. Tetraspanin12 regulates ADAM10-dependent cleavage of amyloid precursor protein. FASEB J. 2009;23:3674–3681. doi: 10.1096/fj.09-133462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gutiérrez-López M. D., Gilsanz A., Yáñez-Mó M., Ovalle S., Lafuente E. M., Domínguez C., Monk P. N., González-Alvaro I., Sánchez-Madrid F., Cabañas C. The sheddase activity of ADAM17/TACE is regulated by the tetraspanin CD9. Cell. Mol. Life Sci. 2011;68:3275–3292. doi: 10.1007/s00018-011-0639-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arduise C., Abache T., Li L., Billard M., Chabanon A., Ludwig A., Mauduit P., Boucheix C., Rubinstein E., Le Naour F. Tetraspanins regulate ADAM10-mediated cleavage of TNF-alpha and epidermal growth factor. J. Immunol. 2008;181:7002–7013. doi: 10.4049/jimmunol.181.10.7002. [DOI] [PubMed] [Google Scholar]
- 48.Yáñez-Mó M., Gutiérrez-López M. D., Cabañas C. Functional interplay between tetraspanins and proteases. Cell. Mol. Life Sci. 2011;68:3323–3335. doi: 10.1007/s00018-011-0746-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhn S., Koch M., Nübel T., Ladwein M., Antolovic D., Klingbeil P., Hildebrand D., Moldenhauer G., Langbein L., Franke W. W., et al. A complex of EpCAM, claudin-7, CD44 variant isoforms, and tetraspanins promotes colorectal cancer progression. Mol. Cancer Res. 2007;5:553–567. doi: 10.1158/1541-7786.MCR-06-0384. [DOI] [PubMed] [Google Scholar]
- 50.Marhaba R., Klingbeil P., Nuebel T., Nazarenko I., Buechler M. W., Zoeller M. CD44 and EpCAM: cancer-initiating cell markers. Curr. Mol. Med. 2008;8:784–804. doi: 10.2174/156652408786733667. [DOI] [PubMed] [Google Scholar]
- 51.Rupp A.-K., Rupp C., Keller S., Brase J. C., Ehehalt R., Fogel M., Moldenhauer G., Marmé F., Sültmann H., Altevogt P. Loss of EpCAM expression in breast cancer derived serum exosomes: role of proteolytic cleavage. Gynecol. Oncol. 2011;122:437–446. doi: 10.1016/j.ygyno.2011.04.035. [DOI] [PubMed] [Google Scholar]
- 52.Stoeck A., Keller S., Riedle S., Sanderson M. P., Runz S., Le Naour F., Gutwein P., Ludwig A., Rubinstein E., Altevogt P. A role for exosomes in the constitutive and stimulus-induced ectodomain cleavage of L1 and CD44. Biochem. J. 2006;393:609–618. doi: 10.1042/BJ20051013. [DOI] [PMC free article] [PubMed] [Google Scholar]