

Abstract
Effective inhibitors of cancer cell migration and invasion can potentially lead to clinical applications as a therapy to block tumor metastasis, the primary cause of death in cancer patients. To this end, we have designed and synthesized a series of thiazole derivatives that showed potent efficacy against cell migration and invasion in metastatic cancer cells. The most effective compound, 5k, was found to have an IC50 value of 176 nM in the dose-dependent transwell migration assays in MDA-MB-231cells. At a dose of 10 μM, 5k also blocked about 80% of migration in HeLa and A549 cells and 60% of invasion of MDA-MB-231 cells. Importantly, the majority of the derivatives exhibited no apparent cytotoxicity in the clonogenic assays. The low to negligible inhibition of cell proliferation is a desirable property of these antimigration derivatives because they hold promise of low toxicity to healthy cells as potential therapeutic agents. Mechanistic studies analyzing the actin cytoskeleton by microscopy demonstrate that compound 5k substantially reduced cellular f-actin and prevented localization of fascin to actin-rich membrane protrusions. These results suggest that the antimigration activity may result from impaired actin structures in protrusions that are necessary to drive migration.
Keywords: thiazole derivatives, synthesis, antimigration, anti-invasion, f-actin, fascin
Metastasis is the major cause of death in cancer patients: nearly 90% mortality has been attributed to metastatic spread of the disease rather than to the primary tumor.1−3 Decades of intensive research have focused on the search for therapeutic solutions targeting cancer cell migration and invasion and angiogenesis.4−7 Metastasis is a complex process, involving multiple steps that include cancer cell motility, intravasation, transit and survival in the circulation, extravasation, and growth at a new site. While in theory inhibition of any of these metastatic stages will prevent the formation of tumors at remote sites, clinically, the window of opportunity to block metastasis may not be as optimal as one might hope.8 For example, stages involving cancer cell survival in the circulation, arrest, and extravasation may not be ideal targets for development of therapeutic solutions as these processes appear to occur relatively fast, in large numbers, and are less vulnerable to drug interference. On the other hand, the growth of cancer cells in secondary sites takes much longer to cause irreversible damage, thus offering a broader time window for the prevention of metastasis.8,9
Small molecule drugs such as matrix metalloproteinases inhibitors10−16 and vascular disrupting agents17−23 have been developed to block metastasis, so far with only limited clinical success. Most chemotherapies target cancer cell proliferation as a means to inhibit dissemination, leading to toxicity to healthy cells as well as acquired resistance in cancer cells. The metastasis-modifying processes, however, may be more effectively influenced by long-term treatment of noncytotoxic drugs such as protease inhibitors, chemokine antagonists, kinase blockers, adhesion modifiers, and anti-inflammation agents.9 A search for improved, more potent, and less toxic drugs for metastasis intervention remains an ongoing effort.
Thiazole is a common structural motif in a large number of anticancer agents,18,24−28 including the clinically used BCR/ABL inhibitor dasatinib for chronic myelogenous leukemia (CML).29 These thiazole derivatives have been reported to inhibit cancer cell growth and proliferation and vasculature formation through a variety of mechanisms and therapeutic targets. In an effort to design and synthesize new thiazole compounds for potential anticancer agents, we have found that a methyl substitution on the thiazole nitrogen would dramatically reduce the antiproliferation activity. However, a subsequent cell motility assay revealed that this structural modification allowed the compounds to inhibit cell migration. One such thiazole derivative (5a) showed a strong ability to block cancer cell migration but exhibited no apparent cellular toxicity in both cell survival and colony formation assays. This finding prompted us to design a series of thiazole derivatives by varying the substitution groups on the thiazole ring. We report here the preparation and biological activity evaluation of 40 such derivatives.
As shown in Scheme 1, the designed derivatives were synthesized following the general procedures as detailed below. The condensation-cyclization of 2-bromo-1-phenylethanone (1a) and 2-bromo-1-(2,4-dimethylphenyl)ethanone (1b) with thiourea afforded near quantitative yield of 2-amino-4-phenylthiazole (2a) and 2-amino-4-(2, 4-dimethylphenyl) thiazole (2b) in refluxing ethanol, respectively.18,30 Following acylation of 2a–c by acyl chloride or direct N,N′-dicyclohexylcarbodiimide-promoted coupling of 2a–c with the carboxylic acids provided the amides 3, and further alkylation of 3 led to the desired derivatives 4 and their corresponding isomers 5.18
Scheme 1. Preparation of a Series of Thiazole Derivatives as Novel Antimigration and Anti-invasion Agents.

Reagents and conditions: (a) Thiourea, ethanol, reflux. (b) Method A: R1CO2H, dicyclohexylcarbodimide, DMAP, CH2Cl2, rt; method B: R1COCl, DMAP, CH2Cl2, 0 °C–rt. (c) (i) NaH, THF, 0 °C–rt; (ii) MeI or EtBr 0 °C–rt.
Interestingly, alkylation of 3 formed two different isomers 4 and 5 depending on which nitrogen on amides 3 to take part in the alkylation. The result showed that the formation ratio of isomers 4 and 5 was clearly linked to the type of R1 groups in the amides 3. If R1 groups were alkyl groups, exclusive isomers 4 (4f, 4g, 4h, and 4i) were acquired, and possible isomers 5 as minor products (<1%) were found only by GC-MS analysis. However, when R1 groups turn into aryl groups, the formation of isomers 5 (5d and 5f) increased significantly to exceed the formation of isomers 4 (4a and 4b) except the ethylation product of 3b. In fact, most of isomers 5 (5a–c, 5e, 5h, 5i, and 5k) were the only purified products obtained, due to negligible amounts of their corresponding isomers 4 as products.
For derivatives 4 and their corresponding isomers 5, the structural differences were such that the derivatives 4 had the thiazole ring moiety, whereas isomers 5 contained the thiazol-2(3H)-yliene ring. The formation of thiazol-2(3H)-yliene ring of isomers 5 considerably undermined the aromaticity of the original thiazole ring and further decreased the deshielding of the thiazole protons by a ring current of π-electrons. Therefore, the thiazol-2(3H)-yliene ring of isomers 5 showed a relatively higher field 1H chemical shift (∼6.5 ppm) than that of the thiazole ring of the corresponding isomers 4 (∼7.1 ppm). The decrease of aromaticity of the thiazol-2(3H)-yliene ring of isomers 5 increased their polarity, as reflected in the longer retention times of derivatives 5 as compared to derivatives 4 in GC-MS analysis. Similarly, the Rf value of derivatives 5 was smaller than that of derivatives 4 in silica gel-based TLC.
To determine the exact structures of derivative 4e and its corresponding isomer 5j, single crystals of 4e and 5j were grown by vapor diffusion of hexane into the dichloromethane solutions of the compounds and analyzed by X-ray crystallography (the detailed crystal data are provided in the Supporting Information). The results are presented in Figure 1. The X-ray single crystal analysis of isomers 4e and 5j further confirmed their respective structures.
Figure 1.

ORTEP plot of molecular structures of 4e and 5j.
To determine the effects of the synthesized thiazole derivatives on cancer cell migration, we performed a transwell migration assay on each compound using an invasive and metastatic breast cancer cell line, MDA-MB-231. When cells were seeded at a density of 2.5 × 104 in media free of serum in the upper chamber but containing 5% FBS in the lower chamber, their ability to migrate in the presence and absence of 10 μM derivatives was measured by counting the total number of cells in the lower chamber after 24 h. As shown in Table 1, of the 40 synthetic derivatives, most displayed moderate or potent antimigration activity, 20 derivatives showed greater than 50% inhibition, and the three most potent derivatives (3g, 5j and 5k) blocked cell migration by over 80%. These results demonstrate that the synthetic derivatives are effective migration inhibitors.
Table 1. List of All Synthetic Derivatives with Migration Inhibition and Colony Formation Data When MDA-MB-231 Breast Cancer Cells Were Treated with 10 μM Derivatives.
| compds | migration inhibition (% of vehicle control) | effect on cell proliferation (colony formation) (% of vehicle control) |
|---|---|---|
| DMSO | 100.0 | 100.0 |
| 2a | 86.0 | 103.6 |
| 2b | 45.5 | 110.9 |
| 3a | 34.5 | 85.9 |
| 3b | 88.0 | 98.8 |
| 3c | 78.0 | 95.0 |
| 3d | 103.0 | 98.6 |
| 3e | 46.5 | 80.9 |
| 3f | 40.9 | 67.5 |
| 3g | 18.6 | 57.3 |
| 3h | 80.8 | 102.8 |
| 3i | 32.3 | 95.5 |
| 3j | 31.3 | 105.2 |
| 3k | 68.2 | 99.8 |
| 3l | 92.7 | 103.4 |
| 3m | 75.2 | 90.7 |
| 3n | 28.3 | 71.5 |
| 3o | 33.7 | 85.0 |
| 3p | 59.2 | 108.8 |
| 4a | 78.2 | 107.7 |
| 4b | 72.2 | 97.7 |
| 4c | 43.5 | 97.1 |
| 4d | 54.5 | 96.5 |
| 4e | 39.2 | 123.4 |
| 4f | 54.3 | 132.4 |
| 4g | 72.7 | 97.5 |
| 4h | 71.4 | 127.5 |
| 4i | 80.0 | 111.1 |
| 4j | 91.5 | 110.4 |
| 5a | 43.3 | 85.0 |
| 5b | 77.6 | 107.3 |
| 5c | 28.1 | 98.2 |
| 5d | 59.7 | 63.8 |
| 5e | 26.5 | 75.4 |
| 5f | 37.1 | 102.3 |
| 5g | 35.9 | 40.9 |
| 5h | 62.0 | 111.5 |
| 5i | 32.1 | 97.7 |
| 5j | 13.0 | 25.2 |
| 5k | 14.3 | 120.9 |
| 5l | 40.6 | 94.0 |
As shown in Table 1, transformed from 2a, 2b, and 2c by condensing with various carboxylic acid or acyl chloride, the amides 3 exhibited widely variable activities from the most active 71.4% inhibition (3g) to a slight (3%) stimulation of migration (3d). The activity variation of the amides 3 suggests that the R1 group may be important but not necessary in conferring the desired antimigration property. The methylation transformation of the amides 3 (3b–f, 3h, 3j, 3o, and 3p) to the desired thiazole derivatives 5 (5b–f, 5h, 5j–l) mostly improves the antimigration activity of the compounds with the exception of 3a (65.5% inhibition) to 5a (56.7% inhibition) and 3g (81.4% inhibition) to 5g (64.1% inhibition). This conversion led to the discovery of the most potent derivatives 5j (87% inhibition, IC50 = 0.189 μM) and 5k (85.7% inhibition, IC50 = 0.176 μM). Upon methylation of the amides 3, the resultant isomers 4 (4a–c, 4e, and 4j) have consistently lower activities than those of the corresponding isomers 5 (5d, 5f, 5g, 5j, and 5l). Additionally, it is noted that replacement of the methyl moiety with an ethyl group in 5a (56.7% inhibition) slightly increased the activity of 5i (67.9%).
Given the potent antimigration efficacy demonstrated by most of the thiazole derivatives, we decided to study the dose response of the most potent derivatives to obtain their IC50 values in suppressing the transwell migration of the MDA-MB-231 cells. As shown in Table 2, the IC50 values for the 10 selected derivatives varied from 2.87 to 0.176 μM.
Table 2. IC50 Values for 10 Most Potent Antimigration Compounds in MDA-MB-231 Breast Cancer Cells.
| compds | IC50 (μM) |
|---|---|
| 3a | 2.49 |
| 3i | 2.87 |
| 3j | 1.29 |
| 3n | 1.01 |
| 3o | 0.839 |
| 4e | 0.366 |
| 5c | 1.12 |
| 5i | 2.08 |
| 5j | 0.189 |
| 5k | 0.176 |
We next performed clonogenic assays on the breast cancer cells treated with the compounds to rule out any indirect effect on cell migration due to cytotoxicity. MDA-MB-231 cells were allowed to grow for 14 days in six-well plates in the presence or absence of the synthetic compounds at 10 μM. The cell toxicity data for all derivatives are listed in Table 1 along with antimigration data for comparison. Figure 2 shows five examples of the proliferation and colony formation images of antimigration derivative-treated colonies as compared to that of DMSO-treated control cells. The five compounds, 2b, 5j, 3n, 4e, and 5k, all inhibited cell migration by over 50% (55–86%); yet, no apparent cell toxicity was observed when cells were treated with the derivatives at the same dose of 10 μM for 2 weeks, with the exception of 5j, which significantly inhibited colony formation of the breast cancer cells by 75% (see also Table 1). A few other antimigration derivatives did exhibit a moderate level of inhibition of the breast cancer cell proliferation. For example, derivative 3g blocked both cell migration (81.4%) and colony formation (42.7%). For these derivatives, cytotoxicity may have partially contributed to their overall antimigration effect.
Figure 2.

Clonogenic assay demonstrates no cytotoxicity of thiazole derivatives that potently inhibit migration of MDA-MB-231 cells in four out of five selected compounds. Images of colony formation are shown for cells treated with (1) DMSO, (2) 2b, (3) 5j, (4) 3n, (5) 4e, and (6) 5k.
On the basis of the potent effects of synthetic thiazole derivatives in blocking migration of MDA-MB-231 cells, we selected 10 derivatives with high antimigration activity but low or negligible cytotoxicity to test their antimigration activity in another metastatic cell line, HeLa. For comparison of possible cancer cell-specific mode of action, we also included 5j, a potent antimigration derivative that also inhibited the proliferation of the triple negative breast cancer cells. Results are summarized in Figure 3. The derivatives exhibited excellent antimigration activity in HeLa cells, as evidenced by the dramatically reduced transwell migration (60–86% inhibition) when treated with 10 μM derivatives. While these derivatives demonstrated similar antimigration efficacies in MDA-MB-231 and HeLa cells, some differences were noted. For example, the derivative 3n showed 30% inhibition of colony formation in MDA-MB-231 cells but had no apparent cytotoxicity in HeLa cells. On the other hand, the derivative 5i was not toxic to 231 cells, but it suppressed the clonogenic capability of HeLa cells by 45% (Figure 3). Interestingly, the derivative 5j was a strong inhibitor of cell proliferation for both HeLa and MDA-MB-231 cells. The derivative that emerged as the most potent antimigration agent with no apparent cytotoxicity in both metastatic breast cancer and cervical cancer cell lines was 5k, achieving over 85% inhibition of transwell migration in both cell lines at a dose of 10 μM. Thus, 5k may be further evaluated for its potential as an antimigration and antimetastatic agent. Additional migration assay of a nonsmall cell lung cancer cell line, A549 also demonstrated that in the presence of 10 μM 5k, and the metastatic lung cancer cells lost nearly 80% of migratory capacity (Figure 4).
Figure 3.

Effect of selected thiazole derivatives on the migration and proliferation of HeLa cells.
Figure 4.
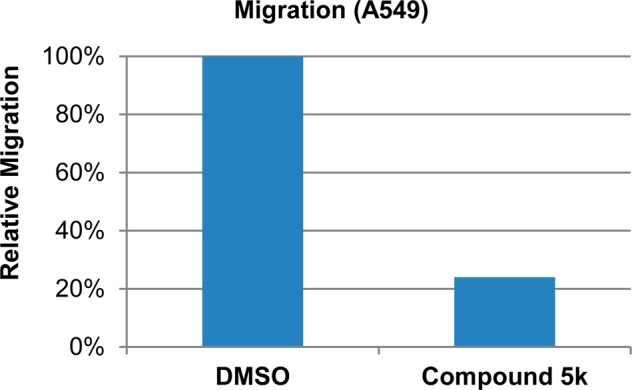
Derivative 5k strongly inhibits migration of A549, a metastatic nonsmall cell lung cancer cell line.
To determine if the synthetic thiazole derivatives block invasion of metastatic cancer cells, we performed matrigel invasion assays of MDA-MB-231 cells treated with 10 selected derivatives. As shown in Figure 5, all 10 derivatives exhibited marked inhibition of matrigel invasion of the breast cancer cells, with percent invasion reduced to approximately 40–60% of the control at a dose of 10 μM. The derivative 5k, the most active antimigration agent without any apparent cytotoxicity, also appears to be the most potent compound in blocking cell invasion.
Figure 5.
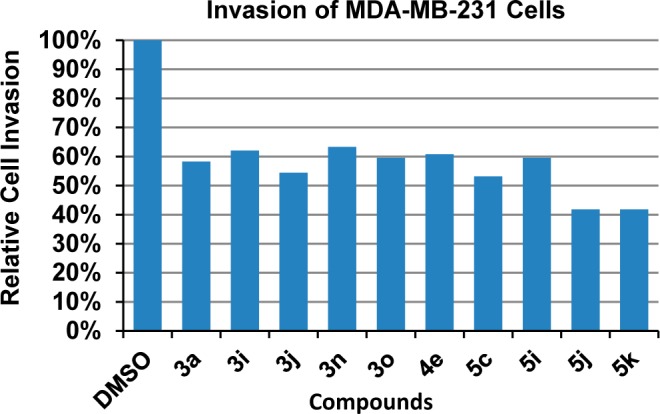
Effect of selected thiazole derivatives on the invasion of MDA-MB-231 breast cancer cells.
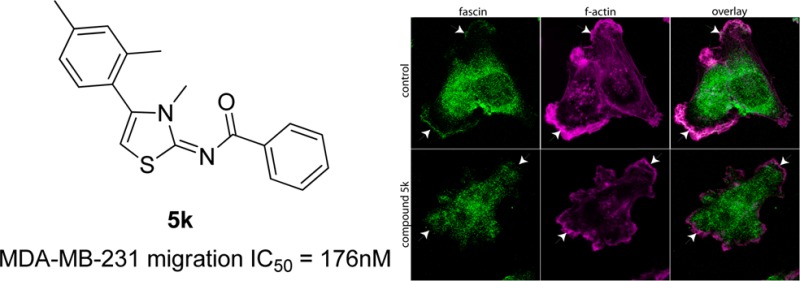
An essential component of migration is protrusion of the cell membrane, which is driven by actin polymerization. Previous studies have shown that a natural compound known as migrastatin blocks actin bundling by binding to the actin regulatory protein fascin, which is linked to migration in cell culture systems, and metastasis in vivo.31−33 While the study by Chen et al. showed that migrastatin blocked the actin bundling activity of fascin using purified proteins in vitro, the effects on actin structures in cells were not tested.31 Thus, we tested the hypothesis that compound 5k interferes with f-actin in membrane protrusions associated with cell motility. MDA-MB-231 cells were serum starved ∓10 μM 5k, then stimulated with serum for 2 h to induce actin-rich membrane protrusions, which were analyzed by fluorescent microscopy. Figure 6 shows that control cells (DMSO-treated) had robust actin-rich membrane protrusions, which were significantly reduced in cells treated with 5k (55% reduction in f-actin intensity; p < 0.01). To further probe for a possible role of fascin in the reduction of actin-rich membrane protrusions, we determined the localization of fascin by immunofluorescence microscopy.
Figure 6.

Derivative 5k strongly suppresses actin-rich membrane protrusions in MDA-MB-231 cells. F-actin staining in (A) control cells (DMSO-treated) or (B) cells treated with 5k and (C) quantitation of f-actin intensity in vehicle- and 5k-treated cells.
Figure 7 shows that in control cells, a pool of fascin is localized within the zone of f-actin in the protrusions. This pool of fascin was notably missing from the actin-rich membrane regions in the cells treated with 5k (indicated by arrowheads.) Thus, our results demonstrate that compound 5k significantly blocks f-actin and is correlated with the absence of fascin in the membrane protrusions, suggesting that its mechanism of action is to perturb the actin dynamics required for tumor cell migration.
Figure 7.

Immunofluorescence microscopic images of localization of fascin (green) and f-actin (magenta) in MDA-MB-231 cells treated with DMSO (vehicle) or synthetic derivative 5k. Colocalization of f-actin and fascin appears as white pixels.
We have designed and synthesized 40 thiazole derivatives as novel antimigration and anti-invasion agents. Structural modification of the substitution groups on the central thiazole ring led to the identification of several potent migration inhibitors that strongly suppressed cell motility in metastatic cancer cells. More importantly, these compounds exhibited no apparent cytotoxicity as they do not inhibit the ability of metastatic cancer cells to form colonies when treated with the migration inhibitors. Thus, our study provides a novel type of small molecule therapeutic agents that aim to block cancer cell migration and invasion without exerting cell toxicity. Furthermore, we provide evidence that the antimigration activity of these compounds may be a result of impaired formation of actin structures in cells, which are known to be necessary for tumor cell migration and metastasis. Further studies to determine the exact antimigration mechanism are underway.
Supporting Information Available
X-ray single crystal data and experimental procedures for the synthesis and characterization of thiazole derivatives, cell culture, in vitro migration assays, clonogenic assays, invasion assays, and fluorescence microscopy. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by NIH RCMI program through Grant 5G12RR026260-03, the Louisiana Cancer Research Consortium (LCRC), Department of Agriculture Grant 58-6435-7-019, and Office of Naval Research Grant N00014-99-1-0763.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chambers A. F.; Groom A. C.; MacDonald I. C. Dissemination and growth of cancer cells in metastatic sites. Nature Rev. Cancer 2002, 2, 563–572. [DOI] [PubMed] [Google Scholar]
- Fidler I. J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nature Rev. Cancer 2003, 3, 453–458. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Weiss L. Metastasis of cancer: A conceptual history from antiquity to the 1990s. Cancer Metastasis Rev. 2000, 19, 193–383. [PubMed] [Google Scholar]
- D’Alise A. M.; Amabile G.; Iovino M.; Di Giorgio F. P.; Bartiromo M.; Sessa F.; Villa F.; Musacchio A.; Cortese R. Reversine, a novel Aurora kinases inhibitor, inhibits colony formation of human acute myeloid leukemia cells. Mol. Cancer Ther. 2008, 7, 1140–1149. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Ueno M.; Kamiya D.; Nishiyama A.; Matsumura M.; Wataya T.; Takahashi J. B.; Nishikawa S.; Nishikawa S.; Muguruma K.; Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [DOI] [PubMed] [Google Scholar]
- Singh R. P.; Raina K.; Sharma G.; Agarwal R. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial–mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clin. Cancer Res. 2008, 14, 7773–7780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers A. F.; MacDonald I. C.; Schmidt E. E; Morris V. L.; Groom A. C. Clinical targets for anti-metastasis therapy. Adv. Cancer Res. 2000, 79, 91–121. [DOI] [PubMed] [Google Scholar]
- Epstein R. J. Maintenance therapy to suppress micrometastasis: the new challenge for adjuvant cancer treatment. Clin. Cancer Res. 2005, 11, 5337–5341. [DOI] [PubMed] [Google Scholar]
- Overall C. M.; Lopez-Otin C. Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nature Rev. Cancer 2002, 2, 657–672. [DOI] [PubMed] [Google Scholar]
- Shi Z.-G.; Li J.-P.; Shi L.-L.; Li X. An updated patent therapeutic agents targeting MMPs. Recent Pat. Anticancer Drug Discovery 2012, 7, 74–101. [DOI] [PubMed] [Google Scholar]
- Hua H.; Li M.; Luo T.; Yin Y.; Jiang Y. Matrix metalloproteinases in tumorigenesis: An evolving paradigm. Cell. Mol. Life Sci. 2011, 68, 3853–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rucci N.; Sanità P.; Angelucci A. Roles of metalloproteases in metastatic niche. Curr. Mol. Med. 2011, 11, 609–622. [DOI] [PubMed] [Google Scholar]
- Raffo D.; Pontiggia O. Simian, M. Role of MMPs in metastatic dissemination: Implications for therapeutic advances. Curr. Pharm. Biotechnol. 2011, 12, 1937–1947. [DOI] [PubMed] [Google Scholar]
- Dormán G.; Cseh S.; Hajdú I.; Barna L.; Kónya D.; Kupai K.; Kovács L.; Ferdinandy P. Matrix metalloproteinase inhibitors: A critical appraisal of design principles and proposed therapeutic utility. Drugs 2010, 70, 949–964. [DOI] [PubMed] [Google Scholar]
- Fingleton B. MMPs as therapeutic targets—Still a viable option?. Semin. Cell Dev. Biol. 2008, 19, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Qiu X.-L.; Li G.; Wu G.; Zhu J.; Zhou L.; Chen P.-L.; Chamberlin A. R.; Lee W.-H. Synthesis and biological evaluation of a series of novel inhibitor of Nek2/Hec1 analogues. J. Med. Chem. 2009, 52, 1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollebecque A.; Massard C.; Soria J. C. Vascular disrupting agents: A delicate balance between efficacy and side effects. Curr. Opin. Oncol. 2012, 24, 305–315. [DOI] [PubMed] [Google Scholar]
- Spear M. A.; LoRusso P.; Mita A.; Mita M. Vascular disrupting agents (VDA) in oncology: Advancing towards new therapeutic paradigms in the clinic. Curr. Drug Targets 2011, 12, 2009–2015. [DOI] [PubMed] [Google Scholar]
- Marrelli M.; Conforti F.; Statti G. A.; Cachet X.; Michel S.; Tillequin F.; Menichini F. Biological potential and structure-activity relationships of most recently developed vascular disrupting agents: an overview of new derivatives of natural combretastatin A-4. Curr. Med. Chem. 2011, 18, 3035–3081. [DOI] [PubMed] [Google Scholar]
- Heath V. L.; Bicknell R. Anticancer strategies involving the vasculature. Nat. Rev. Clin. Oncol. 2009, 6, 395–404. [DOI] [PubMed] [Google Scholar]
- Schwartz E. L. Antivascular actions of microtubule-binding drugs. Clin. Cancer Res. 2009, 15, 2594–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Brancale A.; Ricci A.; Hamel E.; Bortolozzi R.; Basso G.; Viola G. Convergent synthesis and biological evaluation of 2-amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl thiazoles as microtubule targeting agents. J. Med. Chem. 2011, 54, 5144–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy A. J.; Mathews T. P.; Kharel Y.; Field S. D.; Moyer M. L.; East J. E.; Houck J. D.; Lynch K. R.; Macdonald T. L. Development of amidine-based sphingosine kinase 1 nanomolar inhibitors and reduction of sphingosine 1-phosphate in human leukemia cells. J. Med. Chem. 2011, 54, 3524–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli R.; Baraldi P. G.; Salvador M. K.; Preti D.; Aghazadeh Tabrizi M.; Brancale A.; Fu X. H.; Li J.; Zhang S. Z.; Hamel E.; Bortolozzi R.; Porcù E.; Basso G.; Viola G. Discovery and optimization of a series of 2-aryl-4-amino-5-(3′,4′,5′-trimethoxybenzoyl)thiazoles as novel anticancer agents. J. Med. Chem. 2012, 55, 5433–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing T. D.; Metz D. P.; Whittington D. A.; McGee L. R. PI3Kδ and PI3Kγ as Targets for Autoimmune and inflammatory diseases. J. Med. Chem. 2012, 55, 8559–8581. [DOI] [PubMed] [Google Scholar]
- Li W. W.; Wang X. Y.; Zheng R. L.; Yan H. X.; Cao Z. X.; Zhong L.; Wang Z. R.; Ji P.; Yang L. L.; Wang L. J.; Xu Y.; Liu J. J.; Yang J.; Zhang C. H.; Ma S.; Feng S.; Sun Q. Z.; Wei Y. Q.; Yang S. Y. Discovery of the novel potent and selective FLT3 inhibitor 1-{5-[7-(3- morpholinopropoxy)quinazolin-4-ylthio]-[1,3,4]thiadiazol-2-yl}-3-p-tolylurea and its anti-acute myeloid leukemia (AML) activities in vitro and in vivo. J. Med. Chem. 2012, 55, 3852–3866. [DOI] [PubMed] [Google Scholar]
- Schade A. E.; Schieven G. L.; Townsend R.; Jankowska A. M.; Susulic V.; Zhang R.; Szpurka H.; Maciejewski J. P. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008, 111, 1366–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Saha B.; Sawant D.; Kundu B. Synthesis of Novel N-Rich Polycyclic Skeletons Based on Azoles and Pyridines. J. Comb. Chem. 2007, 9, 783–792. [DOI] [PubMed] [Google Scholar]
- Chen L.; Yang S.; Jakoncic J.; Zhang J. J.; Huang X. Y. Migrastatin analogues target fascin to block tumour metastasis. Nature 2010, 464, 1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]; Erratum in:Nature 2011, 476, 240.
- Hashimoto Y.; Kim D. J.; Adams J. C. The roles of fascins in health and disease. J. Pathol. 2011, 224, 289–300. [DOI] [PubMed] [Google Scholar]
- Jayo A.; Parsons M. Fascin: A key regulator of cytoskeletal dynamics. Int. J. Biochem. Cell Biol. 2010, 42, 1614–1617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.