

Abstract

Enhanced activity of the Ser/Thr protein kinase, RSK, is associated with transformation and metastasis, which suggests that RSK is an attractive drug target. The natural product SL0101 (kaempferol 3-O-(3″,4″-di-O-acetyl-α-l-rhamnopyranoside)) has been shown to be an RSK selective inhibitor. However, the Ki for SL0101 is 1 μM with a half-life of less than 30 min in vivo. To identify analogues with improved efficacy we designed a set of analogues based on the crystallographic model of SL0101 in complex with the RSK2 N-terminal kinase domain. We identified an analogue with a 5″-n-propyl group on the rhamnose that has >40-fold improved affinity for RSK relative to SL0101 in an in vitro kinase assay. This analogue preferentially inhibited the proliferation of the human breast cancer line, MCF-7, versus the normal untransformed breast line, MCF-10A, which is consistent with results using SL0101. However, the efficacy of the 5″-n-propyl analogue to inhibit MCF-7 proliferation was only 2-fold better than for SL0101, which we hypothesize is due to limited membrane permeability. The improved affinity of the 5″-n-propyl analogue for RSK will aid in the design of future compounds for in vivo use.
Keywords: Kaempferol 3-O-(3″,4″-di-O-acetyl-α-l-rhamnopyranoside); SL0101; RSK; p90 ribosomal S6 kinase; kinase inhibitor
The family of Ser/Thr protein kinases, p90 ribosomal S6 kinase (RSK), has been implicated in numerous different cancers including breast, lung and prostate cancer.1−3 There are four RSK family members, and of these RSK1 and RSK2 promote metastasis. RSK is an unusual kinase in that it contains two nonidentical functional kinase domains, an N-terminal (NTKD) and a C-terminal (CTKD) kinase domain.4 The NTKD, which has a high sequence homology between family members, belongs to the AGC kinase family and is responsible for phosphorylation of target substrates. We previously found that SL0101 (1) (Figure 1), a kaempferol-α-l-(3″,4″)-diacetylrhamnoside, was a relatively specific inhibitor of the NTKD of RSK and did not inhibit the two most closely related kinases, p70 S6 kinase and mitogen- and stress-activated kinase.5 In in vitro kinase assays with ∼70 kinases, SL0101 (1) was found to partially inhibit Aurora B and PIM 3.6,7 However, these results are not straightforward to interpret because the relative kinase inhibition is dependent on the [ATP] in the assay. In both screens the [ATP] was higher in the RSK assay than with Aurora B and PIM 3, which would result in SL0101 (1) being less effective against RSK than in the assay conditions used for Aurora B and PIM 3.
Figure 1.
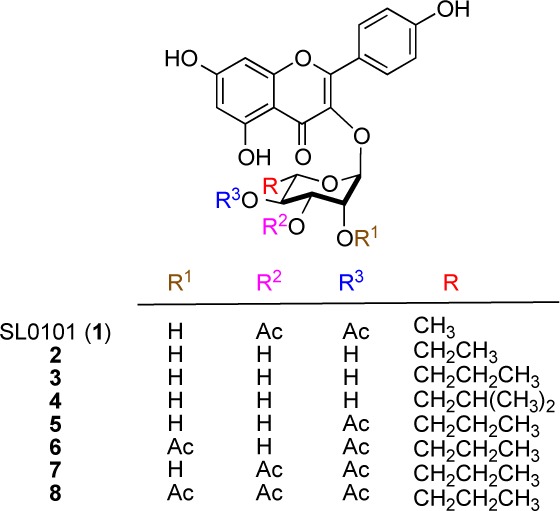
The RSK inhibitor, SL0101 (1), and analogues examined in this study.
An effective inhibitor of RSK in vivo would be invaluable in the study of RSK function in homeostasis and in disease states. To evaluate the suitability of SL0101 (1) for in vivo use we analyzed its pharmacokinetic behavior by intravenous (iv) and intraperitoneal (ip) administration of a single dose into male CD-1 mice. Because of the limited solubility of SL0101 (1) a carrier of 1:1:15 Cremophor:EtOH:phosphate-buffered saline was required. Regardless of the dosing method, the half-life (t1/2) of SL0101 (1) was <30 min (Table 1). More importantly, the maximum concentration achieved was ∼10-fold below that required to inhibit proliferation of the breast cancer cell line, MCF-7, in culture.5 Thus, SL0101 (1) is not suitable for in vivo testing and a medicinal chemistry effort is required to identify analogues with improved pharmacokinetic properties, as well as potency and stability.
Table 1. Pharmacokinetic Analysis of SL0101 (1) in Male CD-1 Micea.
| iv dose (mg/kg) | AUC/D (ng·h·kg/mL/mg) | C0 (ng/mL) | Cmax (ng/mL) | t1/2 (h) |
|---|---|---|---|---|
| 1 (iv) | 38.7 | 291 | 0.15 | |
| 2.5 (ip) | 1023 | 1851 | 0.40 |
AUC/D: area under the curve, extrapolated to infinity and normalized to the dose in mg/kg. C0 (ng/mL): maximum plasma concentration extrapolated to t = 0. Cmax (ng/mL): maximum plasma concentration. t1/2 (h): half-life.
The crystal structure of SL0101 (1) in complex with the RSK2 NTKD has been reported.8 The SL0101 (1) binding pocket overlaps with the ATP binding site of the NTKD but is distinct from it, as it is formed by a substantial structural rearrangement of the N-lobe of the kinase domain. The interaction of SL0101 (1) with RSK is partially stabilized by hydrogen bonds between the protein and phenolic hydroxyls on the kaempferol backbone in which the hydroxyl groups serve as hydrogen bond donors. Previously, we determined that loss of any of these hydroxyl groups substantially decreased the affinity of RSK for SL0101 (1).9 Furthermore, an analogue in which the hydroxyl groups were O-methylated and therefore could not donate hydrogen bonds did not inhibit RSK.9 Thus our previous experience with the SAR of SL0101 (1) analogues is in good agreement with the crystallographic model, which supports its use in designing future analogues.
The crystallographic model of the RSK2 NTKD in complex with SL0101 (1) indicates that the 5″-methyl of the rhamnose only partially fills a hydrophobic pocket.8 We modeled a set of analogues bearing longer aliphatic chains at the 5″ position using the docking function of the Molecular Operating Environment (MOE) program. The RSK2 NTKD in complex with SL0101 (1) was used as a starting point for the calculations. The kinase was processed and the analogues constructed in the binding pocket from the crystallized inhibitor SL0101 (1) using the build function. We performed a docking calculation using the “rigid receptor” presets on both SL0101 (1) and the analogues, and the highest-scoring binding pose as determined by the calculated binding free energy in each case was consistent with the crystal structure of the complex. The results clearly show that the longer aliphatic chains occupy a hydrophobic area in the binding pocket unoccupied by any fragment of SL0101 (1) (Figure 2). Thus, we envisioned a series of C-5″ substituted analogues of SL0101 (2–4, Figure 1). The acetyl groups on the rhamnose contribute substantially to the IC50 for RSK inhibition as deacetylated SL0101 is ∼10-fold less potent than SL0101 (1).9 Therefore, we also envisioned a series of analogues in which the number and positioning of the acetyl groups were varied in combination with the 5″-n-propyl group. Efficacy of SL0101 analogues in cell-based assays can be limited by their solubility10 so we selected the 5″-n-propyl analogue 3 as the parent compound based on its lower LogP compared to 4 (Table 2).
Figure 2.
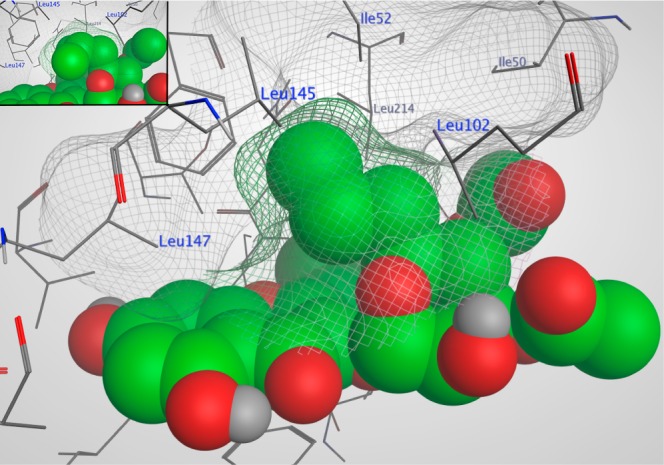
A hydrophobic pocket within the RSK2 NTKD allows for an extended 5″ aliphatic chain on the rhamnose. Molecular docking of analogues 3″,4″-diacetyl versions of 3 and 4 (inset) were performed using the X-ray crystallographic structure of SL0101 (1) in complex with RSK2 NTKD.8
Table 2. In Vitro Potency of SL0101 (1) and Analoguesa.
| RSK2 IC50 (μM) | LogP | p (1) | p (3) | |
|---|---|---|---|---|
| SL0101(1) | 0.99 (0.74–1.32) | 2.11 | ||
| 2 | 1.51 (0.45–5.09) | 1.64 | 0.917 | |
| 3 | 0.71 (0.47–1.09) | 2.20 | 0.211 | |
| 4 | 0.48 (0.17–1.33) | 2.41 | 0.073 | |
| 5 | 1.50 (0.90–2.48) | 2.90 | 0.753 | 0.183 |
| 6 | 0.62 (0.18–2.15) | 3.39 | 0.232 | 0.804 |
| 7 | 0.02 (0.01–0.04) | 3.17 | 0.007 | 0.005 |
| 8 | 1.77 (1.24–2.53) | 3.87 | 0.008 | 0.008 |
IC50: concentration needed for 50% inhibition of RSK2; the 95% confidence interval (CI) is shown in parentheses; n > 2 in quadruplicate. p(1): Student’s t test compared to SL0101 (1). p(3): Student’s t test compared to analogue 3.
Previously, we reported both a traditional carbohydrate11 and a de novo asymmetric approach12,13 to SL0101 (1)14 and several carbohydrate analogues (1, 1a–d, Scheme 1).15 While the carbohydrate approach has some real advantages in terms of convergency, the de novo approach has the advantage of being amenable to the divergent late stage substitution of the pyranone ring.16−20 Because of its inherent ability to incorporate C-5″ substitution, we decided to further develop the de novo approach to pursue the synthesis and evaluation of the rhamno-sugar of SL0101 analogues 2–8.
Scheme 1. De Novo Approach to SL0101 (1).
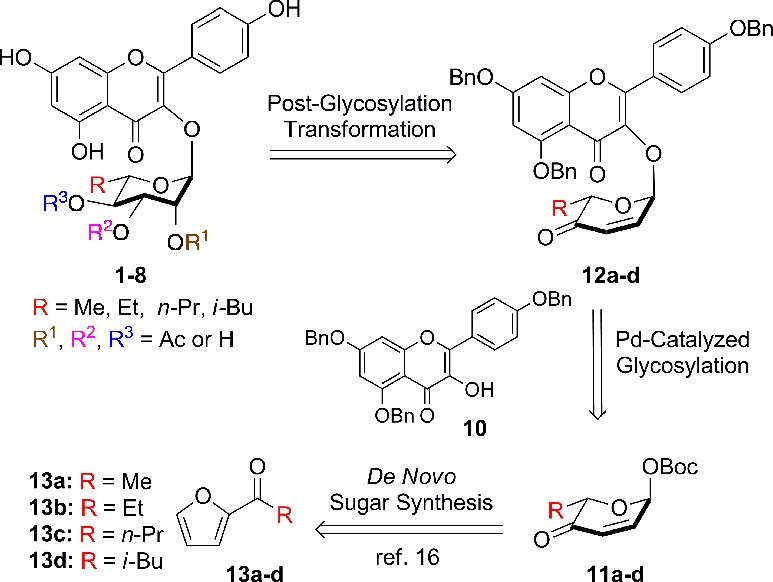
Based on our previous experience with SL0101 (1), we envisioned the desired target molecules 2–8 as being derived from the appropriately substituted pyranones 12a–d (Scheme 2). Key to the overall efficiency of this approach is that the enone served as precursor for the installation of the desired triol functionality in 2–8 for further acylation. Similarly, pyranones 12a–d could be prepared from a palladium-catalyzed glycosylation of aglycon 10 with Boc-pyranones 11a–d serving as the glycosyl donor.21,22 Finally, pyranones 11a–d could be derived from furans 13a–d.23,24 To accomplish this divergent synthetic effort we planned on preparing all the desired target molecules 2–8 by executing multistep parallel reactions on three key intermediates (14b–d, vide infra).
Scheme 2. Retrosynthetic Analysis of SL0101 (1) Analogues.
With the desired coupling partners in hand (10 and 11b–d), we first pursued their coupling and diastereoselective transformation into three structurally divergent allylic alcohols 14b–d. Exposure of 11b–d and 10 to our typical Pd-catalyzed glycosylation procedure (2.5 mol % Pd2(DBA)3·CHCl3 and 10 mol % of PPh3) gave the coupling products 12b–d with complete transfer of anomeric stereochemistry (Scheme 3). A subsequent Luche reduction25−27 (NaBH4 at −78 °C) diastereoselectively converted the pyranones 12b–d into the desired allylic alcohols 14b–d (dr >20:1).
Scheme 3. Synthesis of SL0101 Analogues 2–8.
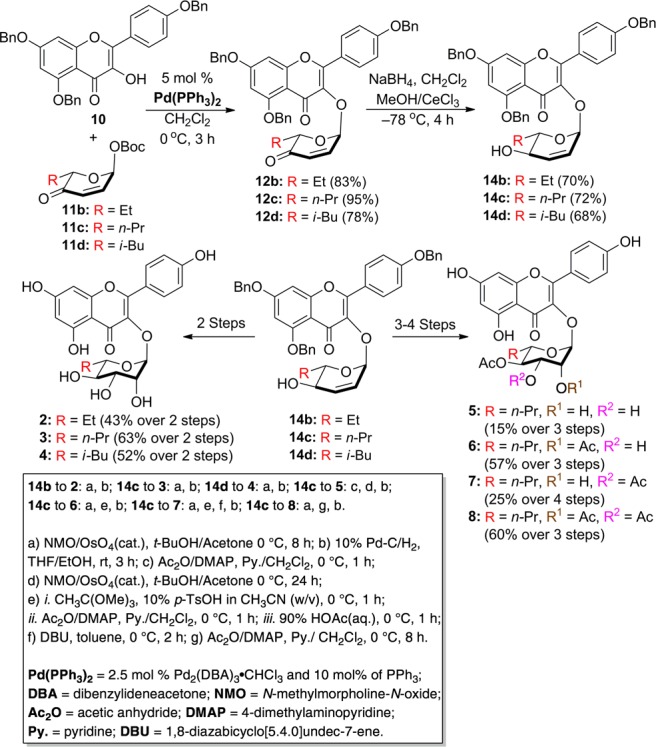
We next pursued the direct conversion of allylic alcohols 14b–d into the desired triols 2–4 with rhamno-stereochemistry. This was accomplished in parallel by exposing 14b–d to typical Upjohn dihydroxylation conditions (NMO/OsO4 in t-BuOH/acetone).28 The crude products from the dihydroxylation reactions were filtered through a pad of silica gel (to remove osmium, N-methylmorpholine and its N-oxide (NMO)) and subjected to typical hydrogenolysis conditions (10% Pd/C, 1 atm of H2). To our delight, the three desired SL0101 (1) analogues 2–4 could be isolated in pure form after careful silica gel chromatography (43–63% yields).
Buoyed by the success, we next pursued the direct conversion of 14c into 5–8, by incorporating selective acylation steps to the previous 2-step sequence. For instance, the allylic alcohol 14c, with an n-Pr-group, was cleanly converted to the C-4 acetylated rhamno-diol 5 (i.e., acylation/dihydroxylation/hydrogenolysis) in 15% yield after silica gel chromatography. By simply switching the order of acylation and dihydroxylation, the triacetate 8 was similarly prepared in a 60% overall yield. In contrast, the synthesis of diacetates 6 and 7 required a selective diacylation using the orthoester/acylation/hydrolysis/hydrogenolysis protocol to give 6 (57% overall yield). Finally, by incorporating an isomerization step into the sequence (i.e., orthoester/acylation/isomerization/hydrogenolysis) C-3/C-4 diacetate 7 could be prepared in a 25% overall yield.
The affinities of the C-5″ substituted analogues for RSK were determined by their ability to inhibit the activity of purified, recombinant RSK2 in an in vitro kinase assay.5 The data were fit using nonlinear regression analysis. There was a trend toward improving the in vitro potency by increasing the chain length, which is consistent with the modeling results indicating that longer chains would be preferred, although the differences did not obtain statistical significance (Table 2). We have reported a lower IC50 for the synthesized SL0101 (1).29 However, the IC50 is relative and dependent on numerous variables including the batch of purified, recombinant RSK2. Therefore, to accurately evaluate the relative potencies of the various analogues each assay was performed in parallel with SL0101 (1).
In the in vitro kinase assay, introduction of a single acetyl group as in analogue 5 increased the IC50, but this difference was not statistically significant (Table 2). Acetyl groups on the 2″, 3″ and 4″ positions (analogue 8) doubled the IC50 in comparison to the 5″-n-propyl (analogue 3) (Table 2). Acetyl groups on the 2″ and 4″ positions (analogue 6) did not alter the IC50. Surprisingly, analogue 7 with acetyl groups on the 3″ and 4″ positions has an IC50 >40-fold lower than SL0101 (1) (Table 2). These results were unexpected because, although previously we had found acetylation to be important, the number or positioning of the acetyl groups was not.9 In the crystal structure of SL0101 (1) in complex with the RSK2 NTKD the acetyl groups are unresolved.8 However, we speculate that the favorable van der Waals interactions between the 5″-n-propyl group and hydrophobic residues in the binding pocket restrict the binding orientation of this set of analogues in such a way that only acetyl groups in the 3″ and 4″ positions can be accommodated without causing unfavorable interactions in the binding pocket.
Analogue 7 was further evaluated for its ability to inhibit the proliferation of the breast cancer cell line MCF-7, in comparison to SL0101 (1). The aqueous solubility of 7 limited the concentrations that could be tested to ≤25 μM. At 25 μM compound 7 was ∼50% more potent at inhibiting proliferation than SL0101(1) (Figure 3). To determine whether analogue 7 was specific for RSK we compared the ability of the compound to inhibit proliferation of MCF-7 versus the immortalized, normal breast line, MCF-10A. We have previously found that a preferential ability to inhibit MCF-7 compared to MCF-10A proliferation indicates specific inhibition of RSK.5,9,10,29 We observed that compound 7 inhibits MCF-7 but not MCF-10A proliferation (Figure 4). These results suggest that compound 7 and SL0101 (1) have similar specificities.
Figure 3.

Efficacy of SL0101 (1) and 7 in MCF-7 cells. Various concentrations of inhibitors were added at time 0, and ATP content was measured after 48 h of treatment. Values are the fold proliferation as a percentage of that obtained with vehicle-treated cells (n = 3 in quadruplicate; bars = SD; *p < 0.05 in a Student’s t test compared to the vehicle. ∧p < 0.05 in a Student’s t test compared to SL0101 (1)).
Figure 4.

Specificity of analogue 7 for inhibition of RSK activity. Various concentrations of 7 were added to MCF-7 or MCF-10A cells, and the assay was performed as described in Figure 3 (n = 3 in quadruplicate; bars = SD; *p < 0.05 in a Student’s t test compared to the vehicle).
To further examine the specificity of analogue 7 we compared the ability of SL0101 (1) and compound 7 to alter the phosphorylation of eukaryotic elongation factor 2 (eEF2). Inhibition of RSK is known to activate EF2 kinase, which phosphorylates eEF2.30 MCF-7 cells were pretreated with inhibitor or vehicle and then stimulated with the mitogen, phorbol myristate acetate (PMA). In the presence of PMA, peEF2 levels decreased as expected as RSK is active and inhibits EF2 kinase. Treatment with SL0101 (1) and compound 7 both increased peEF2 levels compared to the PMA control (Figure 5), which is consistent with inhibition of RSK. We have also found that the levels of the oncogene, cyclin D1, are dependent on RSK activity in MCF-7 cells.31 Consistent with these data SL0101 (1) and compound 7 decreased cyclin D1 levels. As a further comparison of the inhibitory profiles of SL0101 (1) and analogue 7, we immunoblotted the lysates with an antibody that recognizes the (Lys/Arg)X(Lys/Arg)XX(pSer/pThr) motif, where X is any amino acid.4 This motif is recognized by a number of kinases including RSK. Treatment with the inhibitors resulted in a decrease in the intensity of a band at ∼27 kDa. It is not surprising that we only observed a decrease in a single band as PMA is a potent mitogen that will activate many kinases. The intensities of other bands in the immunoblot were similar between SL0101 (1) and analogue 7. Taken together, these results indicate that SL0101 (1) and compound 7 have similar specificities.
Figure 5.
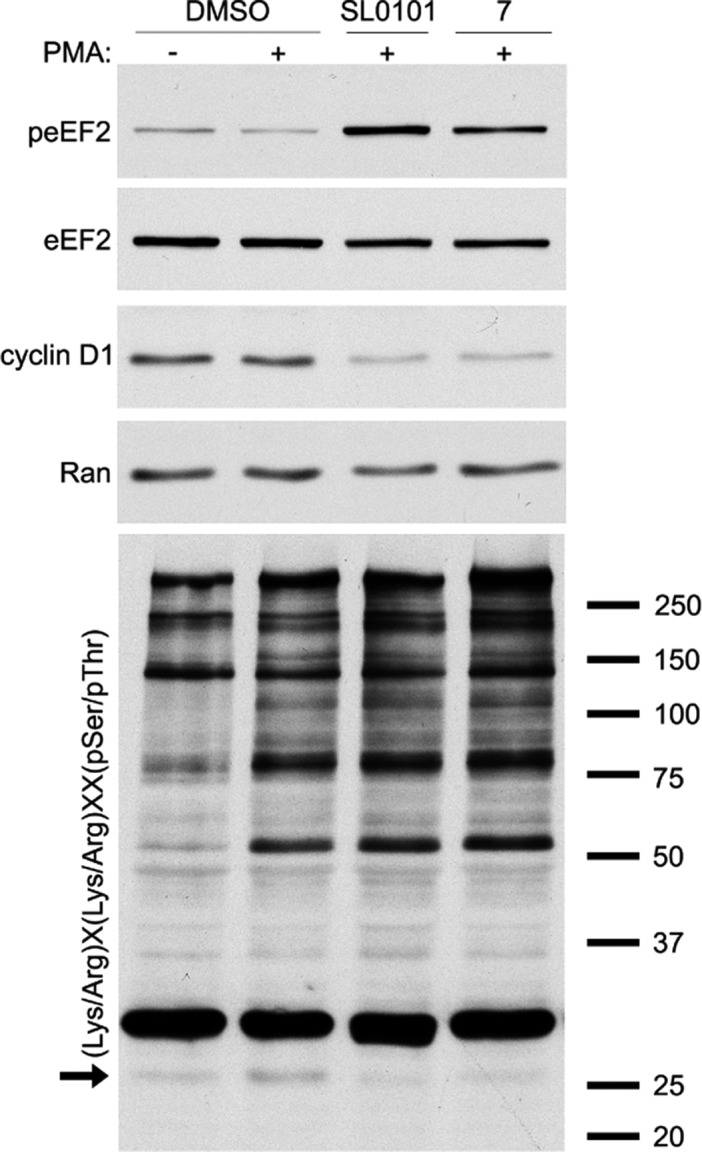
Comparison of compound 7 and SL0101 (1) on altering RSK biomarkers in intact cells. Lysates of MCF-7 cells that were pretreated with inhibitor (SL0101 (100 μM); 7 (25 μM)) and then treated with vehicle (DMSO) or PMA were analyzed by immunoblotting. The arrow indicates a band whose intensity decreases upon treatment of cells with SL0101 (1) and analogue 7.
In summary, we used structure-based design to identify new analogues that improve on the in vitro potency of SL0101 (1). A 5″-n-propyl substituent in combination with 3″- and 4″-acetyl groups (7) on the rhamnose improved the in vitro affinity for RSK by >40-fold compared to SL0101 (1). Analogue 7 specifically inhibits RSK, but its ability to inhibit the proliferation of the breast cancer cell line, MCF-7, is only 2-fold better compared to SL0101 (1), which we hypothesize is due to limited membrane permeability. These studies will provide further guidance in designing a potent SL0101 (1) analogue that can be used in vivo.
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ These authors contributed equally.
This work was supported by the Department of Defense (#W81XWH-11-1-0068 to M.K.H.), NIH (GM084386 to D.A.L. and GM090259 to G.A.O.), and NSF (CHE-1213596).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Eisinger-Mathason T. S.; Andrade J.; Lannigan D. A. RSK in tumorigenesis: connections to steroid signaling. Steroids 2010, 753191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark D. E.; Errington T. M.; Smith J. A.; Frierson H. F. Jr.; Weber M. J.; Lannigan D. A. The serine/threonine protein kinase, p90 ribosomal S6 kinase, is an important regulator of prostate cancer cell proliferation. Cancer Res. 2005, 6583108–3116. [DOI] [PubMed] [Google Scholar]
- Lara R.; Mauri F. A.; Taylor H.; Derua R.; Shia A.; Gray C.; Nicols A.; Shiner R. J.; Schofield E.; Bates P. A.; Waelkens E.; Dallman M.; Lamb J.; Zicha D.; Downward J.; Seckl M. J.; Pardo O. E. An siRNA screen identifies RSK1 as a key modulator of lung cancer metastasis. Oncogene 2011, 30323513–3521. [DOI] [PubMed] [Google Scholar]
- Anjum R.; Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 2008, 910747–758. [DOI] [PubMed] [Google Scholar]
- Smith J. A.; Poteet-Smith C. E.; Xu Y.; Errington T. M.; Hecht S. M.; Lannigan D. A. Identification of the first specific inhibitor of p90 ribosomal S6 kinase (RSK) reveals an unexpected role for RSK in cancer cell proliferation. Cancer Res. 2005, 6531027–1034. [PubMed] [Google Scholar]
- Doehn U.; Hauge C.; Frank S. R.; Jensen C. J.; Duda K.; Nielsen J. V.; Cohen M. S.; Johansen J. V.; Winther B. R.; Lund L. R.; Winther O.; Taunton J.; Hansen S. H.; Frodin M. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 2009, 354511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain J.; Plater L.; Elliott M.; Shpiro N.; Hastie C. J.; McLauchlan H.; Klevernic I.; Arthur J. S.; Alessi D. R.; Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 4083297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utepbergenov D.; Derewenda U.; Olekhnovich N.; Szukalska G.; Banerjee B.; Hilinski M. K.; Lannigan D. A.; Stukenberg P. T.; Derewenda Z. S. Insights into the inhibition of the p90 ribosomal S6 kinase (RSK) by the flavonol glycoside SL0101 from the 1.5 Å crystal structure of the N-terminal domain of RSK2 with bound inhibitor. Biochemistry 2012, 51336499–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. A.; Maloney D. J.; Hecht S. M.; Lannigan D. A. Structural basis for the activity of the RSK-specific inhibitor, SL0101. Bioorg. Med. Chem. 2007, 15145018–5034. [DOI] [PubMed] [Google Scholar]
- Hilinski M. K.; Mrozowski R. M.; Clark D. E.; Lannigan D. A. Analogs of the RSK inhibitor SL0101: optimization of in vitro biological stability. Bioorg. Med. Chem. Lett. 2012, 2293244–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney D. J.; Hecht S. M. Synthesis of a potent and selective inhibitor of p90 RSK. Org. Lett. 2005, 761097–1099. [DOI] [PubMed] [Google Scholar]
- Shan M.; Sharif E. U.; O’Doherty G. A. Total synthesis of jadomycin A and a carbasugar analogue of jadomycin B. Angew. Chem., Int. Ed. 2010, 49499492–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisova S. A.; Guppi S. R.; Kim H. J.; Wu B.; Penn J. H.; Liu H. W.; O’Doherty G. A. A de novo approach to the synthesis of glycosylated methymycin analogues with structural and stereochemical diversity. Org. Lett. 2010, 12225150–5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; O’Doherty G. A. De novo asymmetric syntheses of SL0101 and its analogues via a palladium-catalyzed glycosylation. Org. Lett. 2006, 8225149–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan M.; O’Doherty G. A. Synthesis of SL0101 carbasugar analogues: carbasugars via Pd-catalyzed cyclitolization and post-cyclitolization transformations. Org. Lett. 2010, 12132986–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y.; Wu B.; Qi Z.; Kang S. W.; Rojanasakul Y.; O’Doherty G. A. C5′-alkyl substitution effects on digitoxigenin α-L-glycoside cancer cytotoxicity. ACS Med. Chem. Lett. 2011, 24259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y.; Rojanasakul Y.; O’Doherty G. A. Synthesis and evaluation of the α-D-/α-L-rhamnosyl and amicetosyl digitoxigenin oligomers as anti-tumor agents. ACS Med. Chem. Lett. 2011, 24264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y.; Xin W.; Zhou M.; Stueckle T. A.; Rojanasakul Y.; O’Doherty G. A. Stereochemical survey of digitoxin monosaccharides: new anticancer analogues with enhanced apoptotic activity and growth inhibitory effect on human non-small cell lung cancer cell. ACS Med. Chem. Lett. 2011, 2173–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A. K.; Zhou M.; Azad N.; Elbaz H.; Wang L.; Rogalsky D. K.; Rojanasakul Y.; O’Doherty G. A.; Langenhan J. M. A direct comparison of the anticancer activities of digitoxin MeON-neoglycosides and O-glycosides: oligosaccharide chain length-dependent induction of caspase-9-mediated apoptosis. ACS Med. Chem. Lett. 2010, 17326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; O’Doherty G. The de novo synthesis of oligosaccharides: application to the medicinal chemistry SAR-study of digitoxin. Curr. Top. Med. Chem. 2008, 82114–125. [DOI] [PubMed] [Google Scholar]
- Babu R. S.; O’Doherty G. A. Palladium-catalyzed glycosylation reaction: de-novo synthesis of trehalose analogues. J. Carbohydr. Chem. 2005, 242169–177. [Google Scholar]
- Wu B.; Li M.; O’Doherty G. A. Synthesis of several cleistrioside and cleistetroside natural products via a divergent de novo asymmetric approach. Org. Lett. 2010, 12235466–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. M.; Keranen M. D.; Nguyen H.; Young V. G.; O’Doherty G. A. Syntheses of four D- and L-hexoses via diastereoselective and enantioselective dihydroxylation reactions. Carbohydr. Res. 2000, 328117–36. [DOI] [PubMed] [Google Scholar]
- Li M.; Scott J. G.; O’Doherty G. A. Synthesis of 7-oxa-phomopsolide E and its C-4 epimer. Tetrahedron Lett. 2004, 4551005–1009. [Google Scholar]
- Luche J. L. Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J. Am. Chem. Soc. 1978, 10072226–2227. [Google Scholar]
- Luche J.-L.; Rodriguez-Hahn L.; Crabbe P. Reduction of natural enones in the presence of cerium trichloride. J. Chem. Soc., Chem. Commun. 1978, 14, 601–602. [Google Scholar]
- Gemal A. L.; Luche J. L. Lanthanoids in organic synthesis. 6. Reduction of α-enones by sodium borohydride in the presence of lanthanoid chlorides: synthetic and mechanistic aspects. J. Am. Chem. Soc. 1981, 103185454–5459. [Google Scholar]
- VanRheenen V.; Kelly R. C.; Cha D. Y. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 17231973–1976. [Google Scholar]
- Smith J. A.; Maloney D. J.; Clark D. E.; Xu Y.; Hecht S. M.; Lannigan D. A. Influence of rhamnose substituents on the potency of SL0101, an inhibitor of the Ser/Thr kinase, RSK. Bioorg. Med. Chem. 2006, 14176034–6042. [DOI] [PubMed] [Google Scholar]
- Wang X.; Li W.; Williams M.; Terada N.; Alessi D. R.; Proud C. G. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. EMBO J. 2001, 20164370–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisinger-Mathason T. S.; Andrade J.; Groehler A. L.; Clark D. E.; Muratore-Schroeder T. L.; Pasic L.; Smith J. A.; Shabanowitz J.; Hunt D. F.; Macara I. G.; Lannigan D. A. Codependent functions of RSK2 and the apoptosis-promoting factor TIA-1 in stress granule assembly and cell survival. Mol. Cell 2008, 315722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.