

Abstract
Through pattern recognition receptors the innate immune system detects disruption of the normal function of the organism and initiates responses directed at correcting these derangements. Cellular damage from microbial or non-microbial insults causes the activation of nucleotide-binding domain leucine-rich repeat containing receptors (NLR) in multiprotein complexes called inflammasomes. Here we discuss the role of the NLRP3 inflammasome in the recognition of cellular damage and the initiation of sterile inflammatory responses.
Keywords: caspase-1, necrosis, danger signals
The NLR family
The innate immune system possesses multiple families of germline encoded pattern recognition receptors (PRRs) [1]. These include Toll-like receptors (TLRs), nucleotide-binding domain leucine-rich repeat containing receptors (NLRs), RIG-I-like RNA helicases (RLHs) and C-type lectin receptors (CLRs). These receptors recognize conserved moieties associated with either cellular damage (DAMPs; danger associated molecular patterns) or invading organisms (PAMPs; pathogen associated molecular patterns). Activation of these receptors ultimately leads to the production of cytokines that drive the inflammatory response.
The NLR family of molecules is a recently described group of intracellular receptors with a unique domain architecture consisting of a central nucleotide-binding domain called the NACHT domain that is located between an N-terminal effector domain and a C-terminal LRR (Leucine-rich repeat) domain [2, 3]. The LRR domain serves an autoregulatory role for NLR activation and has been implicated in ligand sensing; however, the mechanism and ligands involved in this interaction remain unknown. The N-terminal domain is either a caspase-recruitment domain (CARD), pyrin domain (PYD) or Baculovirus IAP repeat domain (BIR); the individual domain dictates to which NLR subfamily a receptor belongs and the domain recruits adaptor and effector proteins to the NLR to drive downstream signal transduction.
Activation of NLRP3 results in the formation of a multiprotein complex termed the NLRP3 inflammasome composed of NLRP3, the adaptor molecule ASC and the cysteine protease caspase-1 (Figure 1) [4, 5]. This association and resultant activation of the inflammasome leads to the activation of caspase-1 from its inactive zymogen pro-caspase-1. Active caspase-1 cleaves the pro-forms of the cytokines IL-1β and IL-18 to their active and secreted forms. Caspase-1 may possess additional functions including regulation of glycolysis pathways [6] and unconventional protein secretion [7]; however, in vivo studies demonstrating a role for NLRP3 in these processes are lacking to date.
Figure 1. Model for NLRP3 inflammasome activation by microbes and danger signals.

Activation of the NLRP3 inflammasome results in activation of caspase-1 and the resultant processing of pro-IL-1β into biologically active IL-1β. A wide variety of stimuli including bacterial pore-forming toxins and particulate activators such as silica, asbestos, uric acid, alum and amyloid-β can activate the NLRP3 inflammasome. NLRP3 activating PAMPs and DAMPs induce a K+ efflux and the generation of mitochondrial-derived ROS that play a role in NLRP3 inflammasome activation by an unknown mechanism. Lysosomal damage following crystalline particulate internalization may also influence NLRP3 inflammasome activation through an unknown mechanism that is inhibited by the cathepsin B inhibitor Ca-074-me.
In addition to NLRP3, two other NLR family members have been demonstrated to form inflammasomes and activate caspase-1. The NLRP1 inflammasome is a key mediator of cell death due to anthrax lethal toxin [8] and the NLRC4 inflammasome is activated by numerous Gram-negative bacteria possessing either a type III or type IV secretion system [9–11]. NLRC4 may also interact with another cytosolic NLR, Naip5 to activate caspase-1 in response to cytosolic flagellin [12]. Recent studies have also demonstrated that the cytosolic nucleic acid recognition receptors AIM2 and RIG-I can interact with ASC to form caspase-1 activating inflammasomes [13–17].
NLRP3 in disease pathogenesis
The NLRP3 inflammasome can be activated in response to a wide array of stimuli (Figure 1). These activators lack structural or functional similarity making it unlikely that their activation is through direct interaction with NLRP3. Rather, a common endogenous molecule upon which these pathways converge is likely the actual ligand for NLRP3. Numerous microbes including various bacteria, viruses, fungi, and protozoan parasites can activate the NLRP3 inflammasome (reviewed in [18]). In addition to microbial activators, endogenous danger signals such as ATP, monosodium urate (MSU) and amyloid-β have been demonstrated to activate the NLRP3 inflammasome. It is interesting to speculate that NLRP3, or its evolutionary ancestor, originally served a primary role in host defense against pathogens. But rather than sensing specific conserved PAMPs as the TLRs do, it is capable of detecting a wide swath of divergent pathogens by detecting one of the major consequences of infection, namely, cellular damage. Sequencing of the sea urchin Strongylocentrotus purpuratus genome revealed 222 TLRs and 203 NLRs, demonstrating the importance of these innate immune receptors in lower species such as the echinoderms [19]. As species evolved and vertebrates developed adaptive immune systems some of these early innate NLRs involved in pathogen surveillance have likely been co-opted to serve other functions such as responding to metabolic stress, ischemia and trauma. Recent studies suggest that the NLRP3 inflammasome may play a significant role in metabolic disorders and sterile inflammatory responses including type II diabetes mellitus, gout, Alzheimer’s disease and ischemia [6, 20–23].
Sterile particulate activators of the NLRP3 inflammasome
A number of endogenous and exogenous crystalline molecules have been shown to activate the NLRP3 inflammasome (reviewed in [2]). Uric acid crystals and calcium pyrophosphate dihydrate, the causative agents of gout and pseudogout respectively, were the first crystalline molecules shown to activate the NLRP3 inflammasome [21]. Another endogenous molecule, fibrillar amyloid-β, associated with the pathogenesis of Alzheimer’s disease, also activates the NLRP3 inflammasome in a similar manner [20]. Silica and asbestos particles, which cause the fibrotic lung disorders silicosis and asbestosis respectively, also have been demonstrated to activate the NLRP3 inflammasome [24–26]. Additionally, the adjuvant properties of aluminum hydroxide (alum) have been shown to be dependent upon its ability to activate the NLRP3 inflammasome [27–30].
The mechanism by which the NLRP3 inflammasome is activated remains unknown. However, two events that are common to all activators of the NLRP3 inflammasome are a potassium efflux and the generation of reactive oxygen species (ROS) (Figure 1). Inhibiting the potassium efflux, by increasing extracellular potassium concentrations, results in the abrogation of NLRP3 inflammasome activation [24, 25, 27]. The exact role of the potassium efflux is unclear; however the assembly of the NLRP3 inflammasome may be dependent on a low potassium environment [31]. Similarly, inhibition or scavenging of ROS blocks NLRP3 inflammasome activation (reviewed in [32]). Lysosomal membrane disruption following particulate uptake has also been postulated to play a role in NLRP3 inflammasome activation and is reviewed in detail in this issue by Hornung and Latz [33].
NLRP3 as a sensor of necrotic cell death
Necrotic cells release endogenous DAMPs that alert the innate immune system to tissue damage. Release of ATP from the necrotic cells is a danger signal that activates the innate immune response. ATP binds the purinergic receptor P2X7 triggering the formation of a pannexin-1 hemichannel which results in the activation of the NLRP3 inflammasome [34–36]. The ability of necrotic cells to activate the NLRP3 inflammasome (Figure 2) was recently demonstrated in two independent studies [22, 37]. Iyer et al. showed that macrophages challenged with cells that had undergone specific forms of necrotic cell death (pressure-disruption, complement lysis, hypoxic injury) were capable of activating caspase-1 in an NLRP3-dependent manner [22]. However not all methods of necrosis were capable of activating NLRP3; necrotic cells generated by freeze-thaw or UV irradiation failed to activate caspase-1, highlighting the heterogeneity of different mechanisms of necrotic cell death. The ability of NLRP3 to sense cellular damage could also be seen in an in vivo model of renal ischemic acute tubular necrosis [22]. Both wild-type and NLRP3-deficient mice that were subjected to renal ischemia/reperfusion injury displayed similar acute tubular necrosis following injury. However, the subsequent inflammatory response to this necrotic injury was markedly blunted in mice that lacked NLRP3. This resulted in reduced mortality and preserved renal function in NLRP3-deficeint mice compared to their wild-type counterparts.
Figure 2. Model for NLRP3 inflammasome activation by necrotic cells.
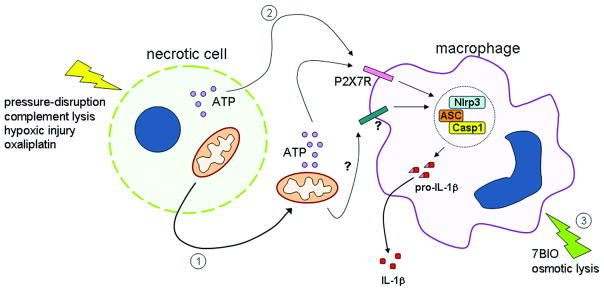
Specific types of cellular damage, such as pressure-disruption or complement lysis, hypoxic injury or treatment with chemotherapeutic agents such as oxaliplatin, result in the release of actively respiring mitochondria (1) or cellular ATP (2). ATP generated by the mitochondria or ATP released from the necrotic cell is capable of activating the NLRP3 inflammasome in macrophages via the P2X7 receptor. Additional unidentified mitochondrial factors may also activate the NLRP3 inflammasome. Cellular damage caused by 7-bromoindirubin-3′-oxime (7BIO) or osmotic lysis results in activation of the NLRP3 inflammasome within the cell undergoing cell death (3). Activation of the NLRP3 inflammasome results in caspase-1 activation followed by processing and secretion of the proinflammatory cytokine IL-1β.
Imaeda and colleagues demonstrated that the mortality associated with acetaminophen-induced hepatotoxicity was partially dependent on NLRP3 [38]. Mice deficient in components of the NLRP3 inflammasome were protected from the lethal effects of acetaminophen-induced hepatotoxicity in vivo and had reduced liver injury compared to wild type mice. Although not directly examined in this study, it is likely that acetaminophen-induced necrosis of hepatocytes, similar to necrosis induced by pressure-disruption and complement, activates the NLRP3 inflammasome in macrophages that encounter these necrotic cells with resultant activation of caspase-1 and processing and secretion of IL-1β. Interestingly, DNA released from damaged hepatocytes was found to stimulate the production of pro-IL-1β and pro-IL-18 through stimulation of TLR9 [38]. This raises the possibility that cytosolic nucleic acid sensors such as RIG-I and AIM2 may also play a role in sterile inflammatory responses to necrotic cell death. In addition, NLRP3 has also been shown to be activated in response to cytoplasmic DNA [39] which may possibly be driving NLRP3 inflammasome activation in response to acetaminophen-induced hepatotoxicity.
Tumor cell death induced by certain chemotherapeutic agents such as anthracyclines and oxaliplatin elicit an immunogenic response that is required for tumor eradication. Ghiringhelli and colleagues found that oxaliplatin treated tumor cells were capable of activating the NLRP3 inflammasome in dendritic cells resulting in the secretion of IL-1β [37]. Importantly, the priming of IFNγ-producing CD8+ T cells by dying tumor cells was also dependent on the NLRP3 inflammasome. The importance of NLRP3 in mediating the adjuvant effects of alum and uric acid has parallels to these new findings that necrotic cells mediate their immunogenicity through NLRP3. Ghiringhelli et al. also found that tumors established in mice deficient in components of the NLRP3-inflammasome had poorer responses to oxaliplatin compared to wild type mice [37].
Both Iyer et al. and Ghiringhelli et al. demonstrated that ATP released from the necrotic cells was responsible for activation of the NLRP3 inflammasome via the P2X7 receptor [22, 37]. Importantly, uric acid, another DAMP that has been postulated to play a role in responses to necrotic cells, was not involved in the ability for necrotic cells to activate the NLRP3 inflammasome. The half-life of extracellular ATP is brief due to efficient degradation by ectoenzymes. Hence, preformed ATP released from the dying cell is likely sensed in close proximity to the necrotic insult. Additionally, we found actively respiring mitochondria are released from necrotic cells and generate ATP that activates the NLRP3 inflammasome, but also importantly this allows the ATP to be carried further from the site of initial insult [22] (Figure 2). Although these findings demonstrate an important role for ATP and the P2X7R in mediating the activation of the NLRP3 inflammasome in response to necrotic cells, it is likely that additional factors released from necrotic cells can also independently activate the NLRP3 inflammasome. This is supported by findings that IL-1β secretion in response to necrotic cells is not completely abrogated in P2X7R-deficeint macrophages and dendritic cells [22, 37]. We also found that unlike NLRP3−/− mice, P2X7R−/− mice retain a neutrophilic influx when challenged with pressure-disrupted necrotic cells intraperitoneally suggesting an NLRP3-dependent inflammatory response independent of P2X7R [22]. In contrast, however, oxaliplatin-treated tumor cells failed to prime T cells for IFNγ production in P2X7R−/− mice [37]. In addition, tumors in P2X7R−/− mice were less responsive to oxaliplatin compared to wild type mice. The reason for the discrepancy for the in vivo requirement of the P2X7R−/− in these two studies is unclear. It is possible that, although the immunogenicity of necrotic cells is predominantly dependent on the P2X7R, the residual IL-1β that is made in the absence of the P2X7R in response to necrotic cells is sufficient to induce neutrophil infiltration to the site of injury. The nature of these factors from necrotic cells that activate NLRP3 independently of the P2X7R remains to be elucidated; action through other purinergic receptors is one strong possibility.
It is established that activation of the NLRP3 inflammasome is a two-step process with the initial priming step delivered by NF-κB activation which also drives pro-IL-1β generation (reviewed in [33]). Generally in vitro studies have provided priming via microbial products acting on TLRs. The initial priming step in vivo has been unclear especially for non-microbial activators of the NLRP3 inflammasome. The recent studies by Iyer et al. [22] and Ghiringhelli et al. [37] show that endogenous DAMPs released concomitantly with cellular injury prime macrophages and dendritic cells for inflammasome activation. This functionality was confirmed by in vitro studies wherein HMGB-1, biglycan and hyaluronic acid were each capable of priming NLRP3 inflammasome activation in response to necrotic cells. The in vivo significance of these studies is underlined as both biglycan and hyaluronic acid expression are upregulated following renal ischemia-reperfusion injury. Consistent with this is the finding that mice deficient in either TLR2 or TLR4, the receptors through which biglycan and hyaluronic acid can activate macrophages [40, 41], have improved outcomes following renal ischemia-reperfusion injury [42–44]. Mice deficient in another cellular receptor for hyaluronic acid, CD44, also display reduced renal injury following ischemia-reperfusion injury [45].
In addition to their role in priming for inflammasome activation, biglycan and hyaluronic acid have themselves been shown to activate the NLRP3 inflammasome. Biglycan is proposed to prime macrophages through TLR2 and TLR4 and then activate the inflammasome by its interaction with the P2X4 and P2X7 receptors [46]. Hyaluronic acid’s ability to activate the NLRP3 inflammasome was dependent on CD44 [47]. Further studies will be required to delineate the contribution of individual endogenous DAMPs in the priming vs. activation of the NLRP3 inflammasome.
Cell disruption leads to endogenous NLRP3 inflammasome activation
Necrosis can also lead to the activation of the NLRP3 inflammasome within the cell undergoing necrosis if components for the inflammasome are present (Figure 2). Following cellular disruption the inflammasome can spontaneously form and acquire the ability to process pro-IL-1β into its mature form [5, 31]. The restoration of potassium, to levels approximating that found within the cytosol of normal cells inhibits this spontaneous inflammasome formation. This suggests the low potassium environment created by potassium efflux from the cell is the requirement for the assembly of the components of the NLRP3 inflammasome [31]. Li and colleagues identified an indirubin oxime derivative, 7-bromoindirubin-3′-oxime (7BIO) that was capable of inducing necrosis with the concurrent activation of the NLRP3 inflammasome [48]. Unlike the sensing of necrotic cells by macrophages and dendritic cells, 7BIO-induced caspase-1 activation was independent of ATP and the P2X7R.
Therapeutic possibilities
Taken together these results have a number of therapeutic implications. Inhibiting NLRP3 inflammasome activation may have beneficial effects in preventing the damage mediated by the sterile inflammatory response in diseases such as renal-, cardiac-, and cerebral-ischemia. In addition, necrosis-induced sterile inflammation in trauma and secondary to infections and sepsis may be modulated by inhibitors of the NLRP3 pathway. The use of the IL-1R antagonist, anakinra, has already been shown to be effective in reducing the adverse events associated with a number of ischemic disease models [49, 50]. Conversely the adjuvant properties of NLRP3 inflammasome activation can be exploited as demonstrated by the increased immunogenicity of chemotherapy-induced tumor cell necrosis [37]. The development of specific antagonists of the NLRP3 inflammasome and an improved understanding of the specific mechanisms that lead to NLRP3 inflammasome activation will be instrumental in developing new therapeutic modalities against the growing number of pathologies associated with inappropriate activation of the NLRP3 inflammasome.
Footnotes
Conflict of interest: The authors declare no financial or commercial conflict of interest.
References
- 1.Trinchieri G, Sher A. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 2.Martinon F, et al. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 3.Pedra JH, et al. Curr Opin Immunol. 2009;21:10–16. doi: 10.1016/j.coi.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agostini L, et al. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F, et al. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 6.Shao W, et al. J Biol Chem. 2007;282:36321–36329. doi: 10.1074/jbc.M708182200. [DOI] [PubMed] [Google Scholar]
- 7.Keller M, et al. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 8.Boyden ED, Dietrich WF. Nat Genet. 2006;38:240–244. doi: 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- 9.Mariathasan S, et al. Nature. 2004;430:213–218. doi: 10.1038/nature02664. [DOI] [PubMed] [Google Scholar]
- 10.Zamboni DS, et al. Nat Immunol. 2006;7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 11.Sutterwala FS, et al. J Exp Med. 2007;204:3235–3245. doi: 10.1084/jem.20071239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lightfield KL, et al. Nat Immunol. 2008 doi: 10.1038/ni.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burckstummer, et al. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- 14.Fernandes-Alnemri, et al. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hornung V, et al. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts, et al. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- 17.Poeck H, et al. Nat Immunol. 2009 [Google Scholar]
- 18.Franchi L, et al. Eur J immunol. 2010;40 doi: 10.1002/eji.200940180. in press. [DOI] [PubMed] [Google Scholar]
- 19.Sodergren E, et al. Science. 2006;314:941–952. doi: 10.1126/science.1133609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halle A, et al. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinon F, et al. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 22.Iyer SS, et al. Proc Natl Acad Sci USA. 2009;106:20388–20393. doi: 10.1073/pnas.0908698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou R, et al. Nat Immunol. 2009 in press. [Google Scholar]
- 24.Dostert C, et al. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cassel SL, et al. Proc Natl Acad Sci USA. 2008;105:9035–9040. doi: 10.1073/pnas.0803933105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornung V, et al. Nat Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eisenbarth SC, et al. Nature. 2008;453:1122–1126. doi: 10.1038/nature06939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, et al. J Immunol. 2008;181:17–21. doi: 10.4049/jimmunol.181.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kool M, et al. J Immunol. 2008;181:3755–3759. doi: 10.4049/jimmunol.181.6.3755. [DOI] [PubMed] [Google Scholar]
- 30.Franchi L, Nunez G. Eur J Immunol. 2008;38:2085–2089. doi: 10.1002/eji.200838549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrilli, et al. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 32.Martinon F. Eur J immunol. 2010;40 doi: 10.1002/eji.200940168. in press. [DOI] [PubMed] [Google Scholar]
- 33.Hornung V, Latz E. Eur J immunol. 2010;40 doi: 10.1002/eji.200940185. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mariathasan S, et al. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 35.Sutterwala FS, et al. Immunity. 2006;24:317–327. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Pelegrin P, Surprenant A. EMBO J. 2006;25:5071–5082. doi: 10.1038/sj.emboj.7601378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghiringhelli F, et al. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 38.Imaeda AB, et al. J Clin Invest. 2009;119:305–314. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muruve DA, et al. Nature. 2008;452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 40.Schaefer L, et al. J Clin Invest. 2005;115:2223–2233. doi: 10.1172/JCI23755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang D, et al. Nat Med. 2005;11:1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 42.Leemans JC, et al. J Clin Invest. 2005;115:2894–2903. doi: 10.1172/JCI22832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu H, et al. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pulskens WP, et al. PLoS ONE. 2008;3:e3596. doi: 10.1371/journal.pone.0003596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rouschop KM, et al. J Am Soc Nephrol. 2005;16:2034–2043. doi: 10.1681/ASN.2005010054. [DOI] [PubMed] [Google Scholar]
- 46.Babelova A, et al. J Biol Chem. 2009;284:24035–24048. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamasaki K, et al. J Biol Chem. 2009;284:12762–12771. doi: 10.1074/jbc.M806084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, et al. J Immunol. 2009;183:1528–1532. doi: 10.4049/jimmunol.0901080. [DOI] [PubMed] [Google Scholar]
- 49.Banwell V, et al. J Stroke Cerebrovasc Dis. 2009;18:269–276. doi: 10.1016/j.jstrokecerebrovasdis.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 50.Wanderer AA. J Pediatr Hematol Oncol. 2009;31:537–538. doi: 10.1097/MPH.0b013e3181acd89d. [DOI] [PubMed] [Google Scholar]