

Abstract
Corticotropin-releasing factor (CRF) and norepinephrine (NE) levels are altered in post-traumatic stress disorder and may be related to symptoms of hyperarousal, including exaggerated startle, in these patients. In animals, activation of both systems modulates anxiety behaviors including startle plasticity, however it is unknown if they exert their actions orthogonally or dependently. We tested the hypothesis that NE receptor activation is required for CRF effects on startle and that CRF1 receptor activation is required for NE effects on startle. The study examined the effects of: 1) α2 agonist clonidine (0.18 mg/kg, ip), α1 antagonist prazosin (0.8 mg/kg), and β1/2 antagonist propranolol (0.8, 8.0 mg/kg) pretreatment on oCRF- (0.6 nmol) induced increases in startle reactivity and PPI disruptions; 2) α2 antagonist atipamezole (1–30 mg/kg) and α1 agonist cirazoline (0.025–1.0 mg/kg) treatment on startle; 3) CRF1 antagonist (antalarmin, 14 mg/kg) pretreatment on atipamezole- (10.0 mg/kg) induced increases in startle. oCRF robustly increased startle and reduced PPI. Pretreatment with clonidine or prazosin, but not propranolol, blocked oCRF-induced increases in startle but had no effect on oCRF-induced disruptions in PPI. Atipamezole treatment increased startle, which was partially attenuated by CRF1 antagonist pretreatment. Cirazoline treatment did not increase startle. These findings suggest that CRF modulation of startle, but not PPI, requires activation of α1 adrenergic receptors, while CRF1 activation also contributes to NE modulation of startle. These data support a bi-directional model of CRF-NE modulation of stress responses and suggest that both systems must be activated to induce stress effects on startle reactivity.
Keywords: CRF, CRH (corticotropin-releasing hormone), Norepinephrine, Startle, PTSD
INTRODUCTION
The acoustic startle reflex is a defensive behavior characterized by reflexive contraction of the skeletal musculature in response to sudden intense acoustic stimuli, and is increased under conditions of threat. Prepulse inhibition (PPI) of the startle reflex refers to suppression of the startle reflex when a startling acoustic stimulus is preceded by a weaker prestimulus. Startle reactivity and inhibition of startle are altered in certain anxiety disorders, including post-traumatic stress disorder (PTSD) (Grillon et al., 1996, 1998; Grillon, 2008; Pole et al., 2009). In PTSD, neurotransmission of corticotropin-releasing factor (CRF), a neuropeptide released in response to stress, may also be altered (Baker et al., 1999; Sautter et al., 2003). CRF levels in cerebrospinal fluid have been found to be higher in PTSD patients compared to control subjects (Baker et al., 1999; Bremner et al., 1997). Given that CRF modulates the startle response (Grillon, 2008; Risbrough and Stein, 2006), one hypothesis is that increases in CRF neurotransmission may contribute to exaggerated startle behaviors (e.g. increased startle magnitude (reactivity) and reduced startle inhibition) observed in PTSD. Identifying the mechanism through which CRF affects startle could therefore be beneficial in providing a target for developing novel compounds to treat anxiety disorders like PTSD.
CRF acts in the brain at two known G-protein-coupled receptors, CRF1 and CRF2 (Hauger et al., 2006). CRF1 activation is required for CRF- or stress-induced increases in startle reactivity and decreases in PPI (Risbrough et al., 2003, 2004, 2009). The central mechanism by which CRF exerts its effect on startle plasticity, however, is unclear. One potential mechanism mediating CRF system regulation of startle and PPI may involve the noradrenergic system (NE). The noradrenergic system, originating primarily in the locus coeruleus (LC), consists of α2 (presynaptic/postsynaptic), α1 (postsynaptic), and α (postsynaptic) receptors. The hypothesis that the NE system may mediate the effects of CRF on startle emerges from observations that CRF terminals synapse with dendrites of noradrenergic neurons in the LC and recent studies have labeled CRF1 receptors in tyrosine-hydroxylase neurons of the LC (Reyes et al., 2006). Additionally, CRF administration (intracerebroventricular (ICV) or directly into LC) increases norepinephrine release in the bed nucleus stria terminalis (BNST) (Curtis et al., 1997; Dunn et al., 2004; Van Bockstaele et al., 1996), a critical site for CRF-induced increases in startle (Lee and Davis, 1997). The BNST is also densely innervated with noradrenergic fibers from the LC (Forray et al., 2000; Koob, 1999) and requires noradrenergic signaling to control fear behaviors (Fendt et al., 2005). Additionally, CRF receptor antagonist treatment or null mutation of the CRF gene blocks stress-induced changes in extracellular NE (Isogawa et al., 2000; Jeong et al., 2000), suggesting that CRF modulates NE release. Increases in NE signaling have been shown to increase startle both in PTSD patients and in some rodent models (Carasso et al., 1998; Davis, 1980; Lahdesmaki et al., 2004; Morgan et al., 1995; Risbrough and Geyer, 2005). These findings support the hypothesis that CRF effects on defensive startle behavior may require activation of the NE system.
Given that CRF increases startle, NE increases startle under certain conditions, and CRF increases NE release, we predicted that blocking NE signaling would reverse the effects of CRF on startle behaviors. To test this hypothesis, we examined the effects of α2 agonist (clonidine) pretreatment on CRF-induced changes in startle. Clonidine decreases NE release by activating inhibitory presynaptic α2 autoreceptors. Second, we examined the contribution of the excitatory α1 or β postsynaptic receptors in CRF effects on startle via pretreatment with an α1 antagonist (prazosin), or a β1/2 adrenergic antagonist (propranolol). To determine if NE receptor activation effects on startle result from CRF receptor activation, we tested the hypothesis that NE activation is sufficient to mimic CRF effects on startle and PPI via treatment with a selective α2 antagonist that increases NE release (atipamezole) and an α1 agonist (cirazoline). Finally, we used a CRF1 antagonist (antalarmin) to determine whether NE signaling–induced changes in startle also require concomitant CRF1 receptor activation, suggesting reciprocal permissive roles of these systems in this behavior.
METHODS
Subjects
Male C57BL/6J mice (2–3 mo; Jackson Laboratories, Bar Harbor, Maine) were housed in a temperature- and humidity-controlled room (12:12 h reverse light/dark cycle; lights off at 08:00 am) (n=7–12/group/experiment). Food and water were available ad libitum. Mice acclimated to the vivarium for at least one week prior to cannulation surgery. All testing occurred between 10:00 am and 6:00 pm and was in accordance with the guidelines of the National Institutes of Health “Guide for the Care and Use of Laboratory Animals”, as approved by UCSD.
Surgery and histological verification
Surgery and histological verification are as described previously (Vinkers et al., 2007). A 23 gauge 7 mm unilateral guide cannula was placed above the lateral ventricle (flat skull; anteroposterior, -0.1 mm; mediolateral, ±1.1 mm; dorsoventral, -1.5 mm below dura). Mice recovered for >5 days before testing. A 30-gauge 8-mm injector was used for drug infusions. After study completion, mice were anesthetized and cannulae placement was checked via dye infusion (1.5 μl). Nine mice were removed from analysis due to incorrect placement.
Apparatus
Startle and PPI testing were performed in ventilated, dark, sound-attenuating commercial startle chambers (39x38x58 cm, SR-LAB system, San Diego Instruments, San Diego, CA) as described previously (Risbrough et al. 2003). A loudspeaker provided continuous broadband 65-dB background noise and all acoustic stimuli. Beginning at stimulus onset, 65 consecutive 1-ms readings were recorded to obtain the average magnitude of the mouse’s startle response.
Experiment 1: Time course and dose response of oCRF
Drugs
In order to identify a behaviorally active dose and timepoint of the CRF1 preferring agonist (oCRF) for use in Experiment 2, mice (n=9–10/group) were treated with 0, 0.06, 0.2, or 0.6 nmol oCRF (0.28–2.8 μg/5 μl, ICV) (Bachem, Torrance, CA). Artificial cerebrospinal fluid (aCSF) was the vehicle. Doses were based on previous studies using the non-selective CRF receptor agonist rat/human (r/h) CRF which dose-dependently increased startle and disrupted PPI (Risbrough et al., 2003, 2004).
Behavioral Testing
Mice were placed in the startle apparatus immediately following drug infusion (“Time 0”). Startle testing was repeated hourly for 6 hours post-infusion, with mice returned to the home cage for 40 min after each test (session was 20 min). The startle session began with a 2-min acclimation period, followed by presentation of four 120 dB pulse trials (presented at start of the session to achieve a stable level of startle reactivity for the remainder of the session). These trials were immediately followed by a block of trials in which the prepulse intensity varied (10 startle pulse-alone trials, each at 120 dB, 40 ms duration, and 30 prepulse+pulse trials). The prepulse+pulse trials were evenly divided among the following trial types (10 each): a 69, 73, or 77 dB prepulse (20 ms) preceding a 120 dB pulse stimulus (40 ms). The time between the prepulse and pulse onset was 100 ms. This block was immediately followed by another block of trials in which the inter-stimulus interval (ISI) between the prepulse and pulse was varied. During the ISI block, 7 startle pulse trials, each at 120 dB, were presented. Additionally, 73 dB prepulses preceded a 120 dB stimulus by 20, 120, 360 ms (four trials of each ISI duration). The average intertrial interval for all trial types throughout the session was 15 sec (range: 6–21 sec). All trial types within the prepulse intensity and ISI trials were presented in a pseudorandom order. The average startle magnitude over the record window (65 ms) was used for all data analysis.
Data Analysis
To assess CRF effects over time, startle magnitude in response to the 120 dB pulses was averaged over each session. A 2-way analysis of variance (ANOVA) was used with oCRF treatment (0, 0.06, 0.2, 0.6 nmol) as the between subject and Time post-infusion (0–6 hours) as the within subject factors. % PPI was calculated using the formula: 100-(((average startle of the prepulse+pulse trials)/average startle in the pulse alone trial)*100). Initially a 3-way ANOVA, with oCRF treatment as the between subject factor and prepulse intensity (69, 73, 77 dB) or ISI (20, 120, 360 ms) and post-infusion time as within subject factors was conducted. Prepulse intensity did not interact with the other factors and thus was removed from the analysis. Tukey’s post-hoc analyses followed significant main or interaction effects.
Experiment 2: The role of α2, α1, and β1/2 noradrenergic receptors in CRF-induced alterations in startle plasticity
Drugs
Experiment 2A: α2 agonist (clonidine) effects in oCRF-treated mice
Mice (n=9–11/group) received 0.6 nmol oCRF (2.8 μg/5 μl, ICV) or aCSF vehicle 2 hours before testing. They also received the α2 agonist clonidine (0.09 mg/kg; 5 ml/kg, IP, Tocris, Ellisville, MO) or saline 30 min before and 60 min after oCRF infusion (0.18 mg/kg total dose). Dose and timing of oCRF was based on Experiment 1 (0.6 nmol has large effects on startle and PPI 2 hours post-infusion; Fig 1). Two injections of clonidine were given to ensure a lasting effect of clonidine over the 2-hr pretest and testing interval (Risbrough et al., 2003, 2004).
Figure 1.

(a) Effect of CRF on acoustic startle responding to 120 dB pulses in C57BL/6J mice. Mice received ICV injections of 0, 0.06, 0.2, or 0.6 nmol oCRF diluted in aCSF vehicle. Acoustic startle responding was measured hourly from 0 to 6 h post-administration. *p<0.05, 0.2 and 0.6 nmol, compared to vehicle group; #p < 0.05, 0.6 nmol compared to 0.06 nmol group, +p < 0.05, 0.6 nmol compared to vehicle, post-hoc test). (b) Effect of oCRF on % PPI (averaged across 69, 73, 77 dB prepulse trials) at 0–6 hours post administration. oCRF significantly decreased % PPI (*p<0.05, main effect of oCRF).
Experiment 2B: α1 antagonist (prazosin) effects in oCRF-treated mice
Mice (n=8–11/group) received oCRF (0.6 nmol, ICV) or aCSF 2 hours before testing. They received the α1 antagonist prazosin (0.4 mg/kg; 5 ml/kg, IP, Sigma-Aldrich, St. Louis, MO) or vehicle (3% Cremophor in water) 30 min before and 1 hour after oCRF infusion for a total dose of 0.8 mg/kg. The prazosin dose was based on pilot studies and previous reports in mice (Okuyama et al., 1999).
Experiment 2C: β1/2 antagonist (propranolol) effects in oCRF-treated mice
Mice (n=7–11/group) received oCRF (0.6 nmol, ICV) or aCSF 2 hours before testing. The β1/2 antagonist propranolol (4 mg/kg; 5 ml/kg, IP, Sigma-Aldrich, St. Louis, MO) or saline was given 30 min before and 1 hour after oCRF (8 mg/kg total; Goldschmidt et al., 1984; Schank et al., 2008). To ensure results were not due to non-selectivity of propranolol for the β receptor, we also used a 10-fold lower dose of propranolol (0.4 mg/kg/injection, 0.8 mg/kg total) in a separate cohort (n=9–11/group) (Izquierdo and McGaugh, 1985; Saha et al., 1991; Zarrindast et al., 2004). To confirm the antagonist activity of propranolol at β1/2 receptors during the time frame of our startle test, we examined the effect of this same dose and pre-injection time on clenbuterol-induced disruptions in locomotor activity in a separate group of mice (n=7–11/group). Timing of propranolol administration before locomotor testing was identical to that in the startle tests, with propranolol (4 mg/kg) administered 2½ hours and 1 hour before testing. Clenbuterol was administered 30 min before testing. Data were analyzed over the first 30 min of the locomotor test to match the duration of the startle session (see below).
Behavioral Testing
A 65-dB background was presented continuously during the session which lasted 30 min. The session began with a 5 min acclimation period followed by presentation of five 120 dB pulse trials (average intertrial interval, 15 sec) (Block 1). These pulses were followed immediately by startle threshold response trials (Block 2), variable prepulse intensity trials (Block 3), and variable ISI trials (Block 4). Five 120 dB pulse trials were presented at the end of the session (Block 5) to measure startle habituation to 120 dB pulses across the session (i.e. Blocks 1–5). During the threshold response block, 20 startle pulse trials varied in intensity (four each at 80, 90, 100, 110, and 120 dB, 40 ms duration) were presented. Next, the variable prepulse intensity block consisted of 12 startle pulse-alone trials (each at 120 dB) and 30 prepulse+pulse trials. The prepulse+pulse trials were evenly divided among the following trial types (10 each): 68, 71, or 77 dB prepulse (20 ms) preceding a 120-dB pulse stimulus (40 ms). The time between prepulse and pulse onset was 100 ms. During the variable ISI block, 8 startle pulse trials (120 dB) were presented. In addition, 71 dB prepulses preceded a 120-dB stimulus by 25, 50, 100, 200, or 500 ms (four trials each). All trial types were presented in a pseudorandom order.
Data Analysis
A 2 or 3-way ANOVA was used with pretreatment and/or treatment as between subject factors and pulse intensity (80, 90, 100, 110, 120 dB), prepulse intensity (68, 71, 77 dB), ISI (25, 50, 100, 200, 500 ms) or startle block (Blocks 1–5) as within subject factors (for brevity we report only the prepulse intensity block data). Tukey’s post-hoc analyses followed significant main or interaction effects.
Experiment 3: The role of CRF1 receptors in noradrenergic modulation of startle
Drugs
Experiment 3A: α2 antagonist (atipamezole) effects on startle
Mice (n=11/dose) were treated with the α2 antagonist atipamezole (1, 3, 10, 30 mg/kg, 10 ml/kg, SC; Pfizer Health, Reno, NV) or saline 30 min before testing (doses and timing based on Risbrough and Geyer, 2005).
Experiment 3B: α1 agonist (cirazoline) effects on startle
Mice (n=11/dose) were treated with the α1 agonist cirazoline (0.25, 0.5, 1.0 mg/kg, 5 ml/kg, IP) (Sigma, St. Louis, MO) or saline 5 min before testing (Alsene et al., 2006; Barr et al., 2006; Carasso et al., 1998; Menon et al., 1990; Shilling et al., 2004; Varty et al., 1999). Based on Experiment 3B, mice were treated with lower doses of cirazoline (0.025, 0.083, 0.25 mg/kg, 5 ml/kg, IP) or saline 5 min before testing (n=10–11/dose).
Experiment 3C: CRF1 receptor antagonist (antalarmin) effects on NE-induced increases in startle
Mice (n=10–12/dose) were treated with the CRF1 antagonist antalarmin (14.0 mg/kg, 10 ml/kg, IP; Sigma-Aldrich, St. Louis, MO) or vehicle (19% DMSO in saline) 1 hour before testing (Bale and Vale, 2003; Djouma et al., 2006). Atipamezole (10 mg/kg, SC) or saline was administered 30 min before testing.
Behavioral Testing
The startle session used in Experiments 3A-3C was identical to what was used in Experiments 2A-2C except it did not contain an ISI block.
Data Analysis
Data analysis was identical to Experiment 2.
RESULTS
Experiment 1: Time course and dose response of oCRF
The effect of oCRF on average startle magnitude depended upon post-infusion time (oCRFXTime: F(18,198)=2.81, p<0.0002; Fig 1a). Subsequent analysis at each timepoint indicated that oCRF significantly increased startle magnitude at 0, 1, 2, and 3 hours post-infusion. Post-hoc analysis indicated that at 0, 1 and 2 hours post-infusion, 0.2 and 0.6 nmol oCRF significantly increased startle (p<0.05, vs. vehicle). At 1 hour, 0.6 nmol also elevated startle compared to 0.06 nmol (p<0.05). At 3 hours, 0.6 nmol continued to increase startle (p<0.05, vs. vehicle).
oCRF treatment significantly decreased %PPI (oCRF: F(3,33)=4.15, p<0.05) (Fig 1b) over the majority of the testing period (oCRFXTime: F(18,198)=1.40, p=0.13). Inspection of the data suggests that the two highest doses (0.2 and 0.6 nmol) disrupted PPI to the greatest extent at 2 hours post-infusion (Fig 1b). oCRF (0.6 nmol) similarly decreased % PPI at 120 msec ISI (oCRFXISI: F(6,66)=5.96, p=0.0001; p<0.05, 0.6 nmol vs. vehicle, post-hoc test). At the shorter ISI trial (20 msec), oCRF increased % PPI (p<0.05, 0.2 nmol vs. vehicle; p<0.1, 0.6 nmol vs. vehicle, post-hoc data; data not shown). These ISI-dependent effects of CRF on PPI are consistent with previous observation in rats (Conti et al., 2009) and suggest that CRF effects on PPI may be via alterations in the speed of prepulse processing (i.e. shifting the time window of prepulse effects on startle reactivity; Graham, 1975).
Experiment 2: The role of α2, α1, and β noradrenergic receptors in CRF-induced alterations in startle plasticity
Experiment 2A: α2 agonist (clonidine) effects in oCRF-treated mice
During the startle threshold test, oCRF increased startle magnitude to high intensity pulses and this effect was reduced by pretreatment with clonidine, the α2 agonist (IntensityXClonidineXoCRF: F(4,140)=4.32, p=0.005; Table 1). A follow-up 2-way ANOVA at each intensity confirmed a significant ClonidineXoCRF interaction at 120 dB pulse (F(1,35)=4.53, p<0.05). Post-hoc tests indicated that startle magnitude in the oCRF/Veh group was significantly higher relative to all other groups (p<0.05).
Table 1.
Effect of α2 agonist (clonidine), α1 antagonist (prazosin), and β antagonist (propranolol) on CRF-induced changes in startle magnitude and % PPI (Mean ± SEM)
| Startle Intensity (dB) | 80
|
90
|
100
|
110
|
120
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vehicle | oCRF | Vehicle | oCRF | Vehicle | oCRF | Vehicle | oCRF | Vehicle | oCRF | |
|
|
|
|||||||||
| Vehicle | 13 ± 2 | 28 ± 5 | 21 ± 4 | 46 ± 11 | 65 ± 9 | 105 ± 17 | 118 ± 21 | 274 ± 45 | 121 ± 24 | 413 ± 92* |
| Clonidine | 8 ± 1 | 18 ± 4 | 13 ± 2 | 39 ± 7 | 39 ± 6 | 83 ± 21 | 79 ± 9 | 136 ± 23 | 108 ± 16 | 195 ± 23 |
|
|
|
|||||||||
| Vehicle | 14 ± 1 | 44 ± 12+ | 30 ± 6 | 70 ± 17 | 68 ± 12 | 89 ± 26 | 106 ± 17 | 202 ± 27 | 130 ± 23 | 375 ± 57* |
| Prazosin | 18 ± 4 | 22 ± 3 | 31 ± 9 | 32 ± 10 | 67 ± 17 | 60 ± 11 | 106 ± 23 | 188 ± 40 | 124 ± 22 | 194 ± 50 |
|
|
|
|||||||||
| Vehicle | 13 ± 3 | 41 ± 12 | 26 ± 7 | 82 ± 22# | 57 ± 6 | 111 ± 28 | 78 ± 13 | 153 ± 27 | 95 ± 13 | 246 ± 34# |
| Propranolol | 12 ± 2 | 63 ± 3 | 20 ± 4 | 81 ± 16 | 49 ± 7 | 134 ± 25 | 80 ± 17 | 218 ± 34 | 87 ± 12 | 224 ± 32 |
| Prepulse Intensity (dB) | 68
|
71
|
77
|
|||
|---|---|---|---|---|---|---|
| Vehicle | oCRFa | Vehicle | oCRFa | Vehicle | oCRF | |
|
|
||||||
| Vehicle | 35 ± 6 | 16 ± 4 | 47 ± 6 | 29 ± 4 | 61 ± 5 | 53 ± 4 |
| Clonidine | 33 ± 6 | 25 ± 2 | 53 ± 6 | 26 ± 5 | 66 ± 4 | 49 ± 6 |
|
| ||||||
| Vehicle | 24 ± 4 | 0.5 ± 13 | 39 ± 6 | 24 ± 9 | 58 ± 4 | 55 ± 7 |
| Prazosin | 14 ± 5 | 14 ± 5 | 36 ± 6 | 27 ± 7 | 53 ± 4 | 54 ± 8 |
|
| ||||||
| Vehicle | 36 ± 7 | 11 ± 12 | 54 ± 6 | 26 ± 11 | 68 ± 5 | 62 ± 7 |
| Propranolol | 26 ± 6 | 18 ± 5 | 48 ± 6 | 28 ± 7 | 54 ± 10 | 64 ± 4 |
p < 0.05, vs. all other groups at 120 dB
p < 0.05, vs. all other groups at 80 dB
p <0.05, vs. Veh/Veh group only at 90 and 120 dB
Significant main effect of oCRF on PPI at 68 and 71 dB prepulse trials in the propranolol experiment (see text for details)
Note: In the clonidine experiment, there was a significant main effect of oCRF on PPI at all prepulse intensities (see text for details).
oCRF-induced increases in startle magnitude were predominantly present in the initial blocks of the session, which was significantly reduced by clonidine pretreatment (Fig 2a; BlockXoCRF: F(4,140)=6.07, p<0.05; BlockXClonidineXoCRF: F(4,140)=3.05, p<0.05). Follow-up 2-way ANOVA conducted at each block confirmed a significant ClonidineXoCRF interaction at Blocks 1 and 2 (Fs(1,35)>4.39, p<0.05). Post-hoc analyses indicated that startle magnitude in the oCRF/Veh group was significantly higher compared to all other groups during Blocks 1 and 2 (p<0.05, oCRF/Veh vs. Veh/Veh, Veh/clonidine, oCRF/clonidine) (Fig 2a, inset).
Figure 2.

(a) Effect of clonidine on CRF-induced increases in startle in Block 1. Clonidine reversed the startle increase in oCRF treated mice (*p<0.05, oCRF/Veh versus all other groups, post-hoc test). (Inset) Effect of clonidine on CRF-induced increases in startle across the test session. oCRF-induced increases in startle during Blocks 1 and 2 was significantly blocked by clonidine treatment (*p<0.05, oCRF/Veh versus all other groups, post-hoc test). (b) Effect of prazosin on CRF-induced increases in startle in Block 1. Prazosin reversed the startle increase in oCRF treated mice (*p<0.05, oCRF/Veh versus. all other groups, post-hoc test). (Inset) Effect of prazosin on CRF-induced increases in startle across the test session. oCRF-induced increases in startle were significantly blocked by prazosin in Blocks 1 and 2 (*p<0.05, oCRF/Veh versus all other groups, post-hoc test. Startle magnitude in the oCRF/Veh group remained significantly higher compared to Veh/Veh and Veh/Prazosin during Block 5 (+p<0.05, oCRF/Veh versus Veh/Veh, Veh/Prazosin, post-hoc test). (c) Effect of propranolol on CRF-induced increases in startle in Block 1 (*p<0.05, main effect of oCRF). (Inset) Effect of propranolol on CRF-induced increases in startle across the test session. Although oCRF increased startle magnitude, this effect was not reversed by propranolol during any block (*p<0.05, main effects of oCRF, see text for details).
As expected, %PPI increased with prepulse intensity (F(2,70)=97.61, p=0.0001) and decreased with oCRF administration (F(1,35)=17.71, p=0.001). The effect of oCRF on PPI appeared dependent upon intensity and clonidine (IntensityXClonidineXoCRF: F(2,70)=3.63, p<0.05). However, 2-way ANOVAs at each intensity indicated that oCRF effects on PPI were not significantly altered by clonidine pretreatment (Table 1). A similar pattern of PPI results was found from the ISI test (data not shown).
Experiment 2B: α1 antagonist (prazosin) effects in oCRF-treated mice
oCRF-induced increases in startle magnitude were significantly reduced by the α1 antagonist prazosin at specific startle pulse intensities (Table 1; IntensityXPrazosinXoCRF: F(4,140)=3.29, p=0.01). Two-way ANOVAs conducted at each intensity revealed a significant interaction at the 80 and 120 dB pulses (PrazosinXoCRF: Fs(1,38>4.10, ps<0.05). At these intensities, the startle magnitude in oCRF/Veh was significantly higher relative to all other groups (p<0.05, oCRF/Veh vs. Veh/Veh, Veh/prazosin, oCRF/prazosin).
oCRF-induced increases in startle were again greatest in the initial blocks (Fig 2b; BlockXoCRF: F(4,140)=3.44, p=0.01). There was also an overall significant interaction between oCRF and prazosin on startle reactivity (oCRFXPrazosin: F(1,35)=4.33, p<0.05) (Fig 2b, inset). Post-hoc analyses indicated that startle magnitude in oCRF/Veh was significantly greater than all other groups during Blocks 1 and 2 (p<0.05). During Block 5, startle magnitude remained significantly greater in the oCRF/Veh group compared to Veh/Veh and Veh/prazosin (p<0.05) and there was a trend towards a significant difference compared to oCRF/prazosin (p<0.1). oCRF-induced disruption of PPI was not affected by prazosin (Table 1).
Experiment 2C: β1/2 antagonist (propranolol) effects in oCRF-treated mice
The effect of oCRF on startle magnitude was greatest at the 90 and 120 dB pulses (IntensityXoCRF: F(4,128)=6.98, p=0.0000, p<0.05 post-hoc test vs. Veh/Veh) (Table 1). As we have seen throughout, oCRF increases in startle magnitude were highest during the initial blocks (BlockXoCRF: F(4,128)=6.65, p<0.0001, Fig 2c). A significant main effect of oCRF was observed during Block 1 (oCRF: F(1,32)=15.57, p< 0.0004) (Figure 2c). Although the main effects of oCRF were maintained during Blocks 2–5 (Fs(1,32)>8.13, ps<0.008), oCRF-induced startle increases were not reversed by propranolol (β1/2 antagonist) in any block (Fig 2c, inset). oCRF-induced disruptions in PPI (IntensityXoCRF: F(2,64)=8.28, p<0.005; at 68 and 71 dB, main effects of oCRF were significant: Fs(1,32)>4.79, p<0.05), were unaffected by propranolol (Table 1). In a similar experiment using a lower dose of propranolol (0.8 mg/kg total) we again found that oCRF-induced increases in startle reactivity and decreases in PPI were not reversed by propranolol (data not shown).
Conversely, this same dose and timing of propranolol treatment was found to block the effects of clenbuterol, a β adrenergic agonist, on locomotor activity. Clenbuterol decreased distance traveled over a 30 minute period and this effect was fully blocked by propranolol pretreatment (PropranololXClenbuterol: F(1,33)= 4.24, p=0.04). (Mean distance traveled (cm± SEM) for Veh/Veh, Veh/Clen, Prop/Veh, and Prop/Clen groups: 4613.9±172.0, 3555.6±313.4, 4427.8±267.9, 4380.8±225.2, respectively).
Experiment 3: The role of CRF1 receptors in noradrenergic modulation of startle
Experiment 3A: α2 antagonist (atipamezole) effects on startle
Similar to oCRF, atipamezole (an α2 antagonist that increases NE release via blockade of autoreceptors) dose-dependently increased startle magnitude predominantly at high startle intensity trials (Table 2; AtipamezoleXIntensity: F(16,188)=2.58, p=0.001) (30 mg/kg atipamezole at 110 and 120 dB; 10 mg/kg at 120 dB, p<0.05, vs. vehicle, Tukey’s test). Unlike CRF, atipamezole effects were relatively robust across all blocks (Atipamezole: F(4,47)=8.75, p=0.0000; AtipamezoleXBlock: F(12,141)=1.99, p=0.03) (Fig 3a, inset). Post-hoc analysis on each block indicated that compared to vehicle, 30 mg/kg increased startle magnitude during all blocks and 10 mg/kg increased startle during Blocks 2, 3 and 4 (p<0.05). Atipamezole did not affect % PPI (Table 2).
Table 2.
Effect of α2 antagonist (atipamezole) and α1 agonist (cirazoline) on startle magnitude and % PPI (Mean ± SEM)
| Startle Intensity (dB) | Prepulse Intensity (dB) | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Atipamezole (mg/kg)
|
80
|
90
|
100
|
110
|
120
|
68
|
71
|
77
|
| 0 | 14 ± 2 | 31 ± 8 | 79 ± 10 | 105 ± 12 | 127 ± 18 | 17 ± 9 | 46 ± 6 | 64 ± 4 |
| 1 | 16 ± 2 | 28 ± 4 | 79 ± 13 | 120 ± 11 | 151 ± 17 | 23 ± 5 | 45 ± 3 | 62 ± 4 |
| 3 | 14 ± 2 | 33 ± 7 | 84 ± 15 | 148 ± 18 | 175 ± 25 | 19 ± 6 | 41 ± 4 | 59 ± 4 |
| 10 | 16 ± 2 | 28 ± 7 | 79 ± 11 | 170 ± 20 | 216 ± 22* | 24 ± 5 | 48 ± 5 | 68 ± 5 |
| 30 | 23 ± 5 | 44 ± 8 | 77 ± 11 | 174 ± 17+ | 223 ± 25* | 20 ± 6 | 48 ± 7 | 76 ± 3 |
| Cirazoline (mg/kg)a | ||||||||
| 0 | 29 ± 6 | 68 ± 10 | 97 ± 11 | 139 ± 18 | 147 ± 23 | 18 ± 7 | 45 ± 7 | 59 ± 7 |
| 0.25 | 18 ± 4 | 31 ± 5 | 53 ± 7 | 86 ± 8 | 107 ± 11 | 24 ± 9 | 55 ± 5 | 75 ± 3 |
| 0.5 | 11 ± 3 | 21 ± 4 | 42 ± 9 | 65 ± 15 | 71 ± 15 | 24 ± 6 | 44 ± 7 | 67 ± 5 |
| 1.0 | 17 ± 4 | 28 ± 5 | 55 ± 14 | 65 ± 8 | 91 ± 13 | 17 ± 7 | 48 ± 6 | 74 ± 2 |
| Cirazoline (mg/kg) | ||||||||
| 0 | 31 ± 7 | 46 ± 8 | 70 ± 9 | 99 ± 9 | 120 ± 12 | 25 ± 4 | 55 ± 4 | 69 ± 4 |
| 0.025 | 26 ± 6 | 55 ± 12 | 89 ± 13 | 123 ± 17 | 134 ± 24 | 32 ± 7 | 55 ± 5 | 66 ± 6 |
| 0.083 | 19 ± 2 | 53 ± 10 | 92 ± 17 | 124 ± 29 | 125 ± 19 | 28 ± 8 | 53 ± 5 | 69 ± 5 |
| 0.25 | 29 ± 5 | 41 ± 6 | 77 ± 11 | 95 ± 12 | 91 ± 13 | 20 ± 10 | 56 ± 5 | 71 ± 4 |
p < 0.05, vs 0 mg/kg at 120 dB
p < 0.05, vs. 0 mg/kg at 110 dB
Significant main effect of cirazoline (see text for details)
Figure 3.

(a) Dose-response effect of atipamezole on startle magnitude in Block 1. Atipamezole (30 mg/kg) significantly increased startle magnitude (*p<0.05, versus vehicle, post-hoc test). (Inset) Effect of atipamezole on startle across the test session. The highest dose of atipamezole (30 mg/kg) increased startle across the session (*p<0.05, 30 mg/kg relative to vehicle in Block 1, 2, 3, and 4, post-hoc test), while 10 mg/kg increased startle during Blocks 2, 3, and 4. (+p<0.05, 10 mg/kg relative to vehicle in Block 2, 3, and 4, post-hoc test). (b) Dose response (0.25–1.0 mg/kg) of cirazoline. The highest dose of cirazoline (1.0 mg/kg) decreased startle magnitude during Block 1 (*p<0.05, 1.0 mg/kg versus vehicle, post-hoc test). (Inset) Dose response of cirazoline on startle magnitude across the startle session. The highest doses of cirazoline decreased startle magnitude (*p<0.05, 1.0 mg/kg relative to vehicle in Block 1; +p<0.05, 0.5 mg/kg relative to vehicle in Block 2; post-hoc test). (c) When a lower dose range (0.025–0.25 mg/kg) was used, cirazoline had no effect on startle magnitude during Block 1 or across the entire session (inset).
Experiment 3B: α1 agonist (cirazoline) effects on startle (0–1.0 mg/kg)
The α1 agonist cirazoline decreased startle magnitude regardless of pulse intensity (Cirazoline: F(3,37)=10.01, p=0.0001; Table 2). Post-hoc analysis conducted on the average startle response (collapsed across intensity) indicated that each dose of cirazoline decreased startle (ps<0.01, vs. vehicle). Cirazoline-induced decreases in startle were most prominent in the initial blocks (CirazolineXBlock: F(9,111)=2.69, p=0.007) (Fig 3b). Compared to vehicle, 1.0 and 0.5 mg/kg decreased startle during Block 1 and 2, respectively (p<0.05) (Fig 3b, inset). Cirazoline did not affect % PPI (Table 2). No effects on startle or PPI were found with lower cirazoline doses (0.025, 0.083, 0.25 mg/kg) (Fig 3c; Table 2).
Experiment 3C: CRF1 receptor antagonist (antalarmin) effects on NE-induced increases in startle
To determine whether startle increases following NE release are modulated by CRF activation, we explored the effects of antalarmin (CRF1 antagonist) on startle reactivity in atipamezole-treated mice. Atipamezole increased startle magnitude at the high intensity pulses and this effect appeared dependent upon antalarmin pretreatment (IntensityXAtipamezole: F(4,152)=4.42, p=0.002; IntensityXAtipamezoleXAntalarmin: F(4,152)=2.56, p=0.04) (Table 3). A 2-way ANOVA conducted at each intensity confirmed a significant AntalarminXAtipamezole interaction at the 120 dB pulse (F(1,38)=4.34, p=0.04) (Table 3). Tukey’s tests at 120 dB indicated that startle magnitude in the Veh/Atipamezole group was significantly higher relative to Veh/Veh (p< 0.05). The effects of atipamezole appeared to be mildly attenuated by antalarmin as the Antalarmin/Atipamezole group was not significantly different from Veh/Veh, however it was also not significantly different from Vehicle/Atipamezole. To compare with previous experiments, we also conducted an analysis on Block 1 (Fig 4), finding a significant AntalarminXAtipamezole interaction (F(1,38)=7.45, p=0.01) with post-hoc tests indicating a significant startle magnitude increase in the Vehicle/Atipamezole group compared to Vehicle/Vehicle (p<0.05, Tukey’s test). A less conservative planned simple comparison revealed a significant reduction of atipamezole effects by antalarmin pretreatment (One-way t-test between Vehicle/Atipamezole and Antalarmin/Atipamezole: p=0.05). Although the effects of atipamezole on % PPI depended on prepulse intensity (AtipamezoleXIntensity: F(2,76)=3.72, p=0.03), subsequent ANOVAs conducted at each intensity separately revealed no significant main effect of atipamezole (Table 3). Antalarmin also did not affect PPI, and it did not interact with atipamezole (Table 3).
Table 3.
Effect of CRF1 antagonist (antalarmin) on atipamezole-induced changes in startle magnitude and % PPI (Mean ± SEM)
| Startle Intensity (dB) | 80
|
90
|
100
|
110
|
120
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Vehicle | Atipam. | Vehicle | Atipam. | Vehicle | Atipam. | Vehicle | Atipam. | Vehicle | Atipam. | |
|
|
|
|||||||||
| Vehicle | 18 ± 3 | 26 ± 6 | 31 ± 7 | 28 ± 8 | 59 ± 11 | 80 ± 13 | 97 ± 16 | 153 ± 22 | 95 ± 13 | 184 ± 17* |
| Antalarmin | 14 ± 1=2 | 19 ± 4 | 30 ± 5 | 43 ± 11 | 84 ± 11 | 82 ± 11 | 117 ± 18 | 144 ± 13 | 126 ± 23 | 141 ± 17 |
| Prepulse Intensity (dB) | 68
|
71
|
77
|
|||
|---|---|---|---|---|---|---|
| Vehicle | Atipam. | Vehicle | Atipam. | Vehicle | Atipam. | |
|
|
||||||
| Vehicle | 21 ± 8 | 34 ± 9 | 53 ± 5 | 57 ± 7 | 70 ± 4 | 79 ± 4 |
| Antalarmin | 27 ± 7 | 34 ± 8 | 56 ± 5 | 59 ± 7 | 70 ± 4 | 75 ± 4 |
p < 0.05, vs. Veh/Veh at 120 dB
Figure 4.

Effect of antalarmin on atipamezole-induced increases in startle in Block 1. *p<0.05 Vehicle/Atipamezole vs. Vehicle/Vehicle; Tukey’s post-hoc Test. +p<0.05 Vehicle/Atipamezole vs. Antalarmin/Atipamezole, planned simple t-test. (Inset) Effect of antalarmin on atipamezole-induced increases in startle across the session. Overall there was a significant AntalarminXAtipamezole interaction which did not interact with block.
DISCUSSION
We investigated the mechanisms by which CRF and NE signaling interact to modulate defensive startle behaviors. Acute oCRF administration robustly increased startle reactivity and reduced PPI. The α2 agonist (clonidine) and the α1 antagonist (prazosin), but not the β1/2 adrenergic antagonist (propranolol), significantly reduced the effects of CRF on startle, suggesting that α2 and α1 receptors mediate CRF-induced increases in startle reactivity. None of these treatments affected CRF-induced disruptions in PPI. We also found a reciprocal contribution of the CRF1 receptor to NE signaling effects on startle, with α2 antagonist (atipamezole)-induced increases in startle attenuated by CRF1 antagonist (antalarmin) pretreatment. These findings support the hypothesis that NE transmission is required for CRF effects on startle reactivity. These data lend support to a model of dual permissive roles for NE and CRF1 receptor activation for stress-induced increases in startle. These findings also suggest that the neurotransmission mechanism mediating CRF effects on startle reactivity is dissociable from CRF-mediated changes in PPI.
oCRF robustly increased startle in all experiments, consistent with earlier rodent findings wherein r/h-CRF increased startle magnitude (Liang et al., 1992; Risbrough et al., 2003, 2004; Swerdlow et al., 1986, 1989). In rats, CRF effects on startle have been localized to a hippocampal-septal-BNST circuit expressing CRF1 and CRF2 receptors (Davis et al., 1997; Van Pett et al., 2000). In mice, both receptors contribute to CRF effects on startle and PPI (Risbrough et al., 2003, 2004, 2009). Early evidence suggested that NE signaling may be induced by activation of the CRF system. Lee et al. (1994) reported that CRF facilitates norepinephrine release in the hippocampal dentate gyrus. Additionally, CRF1 pharmacological blockade and CRF gene deletion block stress-induced increases in norepinephrine release (Griebel et al., 2002; Jeong et al., 2000). Here we identified NE signaling via α1 receptors as a mechanism required for CRF effects on startle behavior, since clonidine and prazosin prevented blocked CRF-induced increases in startle. Both drugs were effective at the 120 dB stimuli, while prazosin also reversed CRF-induced increases in startle at 80 dB (Table 1). These results hint that α1 and α2 receptors could mediate startle at different points in the startle circuit and/or alter neuronal excitability in different ways (reduced threshold vs. increased firing rate), although further testing is required to test this speculation. Neither clonidine nor prazosin treatment alone significantly affected startle in the absence of CRF (Table 1). Thus, it is unlikely that the blockade of CRF-induced increases in startle following clonidine and prazosin pretreatment reflected a summation of opposing effects.
The β1/2 adrenergic antagonist propranolol had no effect on CRF-induced increases in startle, despite the fact that this pretreatment was clearly behaviorally active in blocking the effects of clenbuterol. Thus, the null effect of propranolol on CRF modulation of startle was not due to incorrect dosing or timing of propranolol treatment. However, it should be noted that the CRF-induced increase in startle in the propranolol experiment, though robust and highly significant, was smaller compared to its effect in the clonidine and prazosin experiments. The possibility that this smaller behavioral window obscured detection of a propranolol effect on CRF-induced increases in startle cannot be excluded. Nonetheless, our propranolol data corroborate previous startle findings that blockade of CRF1, but not β1/2 receptors, reduces stress-induced increases in startle reactivity (Adamec et al., 2007, 2009). Taken together, the present data suggest that α2 and α1 adrenergic receptors modulate the CRF startle effect, while the β receptors may not.
After determining that NE signaling, likely via α1 receptors, is required for the effects of CRF on startle reactivity, we next examined the efficacy of α2 antagonist (atipamezole) and α1 agonist (cirazoline) treatments to increase startle reactivity. These studies were based on the hypothesis that if NE release (via blockade of α2 autoreceptor) or α1 activation mediate CRF effects on startle, then modulation of these receptors may be sufficient to mimic CRF-induced alterations in startle reactivity. We found that atipamezole (which releases NE) but not cirazoline (which selectively activated α1) increased startle. The inability of cirazoline to increase startle corroborates a recent report that α1 agonist treatments (cirazoline, methoxamine, or phenylephrine) do not affect baseline startle magnitude in rats, despite activity in other measures (PPI) (Alsene et al., 2006). Thus, NE release is sufficient to mimic CRF-induced increases in startle, but direct α1 activation is not.
These findings raise the question of how could increased NE release potentiate startle if direct stimulation of α1 receptors has no effect? In humans yohimbine, an α2 antagonist, induces increased CRF release (as measured by increased CRF in cerebrospinal fluid; Vythilingam et al., 2000). If the NE-induced release of CRF in turn activates the CRF1 receptor, then this would explain why atipamezole-induced increases in startle were attenuated by the CRF1 antagonist antalarmin (Experiment 3). Thus atipamezole may induce CRF release in the BNST either via its induction of NE release and subsequent activation of excitatory NE receptors (α1 and/or β) at CRF containing neurons, or via direct antagonist activity of inhibitory α2 receptors at CRF containing neurons (Phelix et al. 1994; Shields et al. 2009, Lorton and Davis, 1987). Our data suggest that atipamezole-induced increases in startle would likely be via direct blockade of inhibitory α2 or indirect activation of excitatory β receptors at CRF containing neurons, as activation of α1 with cirazoline did not increase startle reactivity. These data are also consistent with previous reports suggesting that activation of CRF receptors mediates noradrenergic related increases in anxiety behaviors. For example, α2 antagonists (idazoxan) and α1 agonists (phenylephrine) result in stress-like decreases in exploratory behavior and a non-selective CRF antagonist (α-helical CRF) blocks phenylephrine-induced decreases in exploratory behavior (Berridge and Dunn, 1989). Taken together, the present findings that α2 agonist and α1 antagonist treatment disrupt CRF-induced increases in startle and that CRF1 antagonist treatment attenuates α2 antagonist-induced increases in startle lend support to a model of dual permissive roles for CRF and NE receptor activation for stress-induced increases in startle.
To integrate these findings, we have proposed a potential circuit model of the CRF/NE signaling effects on startle reactivity (Fig 5). Previous reports indicate that the BNST is the site of action for CRF1 effects on startle (Walker et al., 2009). However, unlike our findings following systemic prazosin pretreatment (Expt. 2B), we observed no effect of α1 antagonist (4, 12, 40 μg terazosin) infusions in the BNST on CRF-induced startle increases (Gresack et al., unpublished observations) suggesting that α1 effects on CRF modulation of startle are located elsewhere. It is possible that these effects are via activation of α1 at the pontine reticular formation (Stevens et al. 1994) which mediates the acoustic startle reflex via activation of motor neurons in the spinal cord (Koch, 1999). Intrathecal injections of α1 agonists in rats robustly increases startle reactivity, suggesting that brain stem activation of α1 receptors may be sufficient to increase startle (Astrachan et al., 1983). α1 receptors in the reticular formation are predominantly excitatory by facilitating initial neuronal responsiveness and increasing repetitive firing (Stevens et al. 1994). Hence, CRF receptor activation may modulate startle by activating two separate pathways that converge at the nucleus reticularis pontis caudalis (PRC) of the startle circuit (Lee et al., 1996): the BNST-PRC (via glutamate release at the PRC that is independent of α1 activation) (Dong and Swanson, 2004; Holstege et al., 1985), and the LC-PRC (via NE release and subsequent α1 receptor activation at the PRC; Jones and Yang, 1985; Strazielle et al. 1999) (Fig 5). Because NE release in turn increases levels of CRF (Vythilingam et al., 2000), inducing NE release via an α2 antagonist would activate both of these pathways, while direct stimulation of α1 receptors would not (Fig 5). If both pathways are required to achieve a maximal startle response, this model could explain why we did not observe effects of systemic α1 treatment on startle alone. Further site-specific injections of NE and CRF signaling blockers at the PRC are required to test this hypothesis.
Figure 5.
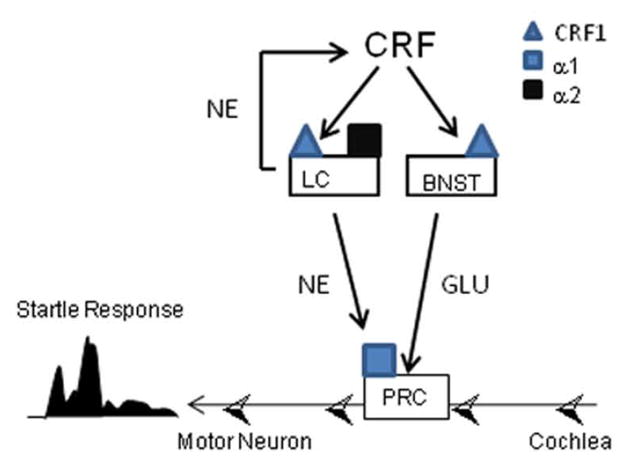
Schematic proposing a circuit model of CRF/NE signaling effects on startle reactivity. In this model, CRF1 receptor activation modulates startle by activating two pathways that converge at the nucleus reticularis pontis caudalis (PRC) of the startle circuit. One pathway is the LC-PRC pathway which requires NE release and α1 receptor activation at the PRC. The second pathway, the BNST-PRC pathway, involves CRF1 receptor activation of glutamatergic neurons projecting to the PRC. See text for additional details. Abbreviations: CRF – corticotropin releasing factor; NE – norepinephrine; LC - locus coeruleus; BNST – bed nucleus stria terminalis; GLU – glutamate; PRC: nucleus reticularis pontis caudalis
In the present study, CRF-evoked disruptions in sensorimotor gating were insensitive to manipulation of the noradrenergic system, since clonidine, prazosin, and propranolol did not affect CRF decreases in PPI. Thus, a dichotomy exists between CRF signaling mechanisms for startle and PPI. Downstream signaling mediators of CRF effects on PPI remains unclear. Neither D1 nor D2 signaling is required for CRF-induced disruption in mice (Vinkers et al., 2007, but see Dirks et al., 2003). Similarly serotonin signaling does not appear to be required for CRF-induced decreases in PPI in rats (Sutherland et al., 2008). CRF does modulate excitatory glutamatergic synapse transmission at forebrain regions that modulate PPI (Liu et al., 2004; Swerdlow et al., 2001), and thus may be a system for future investigation of mechanisms underlying CRF-induced alterations in PPI.
Finally, none of the pharmacological manipulations of the noradrenergic system affected PPI. These findings diverge from previous reports in rats indicating that α1 activation (via cirazoline) decreases PPI (Alsene et al., 2006; Carasso et al. 1998; Swerdlow et al. 2006), and this effect is blocked by α2 activation (via clonidine; Swerdlow et al. 2006). There are few studies of NE effects on PPI in mice. Indeed the present study is the first to our knowledge describing cirazoline effects in mice, which appear to be different than found in rats, perhaps due to species differences in NE receptor function or distribution (Schultz and Daly, 1973, Lorton and Davis, 1987). In mice,, α2c vs. α2a receptor subtypes appear to exhibit opposing roles on PPI α2c and α2a null mutation mice exhibit decreased and increased PPI, respectively (Sallinen et al. 1998; Lahdesmakei et al. 2004). Nonselective activation/blockade of α2 subtypes by clonidine/atipamezole could thus result in overall null effect on PPI in the present study.
The finding that NE release (α2) and α1 receptor activation is required for CRF effects on startle hyperreactivity and that CRF1 activation is required for NE-induced increases in startle may have clinical implications for PTSD. The CRF and noradrenergic systems, both of which influence arousal, have been implicated in the pathophysiology of PTSD (Baker et al., 1999; Bremner et al., 1997; Geracioti et al., 2008). Blockade of α2 receptors selectively increases acoustic startle in combat veterans with PTSD but not combat controls (Morgan et al., 1995). These clinical findings, in conjunction with the present preclinical findings, suggest that PTSD subjects may have increased vulnerability to NE effects on startle, perhaps due to increased CRF signaling (and vice versa). Prazosin has shown some promise in treating sleep disturbances in PTSD (Strawn and Geracioti, 2008) and α2 agonists may be another novel way to treat PTSD symptoms (Krystal and Neumeister, 2009). A current clinical trial is also examining the effects of CRF1 antagonists on PTSD symptomology (clinicaltrials.gov). Our findings support the notion that targeting NE and CRF signaling mechanisms may prove beneficial in specifically treating heightened arousal symptoms associated with PTSD.
Acknowledgments
This research was supported by MH074697. We would like to thank Dr. Mark Geyer for helpful discussions and comments on earlier versions of this manuscript and Ms. Chelsea Wallace for technical assistance.
Footnotes
STATEMENT of INTEREST: None
References
- Adamec R, Fougere D, Risbrough V. CRF receptor blockade prevents initiation and consolidation of stress effects on affect in the predator stress model of PTSD. International Journal of Neuropsychopharmacology. 2009;13:1–11. doi: 10.1017/S1461145709990496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamec R, Muir C, Grimes M, Pearcey K. Involvement of noradrenergic and corticoid receptors in the consolidation of the lasting anxiogenic effects of predator stress. Behavioural Brain Research. 2007;179:192–207. doi: 10.1016/j.bbr.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Alsene KM, Carasso BS, Connors EE, Bakshi VP. Disruption of prepulse inhibition after stimulation of central but not peripheral alpha-1 adrenergic receptors. Neuropsychopharmacology. 2006;31:2150–2161. doi: 10.1038/sj.npp.1300989. [DOI] [PubMed] [Google Scholar]
- Astrachan DI, Gallager DW, Davis M. Behavior and binding: desensitization to alpha 1-adrenergic stimulation of acoustic startle is associated with a decrease in alpha 1-adrenoceptor binding sites. Brain Research. 1983;276:183–187. doi: 10.1016/0006-8993(83)90562-0. [DOI] [PubMed] [Google Scholar]
- Baker DG, West SA, Nicholson WE, Ekhator NN, et al. Serial CSF corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder. American Journal of Psychiatry. 1999;156:585–588. doi: 10.1176/ajp.156.4.585. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. Increased depression-like behaviors in corticotropin-releasing factor receptor-2-deficient mice: sexually dichotomous responses. Journal of Neuroscience. 2003;23:5295–5301. doi: 10.1523/JNEUROSCI.23-12-05295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AM, Powell SB, Markou A, Geyer MA. Iloperidone reduces sensorimotor gating deficits in pharmacological models, but not a developmental model, of disrupted prepulse inhibition in rats. Neuropharmacology. 2006;51:457–465. doi: 10.1016/j.neuropharm.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Dunn AJ. Restraint-stress-induced changes in exploratory behavior appear to be mediated by norepinephrine-stimulated release of CRF. Journal of Neuroscience. 1989;9:3513–3521. doi: 10.1523/JNEUROSCI.09-10-03513.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Licinio J, Darnell A, Krystal JH, et al. Elevated CSF corticotropin-releasing factor concentrations in posttraumatic stress disorder. American Journal of Psychiatry. 1997;154:624–629. doi: 10.1176/ajp.154.5.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carasso BS, Bakshi VP, Geyer MA. Disruption in prepulse inhibition after alpha-1 adrenoceptor stimulation in rats. Neuropharmacology. 1998;37:401–404. doi: 10.1016/s0028-3908(98)00051-3. [DOI] [PubMed] [Google Scholar]
- Conti LH, Sutherland JE, Muhlhauser CM. Interaction between the effects of corticotropin-releasing factor and prepulse parameters on prepulse inhibition in two inbred rat strains and the F1 generation of a cross between them. Behavioural Brain Research. 2009;200:165–172. doi: 10.1016/j.bbr.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis AL, Lechner SM, Pavcovich LA, Valentino RJ. Activation of the locus coeruleus noradrenergic system by intracoerulear microinfusion of corticotropin-releasing factor: effects on discharge rate, cortical norepinephrine levels and cortical electroencephalographic activity. Journal of Pharmacology and Experimental Therapeutics. 1997;281:163–172. [PubMed] [Google Scholar]
- Davis M. Neurochemical modulation of sensory-motor reactivity: acoustic and tactile startle reflexes. Neuroscience and Biobehavioral Reviews. 1980;4:241–263. doi: 10.1016/0149-7634(80)90016-0. [DOI] [PubMed] [Google Scholar]
- Davis M, Walker DL, Lee Y. Roles of the amygdala and bed nucleus of the stria terminalis in fear and anxiety measured with the acoustic startle reflex. Possible relevance to PTSD. Annals of the New York Academy of Sciences. 1997;821:305–331. doi: 10.1111/j.1749-6632.1997.tb48289.x. [DOI] [PubMed] [Google Scholar]
- Dirks A, Groenink L, Westphal KG, Olivier JD, et al. Reversal of startle gating deficits in transgenic mice overexpressing corticotropin-releasing factor by antipsychotic drugs. Neuropsychopharmacology. 2003;28:1790–1798. doi: 10.1038/sj.npp.1300256. [DOI] [PubMed] [Google Scholar]
- Djouma E, Card K, Lodge DJ, Lawrence AJ. The CRF1 receptor antagonist, antalarmin, reverses isolation-induced up-regulation of dopamine D2 receptors in the amygdala and nucleus accumbens of fawn-hooded rats. European Journal of Neuroscience. 2006;23:3319–3327. doi: 10.1111/j.1460-9568.2006.04864.x. [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW. Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. Journal of Comparative Neurology. 2004;468:277–298. doi: 10.1002/cne.10949. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, Palamarchouk V. Brain circuits involved in corticotropin-releasing factor-norepinephrine interactions during stress. Annals of the New York Academy of Sciences. 2004;1018:25–34. doi: 10.1196/annals.1296.003. [DOI] [PubMed] [Google Scholar]
- Fendt M, Koch M, Schnitzler HU. Corticotropin-releasing factor in the caudal pontine reticular nucleus mediates the expression of fear-potentiated startle in the rat. European Journal of Neuroscience. 1997;9:299–305. doi: 10.1111/j.1460-9568.1997.tb01400.x. [DOI] [PubMed] [Google Scholar]
- Fendt M, Siegl S, Steiniger-Brach B. Noradrenaline transmission within the ventral bed nucleus of the stria terminalis is critical for fear behavior induced by trimethylthiazoline, a component of fox odor. Journal of Neuroscence. 2005;25:5998–6004. doi: 10.1523/JNEUROSCI.1028-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forray MI, Gysling K, Andrés ME, Bustos G, et al. Medullary noradrenergic neurons projecting to the bed nucleus of the stria terminalis express mRNA for the NMDA-NR1 receptor. Brain Research Bulletin. 2000;52:163–169. doi: 10.1016/s0361-9230(00)00229-x. [DOI] [PubMed] [Google Scholar]
- Geracioti TD, Jr, Baker DG, Kasckow JW, Strawn JR, et al. Effects of trauma-related audiovisual stimulation on cerebrospinal fluid norepinephrine and corticotropin-releasing hormone concentrations in post-traumatic stress disorder. Psychoneuroendocrinology. 2008;33:416–424. doi: 10.1016/j.psyneuen.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Goldschmidt PL, Frances H, Simon P. Stimulation of beta-adrenergic receptors and spontaneous motor activity in mice. Pharmacology, Biochemistry, and Behavior. 1984;21:177–180. doi: 10.1016/0091-3057(84)90210-7. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Rukstalis M, Lewis MC. Atomoxetine and nicotine enhance prepulse inhibition of acoustic startle in C57BL/6 mice. Neuroscience Letters. 2005;377:85–90. doi: 10.1016/j.neulet.2004.11.073. [DOI] [PubMed] [Google Scholar]
- Graham FK. Presidential Address, 1974. The more or less startling effects of weak prestimulation. Psychophysiology. 1975;12:238–248. doi: 10.1111/j.1469-8986.1975.tb01284.x. [DOI] [PubMed] [Google Scholar]
- Griebel G, Simiand J, Steinberg R, Jung M, et al. 4-(2-Chloro-4-methoxy-5-methylphenyl)-N-[(1S)-2-cyclopropyl-1-(3-fluoro-4- methylphenyl)ethyl]5-methyl-N-(2-propynyl)-1, 3-thiazol-2-amine hydrochloride (SSR125543A), a potent and selective corticotrophin-releasing factor(1) receptor antagonist. II. Characterization in rodent models of stress-related disorders. Journal of Pharmacology and Expimental Therapeutics. 2002;301:333–345. doi: 10.1124/jpet.301.1.333. [DOI] [PubMed] [Google Scholar]
- Grillon C. Models and mechanisms of anxiety: evidence from startle studies. Psychopharmacology. 2008;199:421–437. doi: 10.1007/s00213-007-1019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, 3rd, Davis M, Southwick SM. Effects of experimental context and explicit threat cues on acoustic startle in Vietnam veterans with posttraumatic stress disorder. Biological Psychiatry. 1998;44:1027–1036. doi: 10.1016/s0006-3223(98)00034-1. [DOI] [PubMed] [Google Scholar]
- Grillon C, Morgan CA, Southwick SM, Davis M, et al. Baseline startle amplitude and prepulse inhibition in Vietnam veterans with posttraumatic stress disorder. Psychiatry Research. 1996;64:169–178. doi: 10.1016/s0165-1781(96)02942-3. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS and Neurological Disorders: Drug Targets. 2006;5:453–479. doi: 10.2174/187152706777950684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holstege G, Meiners L, Tan K. Projections of the bed nucleus of the stria terminalis to the mesencephalon, pons, and medulla oblongata in the cat. Experimental Brain Research. 1985;58 :379–391. doi: 10.1007/BF00235319. [DOI] [PubMed] [Google Scholar]
- Isogawa K, Akiyoshi J, Hikichi T, Yamamoto Y, et al. Effect of corticotropin releasing factor receptor 1 antagonist on extracellular norepinephrine, dopamine and serotonin in hippocampus and prefrontal cortex of rats in vivo. Neuropeptides. 2000;34:234–239. doi: 10.1054/npep.2000.0806. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, McGaugh JL. Delayed onset of the amnestic effect of posttraining beta-endorphin: effects of propranolol administered prior to retention testing. European Journal of Pharmacology. 1985;113:105–108. doi: 10.1016/0014-2999(85)90348-6. [DOI] [PubMed] [Google Scholar]
- Jeong KH, Jacobson L, Pacak K, Widmaier EP, et al. Impaired basal and restraint-induced epinephrine secretion in corticotropin-releasing hormone-deficient mice. Endocrinology. 2000;141:1142–1150. doi: 10.1210/endo.141.3.7370. [DOI] [PubMed] [Google Scholar]
- Jones BE, Yang TZ. The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. The Journal of Comparative Neurology. 1985;242:56–92. doi: 10.1002/cne.902420105. [DOI] [PubMed] [Google Scholar]
- Koch M. The neurobiology of startle. Progress in Neurobiology. 1999;59:107–128. doi: 10.1016/s0301-0082(98)00098-7. [DOI] [PubMed] [Google Scholar]
- Koob GF. Corticotropin-releasing factor, norepinephrine, and stress. Biological Psychiatry. 1999;46:1167–1180. doi: 10.1016/s0006-3223(99)00164-x. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Neumeister A. Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience. Brain Research. 2009;1293:13–23. doi: 10.1016/j.brainres.2009.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahdesmaki J, Sallinen J, MacDonald E, Scheinin M. Alpha2A-adrenoceptors are important modulators of the effects of D-amphetamine on startle reactivity and brain monoamines. Neuropsychopharmacology. 2004;29:1282–1293. doi: 10.1038/sj.npp.1300428. [DOI] [PubMed] [Google Scholar]
- Lee EH, Chang SY, Chen AY. CRF facilitates NE release from the hippocampus: a microdialysis study. Neuroscience Research. 1994;19:327–330. doi: 10.1016/0168-0102(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Lee Y, Davis M. Role of the hippocampus, the bed nucleus of the stria terminalis, and the amygdala in the excitatory effect of corticotropin-releasing hormone on the acoustic startle reflex. Journal of Neuroscience. 1997;17:6434–6446. doi: 10.1523/JNEUROSCI.17-16-06434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Lopez DE, Meloni EG, Davis M. A primary acoustic startle pathway: obligatory role of cochlear root neurons and the nucleus reticularis pontis caudalis. Journal of Neuroscience. 1996;16:3775–3789. doi: 10.1523/JNEUROSCI.16-11-03775.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang KC, Melia KR, Miserendino MJ, Falls WA, et al. Corticotropin-releasing factor: long-lasting facilitation of the acoustic startle reflex. Journal of Neuroscience. 1992;12:2303–2312. doi: 10.1523/JNEUROSCI.12-06-02303.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yu B, Neugebauer V, Grigoriadis DE, et al. Corticotropin-releasing factor and Urocortin I modulate excitatory glutamatergic synaptic transmission. Journal of Neuroscience. 2004;24:4020–4029. doi: 10.1523/JNEUROSCI.5531-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon MK, Lloyd RL, Fitten LJ. Antagonism of the hypothermic effect of clozapine in mice by centrally-active alpha 2-adrenergic antagonists and alpha 1-adrenergic agonists. Psychopharmacology. 1990;101:67–72. doi: 10.1007/BF02253720. [DOI] [PubMed] [Google Scholar]
- Morgan CA, 3rd, Grillon C, Southwick SM, Nagy LM, et al. Yohimbine facilitated acoustic startle in combat veterans with post-traumatic stress disorder. Psychopharmacology. 1995;117:466–471. doi: 10.1007/BF02246220. [DOI] [PubMed] [Google Scholar]
- Okuyama S, Sakagawa T, Chaki S, Imagawa Y, et al. Anxiety-like behavior in mice lacking the angiotensin II type-2 receptor. Brain Research. 1999;821:150–159. doi: 10.1016/s0006-8993(99)01098-7. [DOI] [PubMed] [Google Scholar]
- Pole N, Neylan TC, Otte C, Henn-Hasse C, et al. Prospective prediction of posttraumatic stress disorder symptoms using fear potentiated auditory startle responses. Biological Psychiatry. 2009;65:235–240. doi: 10.1016/j.biopsych.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Fox K, Valentino RJ, Van Bockstaele EJ. Agonist-induced internalization of corticotropin-releasing factor receptors in noradrenergic neurons of the rat locus coeruleus. European Journal of Neuroscience. 2006;23:2991–2998. doi: 10.1111/j.1460-9568.2006.04820.x. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Geyer MA. Anxiogenic treatments do not increase fear-potentiated startle in mice. Biological Psychiatry. 2005;57:33–43. doi: 10.1016/j.biopsych.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Geyer MA, Hauger RL, Coste S, et al. CRF1 and CRF2 receptors are required for potentiated startle to contextual but not discrete cues. Neuropsychopharmacology. 2009;34:1494–1503. doi: 10.1038/npp.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risbrough VB, Hauger RL, Pelleymounter MA, Geyer MA. Role of corticotropin releasing factor (CRF) receptors 1 and 2 in CRF-potentiated acoustic startle in mice. Psychopharmacology. 2003;170:178–187. doi: 10.1007/s00213-003-1535-6. [DOI] [PubMed] [Google Scholar]
- Risbrough VB, Hauger RL, Roberts AL, Vale WW, et al. Corticotropin-releasing factor receptors CRF1 and CRF2 exert both additive and opposing influences on defensive startle behavior. Journal of Neuroscience. 2004;24:6545–6552. doi: 10.1523/JNEUROSCI.5760-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risbrough VB, Stein MB. Role of corticotropin releasing factor in anxiety disorders: a translational research perspective. Hormones and Behavior. 2006;50:550–561. doi: 10.1016/j.yhbeh.2006.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha N, Datta H, Sharma PL. Effects of morphine on memory: interactions with naloxone, propranolol and haloperidol. Pharmacology. 1991;42:10–14. doi: 10.1159/000138762. [DOI] [PubMed] [Google Scholar]
- Sallinen J, Haapalinna A, Viitamaa T, Kobilka BK, et al. Adrenergic alpha2C receptors modulate the acoustic startle reflex, prepulse inhibition, and aggression in mice. Journal of Neuroscience. 1998;18:3035–3042. doi: 10.1523/JNEUROSCI.18-08-03035.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautter FJ, Bissette G, Wiley J, Manguno-Mire G, et al. Corticotropin-releasing factor in posttraumatic stress disorder (PTSD) with secondary psychotic symptoms, nonpsychotic PTSD, and healthy control subjects. Biological Psychiatry. 2003;54:1382–1388. doi: 10.1016/s0006-3223(03)00571-7. [DOI] [PubMed] [Google Scholar]
- Schank JR, Liles LC, Weinshenker D. Norepinephrine signaling through beta-adrenergic receptors is critical for expression of cocaine-induced anxiety. Biological Psychiatry. 2008;63:1007–1012. doi: 10.1016/j.biopsych.2007.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz J, Daly JW. Accumulation of cyclic adenosine 3′, 5′-monophosphate in cerebral cortical slices from rat and mouse: Stimulatory effect of α- and β-adrenergic agents and adenosine. Journal of Neurochemistry. 1973;21:1319–1326. doi: 10.1111/j.1471-4159.1973.tb07585.x. [DOI] [PubMed] [Google Scholar]
- Shilling PD, Melendez G, Priebe K, Richelson E, et al. Neurotensin agonists block the prepulse inhibition deficits produced by a 5-HT2A and an alpha1 agonist. Psychopharmacology. 2004;175:353–359. doi: 10.1007/s00213-004-1835-5. [DOI] [PubMed] [Google Scholar]
- Stevens DR, McCarley RW, Greene RW. The mechanism of noradrenergic alpha 1 excitatory modulation of pontine reticular formation neurons. The Journal of Neuroscience. 1994;14:6481–6487. doi: 10.1523/JNEUROSCI.14-11-06481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawn JR, Geracioti TD., Jr Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depression and Anxiety. 2008;25:260–271. doi: 10.1002/da.20292. [DOI] [PubMed] [Google Scholar]
- Strazielle C, Lalonde R, Hébert C, Reade TA. Regional brain distribution of noradrenaline uptake sites, and of α1-, α2- and β-adrenergic receptors in PCD mutant mice: a quantitative autoradiographic study. Neuroscience. 1999;94:287–304. doi: 10.1016/s0306-4522(99)00321-8. [DOI] [PubMed] [Google Scholar]
- Sutherland JE, Page ME, Conti LH. The effect of corticotropin-releasing factor on prepulse inhibition is independent of serotonin in Brown Norway and Wistar-Kyoto rats. Pharmacology, Biochemistry, and Behavior. 2008;89:324–337. doi: 10.1016/j.pbb.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Bongiovanni MJ, Tochen L, Shoemaker JM. Separable noradrenergic and dopaminergic regulation of prepulse inhibition in rats: implications for predictive validity and Tourette Syndrome. Psychopharmacology. 2006;186:246–254. doi: 10.1007/s00213-006-0374-7. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Britton KT, Koob GF. Potentiation of acoustic startle by corticotropin-releasing factor (CRF) and by fear are both reversed by alpha-helical CRF (9–41) Neuropsychopharmacology. 1989;2:285–292. doi: 10.1016/0893-133x(89)90033-x. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. Neural circuit regulation of prepulse inhibition of startle in the rat: Current knowledge and future challenges. Psychopharmacology. 2001;156:194–215. doi: 10.1007/s002130100799. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Vale WW, Koob GF. Corticotropin-releasing factor potentiates acoustic startle in rats: blockade by chlordiazepoxide. Psychopharmacology. 1986;88 :147–152. doi: 10.1007/BF00652231. [DOI] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Colago EE, Valentino RJ. Corticotropin-releasing factor-containing axon terminals synapse onto catecholamine dendrites and may presynaptically modulate other afferents in the rostral pole of the nucleus locus coeruleus in the rat brain. Journal of Comparative Neurology. 1996;364:523–534. doi: 10.1002/(SICI)1096-9861(19960115)364:3<523::AID-CNE10>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. Journal of Comparative Neurology. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Varty GB, Bakshi VP, Geyer MA. M100907, a serotonin 5-HT2A receptor antagonist and putative antipsychotic, blocks dizocilpine-induced prepulse inhibition deficits in Sprague-Dawley and Wistar rats. Neuropsychopharmacology. 1999;20:311–321. doi: 10.1016/S0893-133X(98)00072-4. [DOI] [PubMed] [Google Scholar]
- Vinkers CH, Risbrough VB, Geyer MA, Caldwell S, et al. Role of dopamine D1 and D2 receptors in CRF-induced disruption of sensorimotor gating. Pharmacology, Biochemistry, and Behavior. 2007;86:550–558. doi: 10.1016/j.pbb.2007.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vythilingam M, Anderson GM, Owens MJ, Halasznski TM, et al. Cerebrospinal fluid corticotropin-releasing hormone in healthy humans: effects of yohimbine and naloxone. Journal of Clinical Endocrinology and Metabolism. 2000;85:4138–4145. doi: 10.1210/jcem.85.11.6968. [DOI] [PubMed] [Google Scholar]
- Walker DL, Miles LA, Davis M. Selective participation of the bed nucleus of the stria terminalis and CRF in sustained anxiety-like versus phasic fear-like responses. Progress in Neuropsychopharmacology and Biological Psychiatry. 2009;33:1291–1308. doi: 10.1016/j.pnpbp.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrindast MR, Haidari H, Jafari MR, Djahanguiri B. Influence of beta-adrenoceptor agonists and antagonists on baclofen-induced memory impairment in mice. Behavioral Pharmacology. 2004;15:293–297. doi: 10.1097/01.fbp.0000137211.95623.07. [DOI] [PubMed] [Google Scholar]