

Abstract
In response to decreasing Ca2+ levels in the endoplasmic reticulum, STIM proteins couple with Orai channels in the plasma membrane, leading to Ca2+ influx into the cell. In addition to Ca2+-related endoplasmic reticulum stress, STIM proteins are emerging as general stress sensors that react to multiple stress signals to orchestrate Ca2+ signaling and homeostasis.
STIM proteins are Ca2+-sensing switches that function as dynamic intermembrane communicators, controlling Ca2+ signals and Ca2+ homeostasis. Discovering the role of STIM proteins in mediating Ca2+ signal generation resolved the long-standing question of how depletion of Ca2+ within the endoplasmic reticulum (ER) triggers the opening of plasma membrane (PM) ‘store-operated’ Ca2+ entry channels1,2. The two STIM proteins, STIM1 and STIM2, are ubiquitously expressed, single-spanning ER membrane proteins that operate as mobile mediators of Ca2+ signals1. STIM proteins accurately sense ER luminal Ca2+ levels through their N-terminal Ca2+-binding EF-hand domains and function as switches that are activated by small decreases in luminal Ca2+. In their activated state, STIM proteins are highly dynamic communicators, rapidly aggregating and translocating within the ER membrane into discrete junctions closely juxtaposed with the PM1. Here, the activated STIM proteins couple directly with the highly Ca2+-selective family of Orai channels, tethering and gating the channels to generate Ca2+ entry signals2 (Box 1; Fig. 1). The entering Ca2+ provides spatially restricted and longer-term Ca2+ signals that are crucial in the control of cellular responses, including gene expression and growth1–3.
Box 1. Activation of STIM proteins and coupling to open Orai channels.
At the STIM protein N terminus
The luminal N-terminal Ca2+-sensing domain of STIM1 is a tightly clustered group of short helices comprising EF-hand and sterile α motif (SAM) domains. There are two EF-hands: a ‘canonical’ Ca2+-binding EF-hand (cEF) with a Kd for Ca2+ ranging from 0.2 to 0.6 mM and a ‘hidden’ EF-hand (hEF)12. When Ca2+ is bound, the EF-hand and SAM domains form a tight, stable configuration. When Ca2+ dissociates, the EF-SAM domain unfolds and destabilizes12, leading to rapid oligomerization of STIM1 proteins.
At the STIM protein C terminus
The cytoplasmic C-terminal region of STIM1 contains extensive coiled-coil domains that can span the ER-PM junctional space, estimated to be 10–20 nm13, allowing STIM to couple with and activate PM Orai channels. The C-terminal cytosolic domains of STIM proteins interact with and activate Orai channels. The small ‘CAD’ region of STIM1 (residues 342–448) is sufficient to fully activate Orai channels14. In its resting state, the active CAD domain of STIM1 is obscured by intramolecular electrostatic interactions that fold and bind the active acidic coupling domain on STIM1, preventing it from interacting with the PM. STIM1 activation involves the dissociation of inhibitory intramolecular electrostatic interactions between the coiled-coil CC1 and CAD regions of STIM1. This triggers exposure of the CAD acidic domain, which can then interact with basic sequences on Orai1. In the STIM1 resting state, these acidic residues remain hidden, suggesting that STIM1 has an autoinhibitory intramolecular regulatory mechanism10.
STIM1 has three target interaction sites
A short basic sequence (residues 382–386) within the CAD region interacts electrostatically with an acidic sequence (residues 272– 291) on the C terminus of Orai1; this is the crucial ‘coupling’ interaction required to activate Orai channels10.
A distinct but unknown site on STIM1 likely interacts with the Orai1 N terminus, enhancing the successful interaction10,14.
A lysine-rich, polybasic C-terminal tail on STIM1 binds directly with PM acidic phospholipids and helps anchor STIM1 to the PM independently of Orai channels14.
Figure 1. STIM activation.

(a). STIM proteins in the resting state. Under normal conditions, InsP3R Ca2+ release channels are closed, ER Ca2+ stores are full and STIM proteins with Ca2+ bound to their EF-hands are distributed throughout the ER. There is no coupling with Orai channels in ER-PM junctions, and inactive Orai channels are distributed evenly within the PM. (b) STIM proteins in activated cells. Receptor-induced InsP3 production activates Ca2+ release channels (InsP3Rs). In areas of Ca2 depletion, Ca2+ dissociation from STIM proteins causes their activation, leading to aggregation and translocation into ER-PM junctions, where they become trapped through interactions with PM lipids and Orai proteins. (c) STIM activation and coupling to Orai channels. STIM proteins sense small changes in luminal Ca2+ and function as molecular switches to couple with and activate Orai channels.
STIM proteins as multiple stress sensors
The diminished concentration of Ca2+ within the ER that triggers STIM protein activation is a major stress condition that compromises the luminal protein-folding environment. However, Ca2+ depletion in the ER is not the only stress condition that modulates STIM protein function. STIM1 also functions as an oxidative stress sensor, becoming activated by reactive oxygen species (ROS) and coupling to cause opening of Orai channels independent of ER Ca2+ depletion4. STIM proteins also respond to another major stress: temperature variation. Small temperature increases trigger activation of STIM1, leading to its aggregation and translocation into junctions, independent of Ca2+-store depletion5. At higher temperatures, however, the functional coupling that opens Orai1 channels is blocked; subsequent cooling allows efficient STIM coupling to resume and Orai channels to open.
We consider the activation of STIM proteins in response to each of three conditions of cellular stress: depletion of Ca2+ within the ER lumen, which severely stresses ER function; induction of oxidative stress through ROS; and exposure of cells to rapid temperature changes. In each case, the activation of STIM proteins follows a similar pattern of aggregation and translocation into ER-PM junctions. Within these junctions, activated STIM proteins are able to couple with their targets. STIM proteins have important target proteins in addition to Orai channels, and we also consider the implications of these new targets in STIM-mediated cellular control.
ER Ca2+ stress sensing
The lumen of the ER is a unique environment containing high concentrations of Ca2+ (at least 1,000-fold higher than in the cytosol) and an abundance of Ca2+-binding proteins crucial to the many functions of the ER, including the synthesis of membrane and secretory proteins, folding and post-translational modification of proteins, synthesis of lipids and sterols, as well as Ca2+ signal generation and Ca2+ homeostatic control6. Ca2+-binding proteins in the ER serve both as molecular chaperones to mediate correct protein folding and as buffers to sequester large quantities of Ca2+. Their chaperone function is highly dependent on the amount of luminal Ca2+; decreases in Ca2+ concentration result in the well-described ER stress response, which includes global inhibition of protein synthesis, induction of multiple stress pathways, apoptosis, autophagy and necrosis7. Thus, maintaining high levels of Ca2+ within the ER is crucial, as decreases in luminal Ca2+ cause severe stress, resulting in the activation of the unfolded protein response to mitigate damage from misfolded proteins6.
STIM proteins orchestrate the cellular response to changes in luminal Ca2+. They sense changes in ER Ca2+ and allow cells to adapt and maintain Ca2+ within the ER stores by activating Ca2+ through Orai channels. An intimately associated but distinct role of STIM protein activation is to trigger cytosolic Ca2+ signals that control longer-term cellular functions, including transcription and cell growth. Both STIM1 and STIM2 are ubiquitously expressed, highly homologous proteins varying only at their extreme N and C termini. There are subtle differences in their Ca2+ sensitivities8 and their kinetics of activation9. STIM1 is the major mediator of store-operated Ca2+ signals and becomes rapidly activated when ER Ca2+ is depleted; in contrast, STIM2 has greater sensitivity to Ca2+-store depletion but has a slower activation rate than STIM1 and functions to finely control Ca2+ homeostasis8,9.
STIM proteins are normally widely distributed across the ER. As Ca2+ is depleted from the ER through receptor-induced Ca2+ release, it dissociates and activates STIM proteins, causing them to aggregate and translocate into ER-PM junctions, where they couple to open Orai channels. STIM proteins likely exist as resting dimers and self-associate through interactions within their luminal N termini and their cytoplasmic coiled-coil regions10. As STIM proteins are activated (Box 1; Fig. 1), conformational unraveling occurs and sites on STIM1 become available for interactions with PM lipids or with Orai channels present in the PM. Thus, STIM proteins function as highly controlled molecular switches triggered by small changes in luminal Ca2+. Movement of STIM proteins into ER-PM junctions appears to be diffusion driven, and their accumulation within junctions results from interactions with PM lipids and Orai proteins. Importantly, the activation, aggregation and PM coupling of STIM proteins is entirely reversible: immediately upon replenishment of ER Ca2+, STIM binds Ca2+ at its luminal EF-hand motif and rapidly deactivates, leading to dissociation from the PM and Orai proteins and, finally, diffusion of both STIM and Orai proteins through the junctions to become redistributed within the ER and PM, respectively1.
As accurate sensors of the ER intraluminal Ca2+ environment, STIM proteins are intimately involved in both Ca2+ signaling and Ca2+ homeostasis. Indeed, these two processes are essentially one and the same; complete depletion of Ca2+ within the ER does not occur in living cells, as it would have devastating consequences on the function of the ER. The combination of STIM1 and STIM2, with their distinct sensitivities to luminal Ca2+ changes, accurately detects either very small changes in luminal Ca2+ throughout the ER or larger local changes within specific ER regions containing high densities of Ca2+ release channels (Figs. 1a,b). Thus, the number of STIM proteins moving into ER-PM junctions to activate Orai Ca2+ channels is an integration of all the deviations of Ca2+ that occur throughout the ER. Orai channel gating by STIM proteins results in the entry of just a tiny trickle of Ca2+ into the cytosol by virtue of the exceptionally high Ca2+ selectivity of Orai channels1,2. However, the entry of a few thousand Ca2+ ions is sufficient to mediate significant cytosolic Ca2+ signals in an average-sized cell. The incoming Ca2+ ions can be sequestered using ER Ca2+ pumps to replenish any deficit in luminal Ca2+, protecting the functional integrity of the ER.
Moreover, the ER functions as a Ca2+ reservoir, rhythmically releasing it to generate cytosolic Ca2+ oscillations, which are crucial in providing sustained and precise Ca2+-mediated responses in many cell types3. Indeed, STIM proteins can move in and out of ER-PM junctions in response to local luminal Ca2+ changes occurring during individual oscillations3. The STIM proteins allow Ca2+ entry, which sustains the oscillations and ensures that ER function is not compromised by Ca2+ depletion. Notably, the small amount of STIM-induced entry of Ca2+ within the oscillations can have profoundly important signaling functions. Thus, the spatially restricted Ca2+ entering through STIM-activated Orai channels during oscillations provides a unique ‘Ca2+ signature response,’ which can be crucial in controlling early gene expression and growth responses3,11.
Oxidative stress
The scheme shown in Figure 2 compares the sensing functions and activation mechanisms of STIM proteins in response to Ca2+-store depletion to two other stress conditions—oxidative stress and temperature change. Oxidative stress sensed by STIM proteins causes an initial activation process similar to that induced by Ca2+-store depletion4. The redox state of cells is fundamentally important to their function, and cells use multiple regulatory sensors to maintain redox homeostasis. ROS and reactive nitrogen species trigger important protein modifications via oxidative reactions. Indeed, alteration of cysteine residues by ROS or reactive nitrogen species can have major effects on protein function15. ROS can trigger Ca2+ transients via modulation of inositol-1,4,5-trisphosphate (InsP3)-dependent Ca2+ release as well as through extracellular Ca2+ influx16. Interestingly, STIM1 and STIM2 contain two highly conserved ER luminal cysteines toward the N terminus, and S-glutathionylation of human STIM1 at one of these cysteines (Cys56) results in STIM1 aggregation into PM-associated junctions under conditions where ER Ca2+ content is not depleted4; thus, this modification mimics the effects of Ca2+-store depletion (Fig. 2). Indeed, S-glutathionylation of STIM1 at Cys56 causes a substantial decrease in the binding of Ca2+ to the STIM1 EF-hand motif4, and it is likely that the modification is causing a dissociation of Ca2+ from STIM1 without actual ER Ca2+ depletion.
Figure 2.
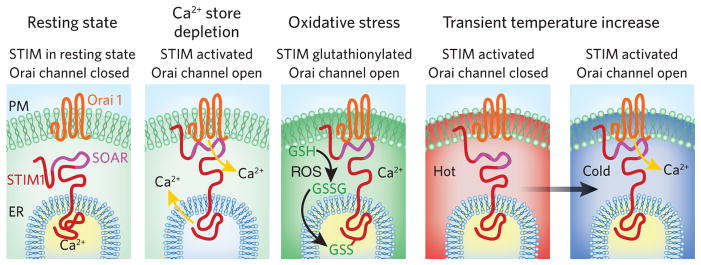
Multiple stress sensing by STIM1. Switching of STIM1 to the activated form can be triggered by either Ca2+ store depletion, oxidative stress or temperature increases. In each case, Ca2+ dissociation from the luminal EF hand likely occurs. For store depletion and activation by ROS, coupling to activate Orai channels occurs. The temperature increase prevents coupling to Orai; upon subsequent cooling, coupling to activate Orai channels occurs. SOAR, STIM-Orai activating region; GSH, glutathione.
S-glutathionylation is a modification of protein thiols formed by a reaction between cysteine residues and the tripeptide glutathione17. It has been implicated in mediating oxidation-induced changes that control a spectrum of responses, including energy metabolism, cytoskeletal assembly, protein folding and stability, ion-channel function and Ca2+ homeostasis17. The effect of glutathionylation on STIM1 activation indicates that STIM1 is important in redox sensing. Considering the close proximity of Cys56 to the EF-SAM domain in STIM1, these findings implicate Cys56 in the control of Ca2+ binding and STIM1 activation. This negative control can be eliminated by either S-glutathionylation or mutation of Cys56. At supraphysiological ROS levels, S-glutathionylated STIM1 induces Orai1-mediated Ca2+ entry, mitochondrial Ca2+ overload and decreased cellular bioenergetics4. In contrast, at physiological ROS levels, elevations in mitochondrial Ca2+ lead to elevated mitochondrial bioenergetics18, though the extent to which S-glutathionylation of STIM1 contributes to this important physiological role remains to be determined.
The S-glutathionylation of proteins is not necessarily a pathophysiological event and may serve as a defense mechanism against the irreversible oxidation of protein thiols15,17, such as the reactions leading to the formation of sulfinic (Cys-SO2H) or sulfonic acids (Cys-SO3H)15. Redox reactions15 and STIM-Orai function19 have been implicated in various disease pathogeneses, including those of cardiovascular diseases, cancer and immune disorders. Redox reaction–dependent disease progression depends on ROS and RNS production rates, endogenous antioxidant levels and duration of exposure. The discovery that STIM1 operates as a ROS sensor suggests that ROS pathogenesis may, in part, be mediated by STIM1-Orai function. STIM1-Orai1 activation in response to ROS overproduction facilitates B-lymphocyte activation, ultimately leading to mitochondrial-dependent cell death4. This activation was observed in response to lipopolysaccharide or IgM stimulation as well as glutathione depletion. Further, ablation of STIM1 markedly attenuated B-lymphocyte cell death in response to all three stimuli4. Conversely, paracrine-derived ROS resulting from immune-cell infiltration of vascular smooth muscle may stimulate STIM1-Orai1 activity and facilitate smooth muscle proliferation in atherosclerosis. Paracrine-derived ROS may also act as a chemoattractant for cancer metastasis16, a process also previously linked with STIM-Orai function19. Thus, characterization of the role of ROS-sensing and ROS-mediated STIM1 modification has enormous importance in a spectrum of pathophysiological conditions.
Temperature sensing
Temperature change is another physical stress that has profound effects on protein structure and protein interactions. Although higher vertebrates are homeothermic, many cell types (for example, circulating blood cells) experience significant temperature differentials, and the pathophysiological consequences of even small temperature changes during fever or hypothermia are profound. Interestingly, temperature alters STIM1 function5; small temperature increases (from 37 °C to above 40 °C) induce STIM1 activation, resulting in aggregation and translocation into ER-PM junctions without Ca2+ store-depletion (Fig. 2). Such changes mirror the actions of Ca2+-store depletion and oxidative stress induced by ROS. However, unlike the effects of store depletion or oxidative conditions, the effects of temperature on the functional interactions between STIM1 and Orai1 are complex. At elevated temperatures (above 40 °C), STIM1-activated coupling to open Orai1 channels is blocked. Exposing cells expressing STIM1 and Orai1 to high temperatures for short periods (1–2 min) ‘prime’ the coupling process. Returning to lower temperatures causes a profound activation of Orai1 channels (Fig. 2), independent of Ca2+-store depletion. Intriguingly, the heat-induced translocation of STIM1 into junctions cannot occur without the lysine-rich, polybasic STIM1 C-terminal domain5. In contrast, STIM1 can still translocate into junctions and activate Orai1 channels in response to Ca2+-store depletion without this polybasic domain. Therefore, the activation of STIM1 by Ca2+-store emptying is not equivalent to its activation by heat.
The explanation for how heat induces STIM1 activation and movement into junctions likely rests on the thermal stability of the EF-SAM domain. In vitro measurements on the degree of folding of the EF-SAM complex revealed an effective temperature causing 50% change (ET50) for STIM1 of ~19 °C in the absence of Ca2+ and ~45 °C in its presence20. This is highly comparable to the ET50 of 43.6 °C measured for heat-induced STIM1 junction formation5. Thus, as temperature increases, the EF-SAM domains of STIM1 likely unfold in a manner similar to that under Ca2+-reduced conditions. However, despite the movement of STIM1 into junctions, Orai1 activation was not observed until the temperature decreased again5. Because heat disrupted the interaction between the CAD domain of STIM1 and Orai1, it was concluded that the inhibition of Orai1 activation is a result of temperature-mediated inhibition of the STIM1-Orai1 coupling process.
The STIM1 response to temperature is complex. As for the activation of STIM1 by ROS, it may be that increases in temperature cause Ca2+ dissociation. The temperature changes that alter STIM activation and coupling fall within the physiological ranges encountered by circulating blood cells, keratinocytes and muscle cells, so STIM can be activated and ‘primed’ by physiologically relevant elevations in temperatures; however, STIM-Orai coupling and Orai activation require a decrease in temperature. This priming and subsequent activation may have particular importance in the responses of immune cells under fever conditions, in which the temperature differentials are amplified.
STIM targets—broader horizons
Although the three Orai channels are the most established STIM targets, additional PM targets have now been reported. Given the ubiquitous expression of STIM proteins among cells, their wide distribution throughout the ER, and their profound and rapid activation and translocation in response to multiple cellular stress conditions, it is not surprising that their interactions extend beyond Orai channels. STIM proteins interact with two additional target channels: TRPC and CaV1.2. In addition, there is evidence suggesting a store-dependent functional interaction between STIM1 and adenylyl cyclase21, although the specifics of this interaction remain uncharacterized.
The most studied of the alternative STIM targets is the TRPC family of channels1,22,23, but despite the breadth of literature examining STIM-TRPC interactions and coregulation1,22, this remains a highly controversial area of study. There is evidence that suggests functional interactions between STIM1 and TRPC channels; likewise, physical associations between STIM1 and several TRPC channel subtypes have been reported22,23. Coexpression and mutational analyses reveal STIM1-dependent TRPC channel activation24, and knockdown strategies show that loss of TRPC expression can decrease SOCE23. TRPC1-knockout animals appear to have normal SOCE within some tissues, although partial loss of this activity was reported in salivary gland acinar cells25. Although knockout of other TRPC channels may have small effects, more definitive studies of their functions concluded that they have no role in store-operated or STIM-activated Ca2+ entry26,27. Recent evidence suggests that PM insertion of TRPC channels may be under the control of STIM and Orai23, providing a potential explanation for some of the confusion regarding the store-dependence of these channels.
An important relationship between STIM proteins and the operation of CaV1.2 (L-type) Ca2+ channels has emerged27,28 (Fig. 3). STIM1 physically associates with L-type channels via a conformational coupling mechanism analogous to STIM-Orai interactions27,28. Indeed, the small Orai-interacting CAD domain of STIM1 also interacts with and modifies the function of CaV1.2, binding to a small region within its C-terminal tail. STIM1 interacts with Orai1 and CaV1.2 channels within the same ER-PM junctions (Fig. 3), and Orai1-induced STIM1 localization increases functional interactions with CaV1.2 channels27. Intriguingly, the interaction between STIM1 and CaV1.2 channels results in channel inhibition27,28 (Fig. 3). Thus, STIM activation in response to store depletion and stress conditions results in the reciprocal control of Orai and CaV1.2 channels. Such STIM-mediated switching between voltage-dependent and voltage-independent Ca2+ signaling may have great significance in controlling the function, growth and development of excitable cells.
Figure 3.

Multiple targets for STIM1. STIM1 interacts with both Orai1 and CaV1.2 channels, forming a large junctional complex within which STIM1 activates Orai1 channels and inhibits CaV1.2 channels. At rest, STIM1 is likely a dimer with its CAD domain (red) masked by internal binding to CC1 (blue; see Fig. 1). Luminal Ca2+ depletion causes STIM1 oligomerization and umasking of CAD acidic residues that bind basic residues on Orai1, trapping and activating its channel. The interaction with CaV1.2 channels requires the same STIM1-CAD domain but has yet to be characterized. Both channel targets appear to coexist within the same ER-PM junctional complex.
Implications and speculations on STIM
The discovery that STIM proteins are Ca2+ sensors provides considerable insight into how communication between the ER and PM controls Ca2+ signal generation. STIM proteins can now be seen as having broader importance and may be considered multimodal stress sensors, controlling Ca2+ signal generation and maintaining Ca2+ homeostasis. The full spectrum of STIM targets has yet to be determined. So far, the targets are predominantly channels, but modification of key signaling enzymes, such as adenylyl cyclase, increases the potential sphere of influence of STIM proteins. So far, STIM targets have all been found within the PM and are modified by transmembrane interactions. Whether STIM proteins exert control over ER membrane proteins is not known.
Investigations of STIM proteins as ROS and heat sensors have focused on Orai1 as the target; the extent to which other PM targets are modulated by ROS or heat remains unknown. STIM proteins are broadly expressed among cell types and throughout the ER. They are also expressed in the PM1, where their Ca2+-sensing motifs are exposed to the exterior of cells. As yet, definitive functions for PM-expressed STIM proteins have not been established. STIM1 or STIM2 animal knockout phenotypes reveal profound changes not only in immune cells but also in muscle, skin and neural cells1,2. Combined STIM1 and STIM2 knockdowns in mice are generally lethal early in life, and it is not yet known what pathophysiologies are affected by the STIM proteins. In conclusion, STIM proteins are central players in multiple stress responses. Future investigations directed toward characterizing the complex physiological and pathophysiological roles of STIM proteins will provide new insights into how stress conditions are sensed by cells and how modification of Ca2+ signals and Ca2+ homeostasis helps cells adjust to stress and avoid consequential damage.
Footnotes
Competing financial interests
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturechemicalbiology/.
References
- 1.Deng X, Wang Y, Zhou Y, Soboloff J, Gill DL. J Biol Chem. 2009;284:22501–22505. doi: 10.1074/jbc.R109.018655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hogan PG, Lewis RS, Rao A. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mancarella S, Wang Y, Gill DL. Curr Biol. 2009;19:R950–R952. doi: 10.1016/j.cub.2009.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawkins BJ, et al. J Cell Biol. 2010;190:391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao B, Coste B, Mathur J, Patapoutian A. Nat Chem Biol. 2011;7:351–358. doi: 10.1038/nchembio.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coe H, Michalak M. Gen Physiol Biophys. 2009;28:F96–F103. [PubMed] [Google Scholar]
- 7.Ron D, Walter P. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 8.Brandman O, Liou J, Park WS, Meyer T. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y, et al. J Biol Chem. 2009;284:19164–19168. doi: 10.1074/jbc.C109.010900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Deng X, Gill DL. Sci Signal. 2010;3:pe42. doi: 10.1126/scisignal.3148pe42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Capite J, Ng SW, Parekh AB. Curr Biol. 2009;19:853–858. doi: 10.1016/j.cub.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 12.Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Cell. 2008;135:110–122. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Liou J, et al. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park CY, et al. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dröge W. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 16.Madesh M, et al. J Cell Biol. 2005;170:1079–1090. doi: 10.1083/jcb.200505022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Cárdenas C, et al. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts-Thomson SJ, Peters AA, Grice DM, Monteith GR. Pharmacol Ther. 2010;127:121–130. doi: 10.1016/j.pharmthera.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 20.Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. J Biol Chem. 2006;281:35833–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- 21.Lefkimmiatis K, et al. Nat Cell Biol. 2009;11:433–442. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 22.Yuan JP, et al. Channels (Austin) 2009;3:221–225. doi: 10.4161/chan.3.4.9198. [DOI] [PubMed] [Google Scholar]
- 23.Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. PLoS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan JP, Zeng W, Huang GN, Worley PF, Muallem S. Nat Cell Biol. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, et al. Proc Natl Acad Sci USA. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeHaven W, et al. J Physiol (Lond) 2009;587:2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, et al. Science. 2010;330:105–109. doi: 10.1126/science.1191086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park CY, Shcheglovitov A, Dolmetsch R. Science. 2010;330:101–105. doi: 10.1126/science.1191027. [DOI] [PubMed] [Google Scholar]