

Abstract
Genome Wide Association Studies (GWAs) have begun to investigate associations between inherited genetic variations and breast cancer prognosis. Here we report our findings from a GWAs conducted in 536 early onset breast cancer patients aged 40 or less at diagnosis and with a mean follow-up period of 4.1 years (S.D=1.96). Patients were selected from the POSH (Prospective study of Outcomes in Sporadic versus Hereditary breast cancer). A Bonferroni correction for multiple testing determined that a p-value of 1.0 × 10−7 was a statistically significant association signal. Following QC we identified 487496 SNPs for association tests in stage-1. In stage 2, 35 SNPs with the most significant associations were genotyped in 1516 independent cases from the same early onset cohort. In stage-2, 11 SNPs remained associated in the same direction (p{less than or equal to}0.05). Fixed effects meta-analysis models identified one SNP associated at close to genome wide level of significance 556 kb upstream of the ARRDC3 locus HR=1.61 (1.33-1.96, p=9.5 × 10-7). Four further associations at or close to the PBX1, RORα, NTN1 and SYT6 loci also came close to genome wide significance levels (p=10-6). In the first ever GWAS for identification of SNPs associated with prognosis in early onset breast cancer patients we report a SNP upstream of the ARRDC3 locus as potentially associated with prognosis (Median follow-up time for genotypes CC=4 years, CT=3 years and TT=2.7 years, Wilcoxon rank sum test CC vs. CT, p=4 × 10-4 and CT vs. TT, P=0.76). Four further loci might also be associated with prognosis
Keywords: Early onset, Breast cancer, Prognosis, Survival analysis and GWAs
Introduction
Breast cancer incidence increases with increasing age. Less than 5% of all breast cancer cases are diagnosed before 40 years of age and less than 20% before 50 years of age (1). Treatments vary according to tumour stage and biological characteristics, age at diagnosis, menopausal status; co-morbidities are also important considerations in deciding what treatment to offer. Early age at breast cancer diagnosis is associated with a worse prognosis although the reasons for this are still imperfectly understood. Tumours in this age group are more likely to have adverse pathological features including a greater proportion of ER negative high grade tumours (2). Despite accounting for these factors, outcomes remain worse for young onset patients, particularly those with ER positive cancers and this may reflect a poorer response to breast cancer treatments in younger patients (3-6). A Swedish familial study demonstrated higher risk of mortality in affected first degree relatives of breast cancer patients which suggests a genetic component to prognosis following disease onset (7). A smaller gap in age at diagnosis between sister-sister pairs compared with mother-daughter pairs in this familial study coupled with poorer prognosis in sister-sister pairs suggests that earlier disease onset in sister-sister pairs could be linked with a greater genetic component for prognosis. Rare high penetrance genetic predisposition genes like BRCA1 are more frequently found to explain young onset breast cancer cases even in the absence of a family history (8, 9). In addition it is becoming clear that the growing number of common genetic variants which contribute to polygenic breast cancer risk, are associated often more strongly with susceptibility to a particular sub-type of breast cancer (10, 11).
Common genetic variants may influence prognosis either by influencing the type of tumour that develops, the host response to tumour or the handling or metabolism of breast cancer directed therapies. Two recent studies developed from genome wide association experiments have failed to identify SNPs which are irrefutably associated with breast cancer prognosis in individuals of Caucasian ancestry (12). The median age at diagnosis in the patients recruited for these GWASs were 66 and 52 years hence these cohorts are largely comprised of later onset breast cancer patients. A more recent two stage GWAS in Chinese breast cancer patients identified two potential associations with breast cancer but the association effects in replication samples were much weaker than in the discovery set and would not satisfy stringent tests for multiple hypothesis correction (13). Studies exploring the association of known risk SNPs with prognosis have hinted at a possible role for genetic variation in clinical outcomes (14, 15).
Here we report a 2-stage Genome Wide Association Study to identify common genetic variants which are associated with breast cancer prognosis by using a discovery set of young onset patients that were enriched for rapid disease progression and long term breast cancer specific survival. We attempt replication in a larger sample of early onset breast cancer patients from the same cohort who were unselected for survival extremes. We also seek replication of the main findings from analysis in early onset patients in relatively later onset breast cancer cases from Helsinki.
Patients, Materials and Methods
Breast cancer patients
Early onset breast cancer cases were selected from the POSH (Prospective study of Outcomes in Sporadic versus Hereditary breast cancer) study, participants were diagnosed with invasive breast cancer and were aged 40 or younger at diagnosis. Recruitments to the POSH cohort were made between January 2000 and January 2008 from oncology clinics across the UK. The vast majority (98%) of patients recruited to the study presented symptomatically. The recruitment, data collection and follow up procedures for the POSH study participants are described in detail elsewhere (16).
Stage 1 discovery dataset
In stage 1, 574 participants from the POSH study were selected for the discovery phase of the analysis aimed at hypothesis generation (17). To enrich the discovery set patients were selected from POSH in two different groups. The first group had ER, PR and HER-2 negative breast cancer (These triple negative patients have worse prognosis and they relapse early after diagnosis). This triple negative breast cancer group was also used to identify risk genes for Breast Cancer susceptibility in a previous study (11). In the second group we specifically enriched the selection for patients with either very short duration of breast cancer specific survival (<2 years, n=48) or relatively long duration of breast cancer specific survival (>4 years, n=125) but no selection based on immunohistochemistry was made in this group. Breast cancer specific survival was used as the definitive end-point for the survival analysis. Enrichment of affected individuals in a genetic association design increases the efficiency of and power to detect genetic effects (18). As all the study participants for this GWAs were derived from a single randomly sampled cohort of early onset patients, any overestimation of effect sizes in stage-1 was balanced out by meta-analysis with unenriched stage-2 samples. This approach is in keeping with a recent GWAS which identified five new breast cancer susceptibility loci by enriching cases by recruiting individuals with family history of breast cancer (10). There were no screen detected breast cancers amongst the POSH cases included in the discovery analysis, all had presented symptomatically. Amongst young onset breast cancer cases a higher than average proportion is likely to be BRCA1/2 gene carriers. Since BRCA1 and BRCA2 pathogenic germ-line mutations are not known to be independently associated with prognosis, BRCA status was not used in making the case selection for this study. The cohort has not yet been systematically tested for germ-line BRCA1/2 mutations but amongst the POSH study stage-1 participants, 27.4% (147 cases) had previously been analysed for BRCA1 and BRCA2 mutations, either as part of other research studies or because testing had been clinically indicated (strong family history). Of those tested 38 (25.8%) had been found to carry clearly pathogenic mutations in BRCA1 or BRCA2.
Stage 2 replication dataset 1
1516 additional young onset cases from the POSH study that had not been selected in the discovery set were genotyped for replication in stage 2. 22.4% of the stage-2 participants were tested for BRCA status. 21.7% of those tested for BRCA status were found to carry pathogenic BRCA 1 and BRCA 2 mutations.
Breast Cancer Patients from Helsinki
The Helsinki samples were collected in Helsinki, Finland and are representative of Breast cancer cases at the recruitment centre during the collection period (1997-1998 and 2000). All the breast cancer cases included had histopathological and Survival data available. Detailed information on data collection and selection of participants has previously been published (19). The mean age at diagnosis was 56.8 years.
Genome wide genotyping
Genotyping for the 574 phase 1 breast cancer cases was conducted using the Illumina 660-Quad SNP array. Genotyping for the samples was conducted in two separate batches at two locations. 274 patients were genotyped at the Mayo Clinic, Rochester, Minnesota, USA and were selected because they were diagnosed with triple negative breast cancer (ER, PR and HER2 negative) (20). 300 POSH patients were genotyped at the Genome Institute of Singapore, National University of Singapore; this group were selected based on either short duration of breast cancer specific survival (<2 years) or for long duration of breast cancer specific suvrival (>4 years).In order to ensure complete harmonisation of the genotype calling, the intensity data available from both these locations in form of *.idat files were combined and used to generate genotypes based on the algorithm available in the genotyping module of Illumina’s Genome Studio software. A GenCall threshold of 0.15 was selected and the HumanHap660 annotation file was used. Genotyping for the replication samples from Helsinki was conducted using the Illumina 550 platform as previously described (21). The intensity data generated was loaded into Illumina’s Genome studio and genotypes were generated using a GenCall threshold of 0.15. HumanHap550-duo v3 annotation file was used.
SNPs were excluded from analysis based on a minor allele frequency (MAF) cut-off of 0.01, genotyping call rate <95% and Hardy-Weinberg equilibrium p-value<0.0001. We used the pairwise Identity-by-state and multidimensional scaling, implemented in Plink (22), to identify POSH and Helsinki participants whose genotypes did not concur with a European ancestry. 28 individuals were excluded based on a non-European ancestry or missing phenotype information in the POSH discovery analysis (Supplementary Figure 1). Three individuals from the 300 samples genotyped at the site in Singapore were excluded from analysis because of call rates lower than 95%. No individuals from the 274 triple negative cohort genotyped at the Mayo clinic were excluded from analysis based on poor call rate. Genotyping accuracy for SNPs or SNP specific call rates were over 99% in samples genotyped at the Singapore and Mayo clinic sites.*.idat files available from the Helsinki participants were fed into Illumina’s Genome Studio software to call genotypes. No ethnic outliers were identified among the 805 Helsinki participants. 7 Helsinki participants were excluded because of SNP call rate (<95%).
Replication genotyping
Genotyping of the 35 best associated SNPs from stage-1 in the 1516 additional young onset cases from the POSH study was performed by KBioscience (23). SNPs were genotyped using the KASPar chemistry, which is a competitive allele-specific PCR SNP genotyping system using FRET quencher cassette oligo (23) .
Statistical Analysis
To generate estimates of pairwise Identity by descent we performed genome wide linkage disequilibrium based pruning using the --indep-pairwise command in plink. SNP data were pruned after choosing an r2 cut-off at 0.5. SNP pruning was initiated using a window of 50 SNPs. Pairwise LD was then calculated and one SNP within each SNP pair characterised by high LD (r2>0.5) was excluded. This process was repeated while choosing smaller SNP windows of 5 SNPs at a time. Multi-dimensional scaling plots were then generated after generating clusters of related individuals based on pairwise IBS distances.
SNP quality control (QC) measures were implemented using Plink. Post-QC, transposed ped (tped) and tfam files were generated for further analysis. We used GenABEL (24) in R.2.14.0 environment to perform survival analysis using Post-QC genome wide SNP data. Cox-proportional hazard models were implemented using the mlreg command in GenABEL . The mlreg command utilizes the survival package which is routinely used for survival analysis in R. ER status was the only covariate used in Cox models. Follow-up time was calculated as the difference between the date of diagnosis of breast cancer and the date of death due to breast cancer or the date of last follow-up if still alive or deceased from a non-breast cancer cause (breast cancer specific survival). The mean difference in time between age at diagnosis and age at registration was 0.78 years (SD=1.16 years).Kaplan Meier plots were generated using STATA v11.0 and IBM SPSS statistics 19. Mantel Haenszel Fixed effects meta-analysis was performed using the metan module in STATA v11.0 (25). Genome wide meta-analysis was performed using MetABEL (24).
Imputation of the POSH GWAS data set was performed using MACH 1.0 (26) based on SNP genotype and haplotype phase data specific for CEU population available from HapMap phase 2 project. Imputed genotypes were analysed using ProABEL (24). A posterior probability of 0.9 was used to output imputed genotypes. QC- measures for imputation data included excluding SNPs based on a minor allele frequency (MAF) cut-off of 0.01, genotyping call rate <95% and Hardy-Weinberg equilibrium p-value<0.0001.
Manhattan and Regional plots
Manhattan and QQ-plots were generated in R using the plot command. Regional plots were generated using LocusZoom (27).
Sample size calculations
Sample size calculations were performed in R.2.14.2 using survSNP package.
Gene Expression variation by SNP
We used Genevar 3.2.0 to study variation in expression levels by SNP genotypes available from the MuTHER pilot project while using NCBI Build 36 /Ensembl 54 as reference (28). Twin pairs were divided into two groups of unrelated individuals. Expression data from Lymphoblastoid cell lines are reported here.
Prediction of transcription factor binding site changes
The putative changes on transcription factor binding sites caused by the variants were predicted in silico with SNPInspector within Genomatix software suite v2.5 (Genomatix Software GmbH). SNPInspector analysis is based on MatInspector (29).
Results
Clinical characteristics of stage-1 and stage-2 participants are summarised in Table 1. Following QC we had SNP genotype data available for 487496 SNPs in stage-1. We had 79% power to detect a hazard ratio (HR) ≥1.50 when studying a SNP with minor allele frequency (maf≥0.10). In survival analysis models no associations were observed to survive a Bonferroni correction and reach a p-value≤10−7. Eight SNPs among the top 50 SNPs achieved a p-values <7.0 × 10−6.
Table 1.
Characteristics of study participants
| Study | Number of breast cancer deaths |
Total number of Breast cancer patients |
Estrogen Receptor (ER) status- Negative (%) |
Average age at Diagnosis (±SD) |
Follow-up time in years (±SD) |
N-stage | M-stage | HER2 status |
|---|---|---|---|---|---|---|---|---|
|
POSH stage-
1 |
236 | 536 | 370 (69.2%) | 35.7 (3.8) | 4.1 (2.0) | N0-248 N1-262 NA-25 535 |
M0-481 M1-50 NA-4 535 |
Negative-369 Positive-92 NA-74 535 |
|
POSH stage-
2 |
468 | 1518 | 423 (27.4%) NA-8 |
35.7 (3.7) | 4.8 (1.4) | N0-645 N1-838 NA-35 |
M0-1470 M1-42 NA-6 |
Negative-756 Positive-399 NA-363 |
|
Helsinki non-
early onset specific participants |
301 | 805 | 230 (30.0%) NA-39 |
56.8 (12.4) | 7.2 (2.9) | N0-338 N1-446 NA-21 |
M0-740 M1-57 NA-8 |
Negative-402 Positive-86 NA-317 |
NA=not available,
HER2= Human Epidermal Growth Factor Receptor 2, N-stage=metastasis to lymph node, M-stage=metastasis
41 of the remaining 42 SNPs achieved p-values at10−5. At the loci on which multiple SNPs were found to be strongly associated with survival we selected the lead SNP for follow-up in stage-2 along with any other SNP(s) from the same locus which were not in high LD with the lead SNP (r2<0.6). Using this strategy we selected 35 of the best 50 associated SNPs (Supplementary Table 1) for genotyping in the stage-2 validation samples. The qq-plot demonstrated deviation of observed log transformed values from the expected log transformed p-values for SNPs associated with p-values ranging from 10−4 to 10−5 (Figure 1).
Figure 1.
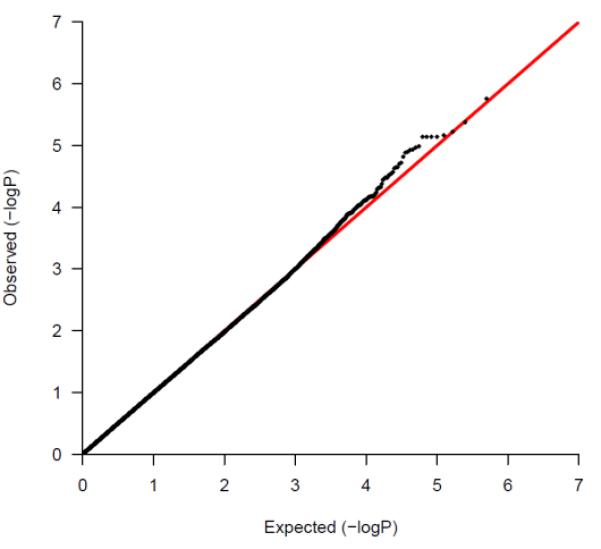
A quantile-quantile (qq plot) of log-transformed observed and expected P-values from the stage-1 analysis:
Stage-2 Results
27 of the 35 SNPs included in the stage-2 genotyping were successfully genotyped and were available for analysis. 1 SNP had greater than 10% duplicate error rate and was excluded from replication analysis. We had 70% power to detect a HR (hazard ratio) ≥ 1.50 in stage 2 analysis. While testing for replication effects of the 27 SNPs, we found 11 SNPs at distinct loci which were associated with prognosis in the same direction as in the stage-1 analysis (Table 2). Replication p-values for these 11 SNPs ranged from 0.05 to 0.005.
Table 2.
Stage Wise association statistics for the eleven SNPs which were associated in stage-2 following discovery in stage-1 analysis:
| SNP (Minor Allele Frequency) |
POSH stage-1 HR (95% CI), p-value |
POSH stage-2 replications HR (95% CI), p-value |
Stage-1 and Stage-2 meta-analysis results |
I2 derived from Cochran’s Q-statistic, p- value for Q-statistic |
|---|---|---|---|---|
| rs421379 (0.05) | 1.98 (1.46-2.70), p=1.2×10−5 | 1.42 (1.11-1.8), p=0.005 | 1.61 (1.33-1.96), p=9.5 × 10−7 | 63.7%, p=0.10 |
| rs3884558 (0.07) | 1.84 (1.40-2.41), p=1.3×10−5 | 1.29 (1.05-1.57), p=0.01 | 1.46 (1.24-1.72), p=3.9 × 10−6 | 76.4%, p=0.04 |
| rs971398 (0.17) | 1.52 (1.23-1.88, p=1.2 × 10−4) | 1.24 (1.05-1.47, p=0.01) | 1.34 (1.18-1.53, p=1.2 × 10−5 | 46.3%, p=0.17 |
| rs7910841 (0.28) | 0.64 (0.51-0.80, p=8.2 × 10−5) | 0.83 (0.72-0.96, p=0.01) | 0.77 (0.68-0.87), p=2.3 × 10−5 | 72.5%, p=0.06 |
| rs12523819 (0.28) | 0.64 (0.51-0.80, p=1.1 × 10−4) | 0.86 (0.74-0.99), p=0.04 | 0.77 (0.88-0.87), p=2.3 × 10−5 | 78.6%, p=0.03 |
| rs3785982 (0.12) | 1.75 (1.36-2.24, p=1.3 × 10−5) | 1.24 (1.03-1.48, p=0.02) | 1.40 (1.21-1.62), p=7.9 × 10−6 | 79.1%, p=0.03 |
| rs2774307 (0.26) | 1.51 (1.24-1.85, p=4.3 × 10−5) | 1.21 (1.05-1.40, p=0.01) | 1.30 (1.16-1.47), p=7.9 × 10−6 | 67.8%, p=0.08 |
| rs10220397 (0.23) | 0.63 (0.50-0.79, p=8.6 × 10−5) | 0.85 (0.73-0.99, p=0.04) | 0.77 (0.68-0.88), p=8.3 × 10−5 | 78.1%, p=0.03 |
| rs303850 (0.42) | 1.48 (1.22-1.79, p=5.2 × 10−5) | 1.13 (1.00-1.29, p=0.05) | 1.23 (1.10-1.36), p=1.5 × 10−4 | 81.1%, p=0.02 |
| rs1387389 (0.36) | 1.47 (1.22-1.77, p=5.0 × 10−5) | 1.20 (1.05-1.37, p=0.007) | 1.28 (1.16-1.43), p=3.8 × 10−6 | 66.6%, p=0.08 |
| rs1513848 (0.07) | 1.87 (1.41-2.46, p=1.0 × 10−5) | 1.25 (1.01-1.55, p=0.04 | 1.45 (1.22-1.72), p=1.6 × 10−5 | 80.2%, p=0.02 |
Stage-1 and Stage-2 meta-analysis
We included the eleven SNPs which remained associated in stage-2 based on consistent direction of effect, in Mantel-Haenszel fixed-effects meta-analysis models (Table-2). The strongest meta-analysis HR was observed at the rs421379 SNP which lies upstream of the ARRDC3 gene (Figure 2, Figure 3) on the long arm of chromosome 5, HR=1.61 (1.33-1.96, p=9.5 × 10− 7). Adjusting for ER-status, N-stage and M-stage slightly reduced the strength of the overall association at this SNP in combined analysis across stage-1 and stage-2 (HR=1.55 (1.27-1.90, p=1.5 × 10−5)). The next best replication signal was observed in an intronic region of the PBX1 (Pre-B-Cell Leukaemia transcription factor-1) gene. The replication p-value for this intronic SNP was second most significant after rs421379 in the two stage meta-analysis, and the overall association at this variant was close to being genome wide significant, HR=1.28 (1.16-1.43, p=3.8 × 10−6). Adjusting for ER-status, N-stage and M-stage did not affect the strength of the association at this variant (HR=1.26 (1.13-1.41, p=3.9 × 10−5)). The above two variants displayed the lowest levels of heterogeneity in meta-analysis. The association observed with a 5′UTR snp at the RORα locus (rs3884558) was also close to the threshold for genome wide significance HR=1.46 (1.24-1.72, p=3.9 × 10−6), although there was modest evidence of heterogeneity in Hazard ratios between stage-1 and stage-2 models (Table 2). Two further associations rs3785982 in the NTN1 gene (HR=1.40 (1.21-1.62, p=7.9 × 10−6) and rs2774307 in the SYT6 gene (HR=1.30 (1.16-1.47, p=7.9 × 10−6)) also came close to genome wide significance. For the five SNPs associated at p≤10−6, we did not observe any eveidence of heterogeneity of effects on survival based on triple negative status of the POSH patients. The heterogeneity I2-statistic for these 5 SNPs ranged from 0-20.6%.
Figure 2.
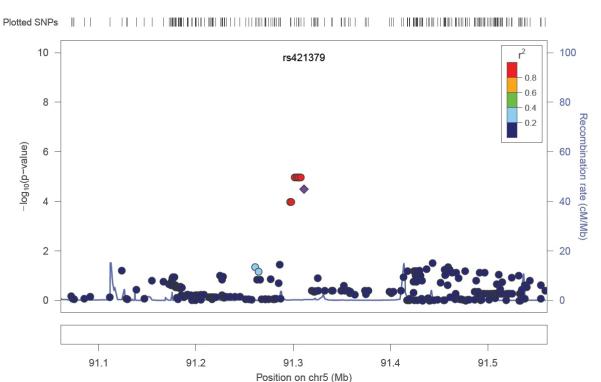
Regional plot of p-values arising from cox-proportional hazard models, 250 kb either side of the rs421379 variant in stage-1. P-values are from imputed and the genotyped SNPs.
Figure 3.
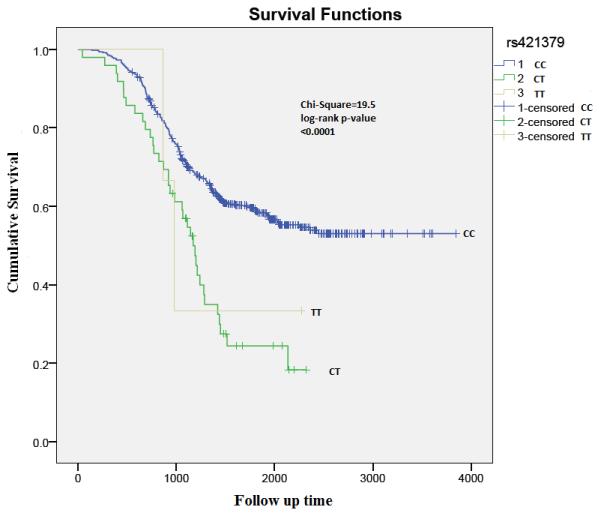
Kaplan Meier analysis plot depicting survival rates by rs421379 genotypes:
Replication attempt in non-age-specific survival analysis
We had 87% power to detect a hazard ratio ≥1.50 when analysing SNPs with Maf≥0.10 in 874 patients available from the Helsinki study. We extracted genotypes from the GWS genotype data of the Helsinki samples for 11 of the 35 SNPs which were associated in the same direction in stage-2 as in stage-1 analysis. Helsinki patients belonged to a relatively higher age group at diagnosis when compared to POSH (Average at diagnosis=56.8, SD=12.4). We found that none of the 11 SNPs which were replicated as associated with prognosis in stage-2 were associated with the same outcome in patients with later onset from Helsinki.
Association scan with imputed SNP data
We imputed SNP genotypes for 2.5 million SNPs based on HapMap Phase 2 data. Imputation analysis did not identify any additional variants which were more strongly associated than the ones we found as most strongly associated using real genotype data 250 kb either side of rs421379, rs3884558 (5′UTR-RORα), rs3785982 (NTN1), rs2774307 (SYT6) and rs1387389 (PBX1) (Figure 2 and Supplementary Figures 2-5).
Associations of the five best associations with clinical predictors
We assessed the associations of all SNPs which were associated at p-value≤10−6 with clinical predictors of breast cancer prognosis. There were five SNPs which were associated at p-value≤10−6, none of these SNPs were associated with ER-status, N-stage or M-stage after performing a Bonferroni correction for number of tests performed (Table 3).
Table 3.
Associations of SNPs associated at p≤10−6 in the two stage meta-analysis with secondary traits linked to breast cancer mortality in stage 1 and stage 2 combined dataset:
| Secondary trait |
rs421379 | rs3884558 | rs3785982 | rs2774307 | rs1387389 |
|---|---|---|---|---|---|
| ER-status | OR=1.13 (95% CI:0.86 to 1.47, p=0.38) |
OR=0.93 (95% CI:0.75 to 1.16, p=0.52) |
OR=1.14 (95% CI:0.94 to 1.39, p=0.54) |
OR=0.96 (95% CI:0.83 to 1.11, p=0.59) |
OR=0.97 (95% CI:0.85 to 1.10, p=0.29) |
| Nodal Status |
OR=1.55 (95%CI:0.96- 2.49, p=0.07) |
OR=1.70 (95% CI:1.14-2.55, p=0.009) |
OR=1.47 (95% CI:1.06- 2.05, p=0.02) |
OR=1.16 (95% CI:0.92-1.45, p=0.20) |
OR=1.20 (95%CI:0.98- 1.46, p=0.05) |
| M-stage | OR=1.22 (95%CI:0.88- 1.68, p=0.24) |
OR=1.42 (95% CI:1.07-1.87, p=0.01) |
OR=1.23 (95% CI:0.97- 1.56, p=0.08) |
OR=1.04 (95% CI:0.88-1.23, p=0.62) |
OR=1.07 (95%CI:0.92- 1.24, p=0.40) |
ER-status= Estrogen receptor status, M-stage=Metastasis stage
Gene Expression variation by SNP
We queried the Genevar 3.2.0 and SNP and CNV annotation database (scandb) to identify Cis or Trans eQTL effects resulting from rs421379. In 156 Lymphoblastoid cell lines sample collected from 78 twin pairs available via the Java based Genevar interface we did not observe an association of rs421379 with expression of ARRDC3 gene (Figure 4). In scandb we observed that rs421379 had trans eQTL effects on expression of RAB34 (p=1× 10−5) and ABCD1 (p=9 × 10−5), but neither of these associations were genome wide significant which could be a result of low sample size available with 30 HapMap-CEU trios available from scandb.
Figure 4.
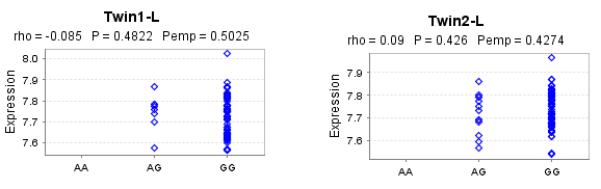
Variation in ARRDC3 expression levels with rs421379 variant. Results are reported from twins from the same pair who were separated by id in two samples named Twin1-L and Twin2-L, which were analysed independently. Rho is the spearman rank correlation (SRC) coefficient, p-value is for SRC and Pemp is the empirical p-value for SRC based on 10,000 permutations:
Discussion
In this manuscript we have reported findings from the first genome wide association study of breast cancer prognosis in early onset breast cancer patients and enriched for poor survival and ER, PR and HER-2 negative cases in discovery stage. Recently two GWASs aimed at identifying risk alleles for poor prognosis were performed in unselected breast cancer patients of Chinese and European ancestries. Azzato et al (12) in their GWAS conducted in 4335 Caucasian breast cancer patients with mean age at diagnosis of 66 years (95% CI=44-83) did not replicate any of their main findings in a large relatively younger cohort of breast cancer patients (Mean age=51 (95% CI=23-69). Azzato et al (12) had taken forward 10 of their most significant findings forward for replication in the SEARCH study. On the contrary Shu et al (13), attempted replication of their top 50 associations in their two stage GWAS in Chinese patients and identified two associations with p-values equal to 1.17 × 10−7 (rs3784099, RAD15L) and 5.75 × 10−6 (rs9934948). We did not find either of these two SNPs (rs3784099, p=0.61 and rs9934948, p=0.25) as associated with prognosis in the stage-1 data used for discovery in this GWAS nor in the Helsinki GWS data.
In our study we attempted replication of 50 of the strongest association signals from stage-1 by selecting the strongest associated SNPs at each of the new discovered loci while excluding any other SNPs which were in relative LD with the best associated SNP at the same locus r2≥0.6. Of the 35 SNPs which were selected from stage-1 for validation in stage-2, 27 SNPs were successfully genotyped in stage-2 and we found 11 of these SNPs to demonstrate nominal to strong replication signals (p-values range: 0.05-5 × 10−3). Such a high replication rate (40.7%) suggests low phenotypic heterogeneity in samples collected between stage-1 and stage-2. Large cohorts of young onset patients with comprehensive treatment and outcome data are uncommon given that less than 5% of all breast cancers are diagnosed before 40 years of age. We were able to enrich our stage-1 samples further with young onset triple negative patients and patients with very short duration of breast cancer specific survival. This allowed us increased statistical power to identify common genetic variants with modest effect sizes (OR≥1.50) in stage-1 data. Despite having a more enriched stage-1 dataset, we did not have sufficient power (<80%) to detect association signals associated with HRs in range of 1.10 to 1.45 in our discovery samples. Future studies in larger early onset cohorts are therefore needed to identify true associations with lower effect sizes than HR≥1.50.
The strongest association signal in our study was observed 596 kb upstream of the ARRDC3 gene. In HapMap we did not find any long range LD between rs421379 and any SNPs at or close to the ARRDC3 locus. The ARRDC3 gene is a member of the arrestin gene family and functions in a novel regulatory pathway that controls the cell surface adhesion molecule, b-4 integrin (ITGb4), a protein associated with aggressive tumour behaviour (30). Furthermore deletion of the region of chromosome 5 containing the ARRDC3 gene is observed more frequently in basal type breast cancer cancers (31). Differential expression levels have also been associated with prognosis in prostate cancer patients (32). The associated SNP rs421379 is located in the 5′ region of the ARRDC3 gene and might affect a transcription binding site and ARRDC3 gene expression, permitting development of a more aggressive, invasive tumour. The associated SNP rs421379 was predicted to disrupt a binding site for Myocyte-specific enhancer factor 2 (MEF2). This transcription factor family consists of four members (MEF2A-D) sharing the binding sequence and MEF2 could regulate ARRDC3 gene expression. Previously, MEF2C has been found to be highly expressed in basal breast cancer along with Notch (33). Later a strong co-expression of Notch1 and MEF2 paralogs has been observed in breast cancer tumour samples from patients with metastatic disease (34).
We did not find a robust association signal between rs421379 and the probe representing variation in ARRDC3 expression (Figure 4). In the SNP and CNV annotation database (scandb) (35), rs421379 is identified to have trans-effects on expression of RAB34 (p=1× 10−5) and ABCD1 (p=9 × 10−5) genes present on chromosomes 17 and X respectively. It should be noted that the association analysis in Genevar 3.2.0 and scandb analysis is based on 154 twins and the 30 HapMap CEU trios respectively as such the statistical power to detect modest effects on gene expression was not high and further given that the gene expression is quantified in lymphoblastoid cell lines these results are not reflective of potential cis-effect of rs421379 in breast cancer related cells.
The second strongest association we observed was at the PBX1 locus (HR=1.28 (1.16-1.43), p=3.8 × 10−6). The protein coded by this gene drives ERα signalling and breast cancer progression through transcriptional programming (36). There was no evidence of substantial heterogeneity in hazard ratios across stage-1 and stage-2 for the SNP associated at the PBX1 locus (Table 2). 11 SNPs according to HapMap Phase 3 data are in high LD (r2>0.8) with the PBX1 SNP we found as strongly associated and all these SNPs were found to be intronic within the PBX1 locus. We also observed strong suggestive evidence for an association at the RORα gene (HR=1.46 (1.24-1.72), p=3.9 × 10−6). It has recently been shown that RORα protein expression is reduced in breast cancer cells and also this lower expression is related to poorer prognosis in breast cancer patients (37). There was some evidence of heterogeneity in hazard ratios between stage-1 and stage-2 patients for the rs3884558 which lies 78.3 kb upstream of the RORα gene (p=0.04). Although there were no SNPs in high LD (r2≥0.8) 2.4 kb beyond rs3884558, SNPs in moderately LD (r2=0.3) are located up to 95.7 kb away from rs3884558 and close to the RORα gene. The SNP rs3884558 was predicted to both disrupt and create multiple transcription factor binding sites. The binding site was predicted to be lost for transcription factors POU2F1, TGIF1, HMGA1/2 and CDX2 and new binding site was predicted to emerge for REV-ERBα, CREB1/2, HMGA1, VBP1 and E4F1. Interestingly REV-ERBα belongs to the same nuclear hormone receptor family as does RORα. Moreover these family members are known to cross-talk and REV-ERBα has been shown to suppress the transcriptional activity of RORα (38).
The two other associations at NTN1 and SYT6 could also be real given NTN1 expression is increased in breast cancer (39) and the replication signal at SYT6 remained strong in post replication meta-analysis (HR=1.30 (1.16-1.47), p=7.9 × 10−6) with no strong evidence of heterogeneity in hazard ratios with the associated variant.
Further studies at population level are needed to confirm the association of the 5 loci associated at p≤10−6 that we have discovered from this two stage GWAS analysis. In future analyses we will study the most strongly associated SNPs from the current study by interrogating additional well characterised and early onset breast cancer cohorts. This will allow us to generate more accurate estimates of gene-survival associations and also allow the implementation of prediction statistics generated using gene score analysis. In addition, further studies are needed to establish beyond doubt the true validity of the remaining SNPs that replicated strongly but were not quite genome wide significant. Published results from biochemical analyses do suggest PBX1, RORα and NTN1 are plausible candidate loci for an effect exerted by the host genotype in altering prognosis. Fine mapping and molecular studies are needed to establish the identity of the causal variant in the intragenic region, 596 kb upstream of ARRDC3 gene. and provide insight into the mechanism of action. Much emphasis currently is on genotyping of somatic mutations in tumours to help refine prognosis and identify treatment targets but this is only a part of the information that influences prognosis. Selecting a well characterised poor prognosis group of patients with high breast cancer specific mortality has been a useful strategy to identifying SNPs that influence prognosis. The ultimate validation of the clinical utility for germ-line genetic variants that influence prognosis will come from genotyping in randomised adjuvant and neo-adjuvant treatment trials. With a clear understanding of the magnitude and mechanism of prognostic SNPs, such genotyping may in future be routinely used in cancer patients to help derive a more complete individualised risk assessment for early relapse and thereby guide treatment choices.
Supplementary Material
Acknowledgments
We would like to thank Nikki Graham (DNA bank) and the staff of the Southampton CRUK Centre Tissue Bank. The POSH study is supported by Breast Cancer Campaign grant number: 2010NovPR62. Funding for the POSH study was also provided by The Wessex Cancer Trust and Cancer Research UK (grant refs A7572, A11699, C22524). We wish to thank Drs. Kristiina Aittomäki, Kirsimari Aaltonen and Karl von Smitten and RN Irja Erkkilä for their help with the Helsinki data and samples. The Helsinki study was financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (132473), the Finnish Cancer Society, The Nordic Cancer Union and the Sigrid Juselius Foundation. Genotyping at the National Institute of Singapore was financially supported by the Agency for Science, Technology and Research (A*STAR), Singapore.
Footnotes
Conflict of Interests: The authors would like to disclose that no potential conflicts of interest exist
References
- 1. http://info.cancerresearchuk.org/cancerstats/types/breast/incidence/#age.
- 2.Gonzalez-Angulo AM, Broglio K, Kau SW, Eralp Y, Erlichman J, Valero V, et al. Women age < or = 35 years with primary breast carcinoma: disease features at presentation. Cancer. 2005;103:2466–72. doi: 10.1002/cncr.21070. [DOI] [PubMed] [Google Scholar]
- 3.Walker RA, Lees E, Webb MB, Dearing SJ. Breast carcinomas occurring in young women (< 35 years) are different. British journal of cancer. 1996;74:1796–800. doi: 10.1038/bjc.1996.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anders CK, Hsu DS, Broadwater G, Acharya CR, Foekens JA, Zhang Y, et al. Young age at diagnosis correlates with worse prognosis and defines a subset of breast cancers with shared patterns of gene expression. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:3324–30. doi: 10.1200/JCO.2007.14.2471. [DOI] [PubMed] [Google Scholar]
- 5.Ahn SH, Son BH, Kim SW, Kim SI, Jeong J, Ko SS, et al. Poor outcome of hormone receptor-positive breast cancer at very young age is due to tamoxifen resistance: nationwide survival data in Korea--a report from the Korean Breast Cancer Society. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2007;25:2360–8. doi: 10.1200/JCO.2006.10.3754. [DOI] [PubMed] [Google Scholar]
- 6.El Saghir NS, Seoud M, Khalil MK, Charafeddine M, Salem ZK, Geara FB, et al. Effects of young age at presentation on survival in breast cancer. BMC cancer. 2006;6:194. doi: 10.1186/1471-2407-6-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartman M, Lindstrom L, Dickman PW, Adami HO, Hall P, Czene K. Is breast cancer prognosis inherited? Breast cancer research : BCR. 2007;9:R39. doi: 10.1186/bcr1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans DG, Howell A, Ward D, Lalloo F, Jones JL, Eccles DM. Prevalence of BRCA1 and BRCA2 mutations in triple negative breast cancer. J Med Genet. 2011;48:520–2. doi: 10.1136/jmedgenet-2011-100006. [DOI] [PubMed] [Google Scholar]
- 9.Robertson L, Hanson H, Seal S, Warren-Perry M, Hughes D, Howell I, et al. BRCA1 testing should be offered to individuals with triple-negative breast cancer diagnosed below 50 years. British journal of cancer. 2012;106:1234–8. doi: 10.1038/bjc.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnbull C, Ahmed S, Morrison J, Pernet D, Renwick A, Maranian M, et al. Genome-wide association study identifies five new breast cancer susceptibility loci. Nature genetics. 2010;42:504–7. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haiman CA, Chen GK, Vachon CM, Canzian F, Dunning A, Millikan RC, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nature genetics. 2011;43:1210–4. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azzato EM, Pharoah PD, Harrington P, Easton DF, Greenberg D, Caporaso NE, et al. A genome-wide association study of prognosis in breast cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2010;19:1140–3. doi: 10.1158/1055-9965.EPI-10-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shu XO, Long J, Lu W, Li C, Chen WY, Delahanty R, et al. Novel genetic markers of breast cancer survival identified by a genome-wide association study. Cancer research. 2012;72:1182–9. doi: 10.1158/0008-5472.CAN-11-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tapper W, Hammond V, Gerty S, Ennis S, Simmonds P, Collins A, et al. The influence of genetic variation in 30 selected genes on the clinical characteristics of early onset breast cancer. Breast cancer research : BCR. 2008;10:R108. doi: 10.1186/bcr2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasching PA, Pharoah PD, Cox A, Nevanlinna H, Bojesen SE, Karn T, et al. The role of genetic breast cancer susceptibility variants as prognostic factors. Hum Mol Genet. 2012;21:3926–39. doi: 10.1093/hmg/dds159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eccles D, Gerty S, Simmonds P, Hammond V, Ennis S, Altman DG. Prospective study of Outcomes in Sporadic versus Hereditary breast cancer (POSH): study protocol. BMC cancer. 2007;7:160. doi: 10.1186/1471-2407-7-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eccles D, Gerty S, Simmonds P, Hammond V, Ennis S, Altman DG, et al. Prospective study of Outcomes in Sporadic versus Hereditary breast cancer (POSH): study protocol. BMC cancer. 2007;7:160. doi: 10.1186/1471-2407-7-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K, Li WD, Zhang CK, Wang Z, Glessner JT, Grant SF, et al. A genome-wide association study on obesity and obesity-related traits. PloS one. 2011;6:e18939. doi: 10.1371/journal.pone.0018939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fagerholm R, Hofstetter B, Tommiska J, Aaltonen K, Vrtel R, Syrjakoski K, et al. NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nature genetics. 2008;40:844–53. doi: 10.1038/ng.155. [DOI] [PubMed] [Google Scholar]
- 20.Haiman CA, Chen GK, Vachon CM, Canzian F, Dunning A, Millikan RC, et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor-negative breast cancer. Nature genetics. 2011;43:1210–4. doi: 10.1038/ng.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Humphreys K, Heikkinen T, Aittomaki K, Blomqvist C, Pharoah PD, et al. A combined analysis of genome-wide association studies in breast cancer. Breast Cancer Res Treat. 2011;126:717–27. doi: 10.1007/s10549-010-1172-9. [DOI] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. http://www.kbioscience.co.uk/genotyping/genotyping_chemistry.html.
- 24. http://www.genabel.org/
- 25.Harri R, Bradburn M, Deeks J, Harbord R, Altman D, Sterne J. metan: fixed- and random-effects meta-analysis. The Stata Journal. 2008;8:3–28. [Google Scholar]
- 26. http://www.sph.umich.edu/csg/abecasis/MACH/index.html.
- 27.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–7. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, et al. The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS genetics. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 30.Draheim KM, Chen HB, Tao Q, Moore N, Roche M, Lyle S. ARRDC3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene. 2010;29:5032–47. doi: 10.1038/onc.2010.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adelaide J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F, et al. Integrated profiling of basal and luminal breast cancers. Cancer research. 2007;67:11565–75. doi: 10.1158/0008-5472.CAN-07-2536. [DOI] [PubMed] [Google Scholar]
- 32.Huang CN, Huang SP, Pao JB, Chang TY, Lan YH, Lu TL, et al. Genetic polymorphisms in androgen receptor-binding sites predict survival in prostate cancer patients receiving androgen-deprivation therapy. Ann Oncol. 2012;23:707–13. doi: 10.1093/annonc/mdr264. [DOI] [PubMed] [Google Scholar]
- 33.Lim E, Wu D, Pal B, Bouras T, Asselin-Labat ML, Vaillant F, et al. Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways. Breast cancer research : BCR. 2010;12:R21. doi: 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pallavi SK, Ho DM, Hicks C, Miele L, Artavanis-Tsakonas S. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 2012;31:2895–907. doi: 10.1038/emboj.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, et al. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26:259–62. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magnani L, Ballantyne EB, Zhang X, Lupien M. PBX1 genomic pioneer function drives ERalpha signaling underlying progression in breast cancer. PLoS genetics. 2011;7:e1002368. doi: 10.1371/journal.pgen.1002368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong G, Wang C, Evers BM, Zhou BP, Xu R. RORalpha suppresses breast tumor invasion by inducing SEMA3F expression. Cancer research. 2012;72:1728–39. doi: 10.1158/0008-5472.CAN-11-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forman BM, Chen J, Blumberg B, Kliewer SA, Henshaw R, Ong ES, et al. Cross-talk among ROR alpha 1 and the Rev-erb family of orphan nuclear receptors. Mol Endocrinol. 1994;8:1253–61. doi: 10.1210/mend.8.9.7838158. [DOI] [PubMed] [Google Scholar]
- 39.Ramesh G, Berg A, Jayakumar C. Plasma netrin-1 is a diagnostic biomarker of human cancers. Biomarkers. 2011;16:172–80. doi: 10.3109/1354750X.2010.541564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.