

Abstract
Firm neutrophil (PMN)-endothelial (EC) adhesion is crucial to the PMN-mediated hyperinflammation observed in acute lung injury. Hypertonic saline (HTS) used for resuscitation of hemorrhagic shock has been associated with a decreased incidence of PMN-mediated lung injury/acute respiratory distress syndrome. We hypothesize that physiologically accessible hypertonic incubation (170mM vs. 140mM, osmolarity ranging from 360-300 mOsm/L) inhibits pro-inflammatory activation of human pulmonary microvascular endothelial cells (HMVECs). Pro-inflammatory activation of HMVECs was investigated in response to TNFα including IL-8 release, ICAM-1 surface expression, PMN adhesion, and signaling mechanisms under both isotonic (control) and hypertonic conditions. Hyperosmolarity alone had no effect on either basal IL-8 release or ICAM-1 surface expression, but did lead to concentration-dependent decreases in TNFα–induced IL-8 release, ICAM-1 surface expression, and PMN:HMVEC adhesion. Conversely, HTS activated p38 mitogen-activated protein kinase (MAPK) and enhanced TNFα activation of p38 MAPK. Despite this basal activation, hyperosmolar incubation attenuated TNFα stimulated IL-8 release and ICAM-1 surface expression and subsequent PMN adherence, while p38 MAPK inhibition did not further influence the effects of hyperosmolar conditions on ICAM-1 surface expression. In addition, TNFα induced NF-kB DNA binding, but HTS conditions attenuated this by 31% (p<0.01). In conclusion, HTS reduces PMN:HMVEC adhesion as well as TNFα-induced pro-inflammatory activation of primary HMVECs via attenuation of NF-kB signaling.
Keywords: ARDS, neutrophils, NF-kB, p38 MAPK, HMVECs, IL-8, ICAM-1, hypertonic saline
INTRODUCTION
The leading cause of late death in severe postinjury patients is multiple organ failure (MOF), to which the lung is the sentinel organ (1). After traumatic shock the neutrophils (PMNs) in circulation are primed by pro-inflammatory mediators changing the PMN phenotype to “adherent.” When these primed PMNs encounter adhesion molecules on the surface of activated endothelium, they become firmly adherent and in the lung this is called pulmonary sequestration, setting the stage for acute lung injury (ALI) and the acute respiratory distress syndrome (ARDS) (2). Given the current understanding that PMN-mediated hyperinflammation is integral to early post-injury ALI/ARDS, there is interest in immunomodulatory resuscitation therapies against traumatic shock.
Hypertonic saline (HTS) was initially employed as a resuscitation fluid because the reduced volumes of infusion could meet the logistics of resuscitation in the field (military and civilian) while maintaining favorable hemodynamic effects over isotonic saline (3, 2). In addition, studies in animal models (4-6) and patients (7) have also linked the immunomodulatory effects of HTS following resuscitation, demonstrating that HTS may decrease ALI by suppressing PMN sequestration and activation following hemorrhagic shock.
Hyperosmolarity [≥200 mOsm/L] activates the p38 mitogen-activated protein kinase (MAPK), an enzyme crucial in signal transduction during inflammatory states such as neutrophil activation (8, 9). In PMNs we have found evidence for HTS-mediated disruption of p38 MAPK signaling (9, 10). P38 MAPK also interacts with nuclear factor kB, (NF-kB) - a transcriptional factor involved in cell survival responsible for expression of genes including proinflammatory ones (11).
P38 MAPK activation, in turn, is required for maximal ICAM-1 surface expression in human pulmonary microvascular endothelial cells (HMVECs) stimulated by tumor necrosis factor α (TNFα) (12), a pro-inflammatory cytokine implicated in the early pathogenesis of ALI/ARDS (13). HMVECs constitutively express the TNFα receptor (TNFR1) which upon TNFα ligation initiates a series of signaling events, activating p38 MAPK, NF-kB (14), and leads to phosphorylation of inhibitory kb (I-kB), an inhibitory NF-kB protein. By undergoing phosphorylation, I-kB is targeted for proteolytic degradation, thus disengaging from NF-kB and allowing it to translocate to the nucleus to carry out its transcriptional duties. Importantly, pulmonary PMN accumulation and the increase in microvascular permeability were abrogated in TNFα knockout mice, as well as TNFR1 knockout mice (15). Notably, the synthesis and release of interleukin-8 (IL-8), a proinflammatory chemokine, and ICAM-1 surface expression are transcriptionally regulated by NF-kB dimers (16). Any of these signal transduction steps could, in principle, be affected by hypertonic conditions.
Pulmonary sequestration of PMNs is dependent upon pro-inflammatory activation of the pulmonary endothelium, and this is a required step towards development of ALI/ARDS(2, 26). Because HTS inhibits PMN-mediated ALI/MOF in animal models, we hypothesize that hyperosmolarity attenuates TNFα-stimulated activation of NF-kB, and thus, decreased IL-8 synthesis and release and a dimunition of ICAM-1 surface expression on HMVECs, resulting in decreased PMN-EC adhesion.
MATERIALS AND METHODS
Materials
HMVECs, EC basal media and growth factors, and trypsin solutions were purchased from Lonza (Walkersville, MD). Tumor necrosis factor-α (TNFα), sodium chloride (NaCl), sorbitol, and dimethylamiloride (DMA) were purchased from Sigma (St. Louis, MO). FITC labeled and non-labeled anti-CD54 (ICAM-1) mAb (84h10) and isotype IgG were purchased from Immunotech (Marseille, France). SB203580 was purchased from Calbiochem (San Diego, CA).
Endothelial Cell Culture
After HMVECs were grown on 12-well plates to 80-100% confluence, the cells were placed in media supplemented with NaCl such that the final Na+ concentration was 140, 150, 160 or 170 mM resulting in media osmolarity of 300, 320, 340 or 360 mOsm/L, respectively. Similarly, sorbitol was added on separate plates in a similar fashion to achieve the same osmolarities. Immediately after changing the osmolar conditions, the endothelial cells (ECs) were then stimulated with or without TNFα (10 U/mL) for 6 hrs at 37°C in 5% CO2. Preliminary time course studies identified 6 hrs to be the adequate stimulation time for significant IL-8 and ICAM-1 upregulation (data not shown). To assess the involvement of p38 MAPK, the specific inhibitor SB203580 was used. HMVECs were incubated with 10-6 M SB203580 for 30 min and then treated as previously described with 300 or 360 mOsm/L media (17). Dimethylsulfoxide vehicle controls demonstrated no effects on ICAM-1 upregulation following TNFα stimulation. After trypsinization, the cells were transferred to round bottom tubes. HMVECs from both untreated and treated groups were found to be >98% viable by trypan blue staining.
ICAM-1 Surface Expression
ICAM-1 surface expression was assessed with antigen:antibody interactions via flow cytometry as previously described (12). The round bottom tubes were centrifuged at 200g for 7 min. at 37°C and the cells were resuspended in 50 μL of 4°C Krebs Ringers Phosphate Dextrose buffer (pH 7.35). The saturating concentration of FITC-labeled anti-CD54 mAb (ICAM-1) was determined (100μg/ml, 2μg/1×106 cells), and added to each tube and incubated for 30 min. at 4°C with proper buffer-treated and isotype controls to account for non-specific antibody binding. The cells were then washed with 500 μL of 4°C phosphate buffered saline (PBS) + 0.1% azide, and resuspended in 1% paraformaldehyde and PBS + 0.05% azide. Fluorescence was measured by flow cytometry performed on the cell samples. All samples were quantified by mean fluorescence intensity (MFI), as a measure of ICAM-1 surface expression. Data are expressed as mean±SEM of 5 experiments.
ICAM-1 Cellular Expression
In order to distinguish whether HTS prevented the actual expression of ICAM-1 at the cell surface or ICAM-1 protein synthesis, we measured ICAM-1 surface expression by flow cytometery, or by permeabilizing paraformaldheyde fixed cells (2%, for 30 min. at room temp) with methanol/acetone, and quantitating the cellular fluorescence on a digital deconvolving microscope (Zeiss Axiovert, running Slidebook 4.2 software) (10, 21, 31). The total intensity of 10 fields/group, (obtained from 3 replicate experiments), was divided by the number of cells/field.
PMN Isolation
PMNs were isolated as previously described (18) by using dextran sedimentation, Ficoll-Hypaque gradient centrifugation, and hypertonic lysis of contaminating red blood cells with a final concentration of 1.25×107 cells/ml. The cell population was determined to be >99% PMNs and >98% viable by Trypan blue exclusion.
PMN Adhesion Assay
PMN adhesion to HMVECs was determined by a modified McClay adhesion assay as previously reported (19, 20). Briefly, HMVECs were grown to confluence in 48-well plates and then exposed to media containing various osmolarities ranging from 300 mOsm (control) to 360 mOsm/L adjusted by the addition of Na+ or sorbitol as mentioned previously. These cells were then incubated with TNFα (5 or 10 ng/mL) for 6 hrs. Control wells contained HMVECs bathed in 300 mOsm/L media without TNFα stimulation. PMNs (1.25 × 106 cells) were then added to each well and incubated for 1 hour. Non-adherent PMNs were removed by thoroughly rinsing the wells twice with warm Hank's balanced saline solution and inverted centrifugation at 200g for 5 minutes. Adherent EC-PMN complexes were lysed using 1% Triton-100 and centrifuged at 5,000g for 5 minutes to remove residual particulate from the supernatant. MPO release was determined spectrophotometrically by optical density changes after a five minute incubation of the supernatants with the specific substrate MPO substrate O-dianiside as reported previously (21). Data are expressed as mean±SEM of 5 experiments.
IL-8 ELISA
HMVECs were grown to confluence in 12-well plates placed in media containing either 140 or 170mM Na+ (300 or 360 mOsm/L), and then stimulated with TNFα (5 or 10 ng/mL) for 6 hrs. Supernatants were removed and stored at -80°C until IL-8 was measured via commercial ELISA (R&D Diagnostics). Data are expressed as mean±SEM of 5 experiments.
p38 MAPK Phosphorylation
Levels of protein phosphorylation were measured using a cell-based colorimetric ELISA system comparing dual phosphorylated (T180/Y182) to total p38 MAPK allowing for more accurate quantification of phosphoprotein levels (SABiosciences, Frederick, MD, USA). HMVECs were grown to confluence in 12-well plates placed in media containing either 140 or 170m M Na+ (300 or 360 mOsm/L), and then stimulated with TNFα (10 ng/mL) for 0, 15, 30 or 60 min. The cells were washed in lysis buffer containing 80% Tris-SDS-BME SepraSol from Integrated Separation Systems (Natick, MA), with 3.2 mM sodium orthovanadate, 16 mM nitrophenolphosphate, 16 mM phenylmethanesulfony fluoride, and 16 ug/ml leupeptin from Sigma (St. Louis, MO). The effects of 170 mM Na+ alone was tested by placing cells in this hypertonic media for 0, 15 30 or 60 min before lysis buffer was added. Data are expressed as mean±SEM of 5 experiments.
I-kB Phosphorylation
Levels of protein phosphorylation were measured using a cell-based colorimetric ELISA system comparing dual phosphorylated (S32, S36) to total I-kB allowing for more accurate quantification of phosphoprotein levels (SABiosciences, Frederick, MD, USA). HMVECs were grown to confluence in 12-well plates placed in media containing either 140 or 170mM Na+ (300 or 360 mOsm/L), and then stimulated with TNFα (10 ng/mL) for 0, 15, 30 or 60 min. The cells were washed in lysis buffer containing 80% Tris-SDS-BME SepraSol from Integrated Separation Systems (Natick, MA), with 3.2 mM sodium orthovanadate, 16 mM nitrophenolphosphate, 16 mM phenylmethanesulfony fluoride, and 16 μg/ml leupeptin from Sigma (St. Louis, MO). Data are expressed as mean ±SEM of 5 experiments.
Nuclear NF-kB DNA Binding
A nuclear kB consensus binding kit was obtained from Active Motif (Carlsbad, CA) and used according to the manufacturer's instructions. Briefly, following cell lysis after TNF stimulation for 1 hour, nuclear pellet was prepared by centrifugation; lysed, and the nuclear NF-kB was added to -kB consensus DNA, coated on 96-well plates. After washing, -kB-DNA bound NF-kB was sandwiched with a polyclonal Ab recognizing the p65 Rel A subunit which was conjugated to a colorimetric enzyme. Data are expressed as mean ±SEM of 5 experiments.
Statistical Analysis
Data are reported as the mean±standard error of the mean (SEM) for all groups tested. Analysis of variance (ANOVA) was performed with post-hoc analysis using Scheffe's test for multiple comparisons contingent upon the equality of variance. Statistical significance was interpreted as p <0.05.
RESULTS
ICAM-1 surface expression on HMVECs
To assess the effects of hyperosmolarity alone on ICAM-1 surface expression, Na+ or sorbitol concentration was increased to adjust the osmolarity to 300, 320, 340, or 360 mOsm/L. Increasing osmolarity alone with Na+ or sorbitol from 300 mOsm/L to 360 mOsm/L did not affect the basal ICAM-1 surface expression, as measured using fluorescently-labeled antibodies to ICAM-1 and flow cytometry (Figures 1).
Figure 1.

Hypertonic saline or sorbitol does not induce expression of ICAM-1 over a physiological range of osmolarities. HMVECs were incubated in media with varying Na+ or sorbitol concentrations to increase osmolarity from 300 to 320, 340, or 360 mOsm/L for 6 hrs. Control is represented by the 300 mOsm/L treated cells. Panel shows ICAM-1 surface expression measured by flow cytometry. Data are expressed as mean±SEM of 5 experiments.
We next examined whether TNFα stimulated ICAM-1 surface expression was affected by hyperosmolar conditions. The media Na+ or sorbitol concentration was changed to adjust the osmolarity from 300, 320, 340 or 360 mOsm/L, before the HMVECs were stimulated with TNFα. Flow cytomtery showed that in the presence of HTS (Figure 2a), TNFα stimulated ICAM-1 surface expression was attenuated by increasing osmolarity in a dose-dependent fashion. Significant inhibition was achieved at 340 and 360 mOsm/L (56.5% and 37.8% of 300 mOsm/L of control, respectively, p <0.05). Similarly, increasing the osmolarity with sorbitol significantly attenuated TNFα stimulated ICAM-1 surface expression (Figure 2a) at 320, 340, and 360 mOsm/L (81.6%, 66.9% and 55.3% of control, respectively). In order to distinguish whether HTS prevented the actual expression of ICAM-1 at the cell surface or decreased ICAM-1 synthesis, we measured ICAM-1 expression by methanol/acetone permeabilized, paraformaldheyde fixed cells (represented in Fig 2b) was quantitated on a digital deconvolving microscope (Zeiss Axiovert, running Slidebook 4.2 software) (10, 21, 31).. The total intensity of 10 such fields/group, (obtained from 3 replicate experiments), was divided by the number of cells/field (>33 cells/group), to obtain the statistics shown in Fig 2c. By showing both a decrease in surface ICAM-1 (by flow cytometry, Fig 2a) and total cellular expression of ICAM-1 (by quantitative microscopy, Fig 2c), we are able to conclude that the actual protein synthesis was decreased, as opposed to stored or newly synthesized ICAM-1 being prevented to translocate to the cell surface due to hypertonicity.
Figure 2.

Hypertonic saline or sorbitol inhibits TNFα-stimulated ICAM-1 surface expression(top panel). ICAM-1 expression at the cell surface is measured by flow cytometry. Data are expressed as mean±SEM of 5 experiments. HMVECs were incubated in media with varying Na+ or sorbitol concentrations to increase osmolarity from 300 to 320, 340,or 360 mOsm/L and then stimulated with TNFα (10 ng/ml) for 6 hrs. Control is represented by the 300 mOsm/L treated cells. *denotes difference from 300 mOsm/L control (p<0.05).
Middle panel represents total ICAM-1 surface expression in permeabilized cells as measured by quantitative wide field microscopy. Green channel shows cellular glycoproteins stained with WGA, red shows ICAM and blue shows nuclei. The images show representative cells at at 6 hours. (A)Control untreated cells, (B) TNFα treated for 6 hours, (C) cells treated with HTS alone (360 mOsm/L), and (D) cells treated with TNFα + HTS (360mOsm/L), also at 6 hours. Bottom panel shows total cellular ICAM obtained from compiling quantitative fluorescence microscopy of 10 fields/group (such as shown in Fig 2b) totaling >33 cells/group. *denotes difference from isotonic TNFα treated group (p<0.05)
IL-8 secretion
After 6 hours, cells stimulated with either 5 or 10 ng/ml of TNFα, produced about 4000±1000 ng/ml of IL-8. At the highest physiological dose tested, incubation with HTS 170 mM Na+ (360 mOsm/L) reduced IL-8 secretion by 53.7±13%and 43.4±10%, respectively (Figure 3).
Figure 3.

Hypertonic saline inhibits TNFα stimulated IL-8 secretion. HMVECs were incubated in media with 140 or 170 mM [Na+] to increase osmolarity from 300 to 360 mOsm/L, and then stimulated with TNFα (5, or 10 ng/ml) for 6 hrs. Control is represented by the 300 mOsm/L treated cells. IL-8 secretion in supernates was measured by ELISA. Data are expressed as mean ±SEM of 5 experiments. *denotes differences from TNF stimulations at 300 mOsm/L (p<0.01).
PMN-EC adhesion
The goal of this adhesion assay was to determine whether the effect of hyperosmolarity on ICAM-1 surface expression had functional significance. Platelet activating factor (PAF) priming of PMNs leads to a rapid conformational change in the β2-integrin (CD11b/CD18) phenotypically altering the PMN such that it is now adhesive (24). PAF also is an effective chemoattractant and induces increased CD11b/CD18 surface expression, the ligand to ICAM-1, which may also participate in adhesion as well as margination to the site of inflammation in the lung. PAF-primed PMN adhesion (assessed by MPO activity remaining despite thorough washing)) to TNF treated HMVECs (5 or 10 ng/ml) was inhibited by pretreatment with Na+ at 360 mOsm/L by71.4% and 63.3%, respectively) compared to control (300 mOsm/L) (Figure 4), Blockade with an anti-ICAM-1 mAb lead to inhibition of adhesion to baseline levels (data not shown).
Figure 4.
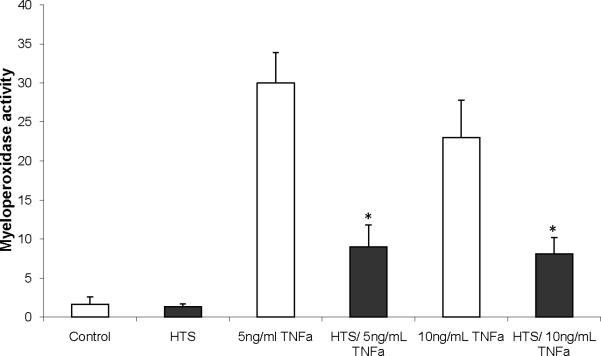
Hypertonic saline (170 mM) attenuates PMN adhesion to HMVEC. PMNs were stimulated with PAF then co-cultured with HMVEC that were incubated in media with varying Na+ or sorbitol concentrations to increase osmolarity from 300 to 360 mOsm/L and then stimulated with TNFα (5, or 10 ng/ml) for 6 hrs. Controls are represented by the 300 mOsm/L treated cells. Adhesion is expressed by MPO activity remaining in the experimental wells. Data are expressed as mean ±SEM of 5 experiments. * indicates significant difference (p<0.01) from TNF stimulations at 300 mOsm/L.
Phosphorylation of total p38 MAPK
Levels of protein phosphorylation were measured using a cell-based colorimetric ELISA system comparing dually phosphorylated to total p38 MAPK allowing for more accurate quantification of phosphoprotein levels (SABiosciences, Frederick, MD, USA). In unstimulated HMVECs, the hyperosmolar (HTS) media of 360 mOsm/L modestly activated p38 MAPK (Figure 5) compared to the 300 mOsm/L control over a time course up to 60 mins, with maximal dual phosphorylation of p38 MAPK occurring at 15 mins. TNFα activation of p38 MAPK in 300 mOsm/L media also demonstrated maximal dual phosphorylation at 15 mins, which decreased in a time-dependent fashion. In addition, TNFα activation of p38 MAPK in the presence of 360 mOsm/L media also led to enhanced dual phosphorylation at 30 mins.
Figure 5.

HTS (170 mM) induces and enhances TNFα (10 ng/ml) stimulated dual phosphorylation (T180/Y182) of p38 MAPK, normalized to total p38 MAPK, determined using ELISA. HMVECs were incubated in media with either 140 mM or 170 mM [Na+] (300 mOsm/L or 360 mOsm/L) and stimulated with TNFα for 0, 15, 30, or 60 min. * indicates statistical difference (p<0.01) from matched isotonic time point. Data are expressed as mean ±SEM of 5 experiments
Inhibition of p38 MAPK
The specific p38 MAPK inhibitor, SB203580, was used to determine whether blockade of p38 MAPK influenced the effects of HTS on ICAM-1 surface expression (Figure 6). SB203580 attenuated TNFα-stimulated ICAM-1 surface expression by 33.8%±3.3% of maximal expression in 300 mOsm/L media. Consistent with previous findings, TNFα stimulated ICAM-1 surface expression was attenuated significantly in 360 mOsm/L media (58.2±5.5% of maximal). The addition of SB203580 to this hyperosmolar media resulted in a similar attenuation (53.5±7.8% of maximal). Thus, p38 MAPK inhibition had no significant additive effect on HTS attenuation of stimulated ICAM-1 expression.
Figure 6.
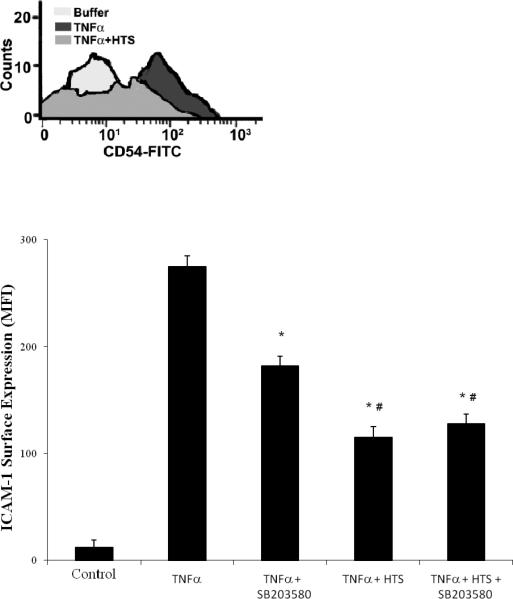
p38 MAPK inhibition reduces TNFα stimulated ICAM-1 surface expression at normal 140mM [Na+] but does not add to the inhibition provided by HTS (170 mM). HMVECs were pretreated with SB203580 (1μM) in media with either 140 mM or 170 mM [Na+] (300 mOsm/L or 360 mOsm/L) then stimulated with TNFα for 6 hrs. Control is represented, by quiescent cells in 300 mOsm/L media. ICAM-1 surface expression is measured by flow cytometry with the panel showing ICAM-1 positive counts from a representative experimental run. Data are expressed as mean±SEM of 5 experiments. * denotes (p<0.05) difference from TNFα–stimulated HMVECs and # denotes difference from SB203580 pretreated cells, in 300 mOsm/L media (isotonic).
Phosphorylation of total I-kB
Levels of protein phosphorylation were measured using a cell-based colorimetric ELISA system comparing dually phosphorylated (S32, S36) to total I-kB (SABiosciences, Frederick, MD, USA). TNFα (10 ng/ml) activation in 300 mOsm/L media demonstrated a peak of dual phosphorylated I-kB at 15 min that declined over time (Figure 7). However, TNFα activation of I-kB in the presence of 360 mOsm/L media lead to depressed dual phosphorylation of the I-kB pool, at 15 min. HTS alone at 360 mOsm/L did not induce activation of I-kB at any time point measured (data not shown).
Figure 7.
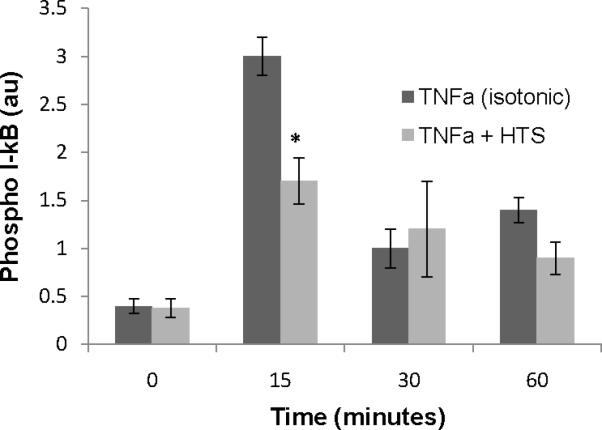
HTS (170 mM), decreases TNFα (10 ng/ml) induced I-kB phosphorylation (Ser 32/36) and consequent degradation (total). HMVECs were stimulated with TNFα for 0, 15, 30, and 60 mins in media with either 140 mM or 170 mM [Na+] (300 mOsm/L or 360 mOsm/L, respectively). Phospho I-kB and total were measured in cell lysates determined by ELISA. Cells incubated with 170 mM [Na+] (360 mOsm/L) alone for up to 60 mins were not different from isotonic cells (300mOsm) at time 0. Data are expressed as mean ±SEM of 5 experiments. * indicates difference (p<0.01) from matched isotonic time point.
Inhibition of NF-kB translocation to the nucleus
At control levels of 140mM [Na+], TNFα induced a 3.6-fold increase of NF-kB over the course of 60 mins in the nuclear fraction, detected as p65 Rel A captured on plate bound consensus -kB DNA oligonucleotide (Figure 8). In HMVECs incubated with HTS (170 mM), TNFα stimulated NF-kB translocation was decreased at both 30 and 60 mins.
Figure 8.

HTS (170 mM) decreases TNFα induced NF-kB nuclear translocation and DNA binding (40.4% and 30.6% for 30 and 60 mins, respectively; p<0.01). HMVECs were stimulated with TNFα for 0, 15, 30, 60 mins in media with either 140 mM or 170 mM [Na+] (300 mOsm/L or 360 mOsm/L, respectively). Nuclear fractions were isolated from cell lysates and p65 Rel A binding to the kB sequence determined by ELISA. Cells incubated with 170 mM [Na+] (360 mOsm/L) alone for up to 60 mins were not different from isotonic cells (300mOsm) at time 0. Data are expressed as mean ±SEM of 5 experiments. * indicates p<0.01 compared to matched time groups.
DISCUSSION
The lung is the first organ to be involved in MOF, which is the leading cause of late death in severe postinjury patients (1). The pathogenesis of postinjury ARDS may be conceptualized as a two-event model (2). The first event, provoked by the initial traumatic insult, results in priming of PMNs and activation of pulmonary ECs leading to PMN-EC adhesion and sequestration (21). A second event (e.g. surgery, sepsis) during this vulnerable period is precipitated by unbridled inflammation characterized by activation of sequestrated PMNs resulting in lung tissue injury manifested as ALI/ARDS. The PMN has been established as a major effector cell in this hyperinflammatory paradigm, and in vivo hemorrhagic shock models demonstrate that HTS attenuates PMN sequestration in the lung and subsequent ARDS (4, 6, 18). Other investigations have revealed that HTS inhibits inflammatory responses of isolated PMN stimulated by diverse signals such as PAF(9), fMLP(34), LTB4 (18) or mesenteric lymph (30). We therefore proceeded to study the effects of HTS on HMVECs. The importance of EC activation is supported by studies demonstrating that PMN-mediated ALI requires endotheliaI ICAM-1 surface expression (25, 26). The data presented here shows the effects of HTS on HMVECs, specifically the molecular mechanism whereby HTS decreases the PMN-EC interaction.
Recent studies have established that ALI/ARDS are mostly localized to the capillary beds which are lined with the microvascular EC (14). Both ICAM-1 surface expression and IL-8 synthesis and secretion by the EC appear integral to ischemia/reperfusion induced inflammatory injury of various organs including the lung (27, 28). Although recognizing the importance of other EC adhesion molecules (i.e.vascular cell adhesion molecule-1), ICAM-1 appears pivotal in the development of ALI (26). The β2-integrin-ICAM-1 interactions are important not only for PMN adherence but also in signaling for cytotoxic potential (20, 29). We used primary HMVECs to assess the effect of HTS not only on the expression of model readouts (IL-8, ICAM-1) but also at the level of functional adhesion; whereas most other investigators have used human umbilical vein endothelial cells, which have little relevance postnatally. To simulate a postinjury state, PMNs were primed with PAF to upregulate CD-11b for our in vitro adhesion assay. We focused our HTS concentrations on the range of physiologically plausible levels and examined whether its effects are due to 1. ionicity, or 2. osmolarity. That requires determining whether 3. the effects of HTS are truly intracellular and recognizing 4. the distinct effects caused by very high (170mM) versus moderate (<170mM) physiological concentrations. A value of [Na+] 170mM was chosen based on studies where (9, 30, 31) clinical infusions elevated circulating [Na+] to no more than 170mM, (360 mOsm/L) and for less than 1 hr. As previously stated, HMVEC ICAM-1 adhesion to the β2-integrin is necessary for PMN-mediated tissue injury. Since HTS can inhibit PAF primed CD11b expression (9), we used HTS only on the HMVEC to interrogate the effects of hyperosmolar conditions on adherence to the vascular endothelium..
The presented data shows that increasing [Na+] alone does not alter basal IL-8 synthesis or ICAM-1 surface expression on HMVECs (Figure 1). However, following TNFα stimulation (to mimic an initial inflammatory event), NF-kB activation, IL-8 secretion, ICAM-1 surface expression and PMN-EC adhesion were all attenuated by HTS at 160 and 170mM (340 and 360 mOsm/L, Figure 2).
In order to investigate the mechanism for the inhibitory effects of HTS, the TNFα-mediated signaling pathways were examined. The Na+/H+ exchange pump is a membrane pump activated upon changing Na+ concentrations (32). Blockade of NHE-1 by the amiloride drug DMA (22) had no effect on the ability of HTS to attenuate ICAM-1 surface expression (data not shown). Because sorbitol also blocked ICAM-1 surface expression at equivalent HTS osmolarity, it is unlikely that NHE-1 has a role in the observed decrease ICAM-1 surface expression, and the mechanism likely involves osmolar, not ionic, inhibition of signal transduction. Indeed, sorbitol also had a concentration-dependent attenuation of ICAM-1 surface expression.
We then investigated whether decreased IL-8 and ICAM-1 expression following TNFα stimulation under hyperosmolarity correlated with decreased primed-PMN adhesion. Hyperosmotic conditions induced by increased Na+ or sorbitol concentration attenuated PMN adhesion to HMVECs by both residual MPO (Figure 4) and 51Cr counting (data not shown). These findings are consistent with the observation that HTS resuscitation results in less PMN sequestration in the lungs when compared to Lactated Ringer's (4). Although clinical trials have not shown a superiority of HTS resuscitation compared to crystalloids, they were either not sufficiently powered, or had the trial stopped early to conclude any definitive diagnosis (33). Thus, the debate continues on HTS as a fluid of resuscitation. HTS may be beneficial in postinjury resuscitation by attenuating stimulated IL-8 secretion and ICAM-1 surface expression and, most importantly PMN adhesion thereby decreasing subsequent tissue injury.
P38 MAPK activation is necessary for maximal lCAM-1 surface expression in HMVEC stimulated with TNFα (12). The presented data here demonstrates that in these primary HMVECs, HTS augments p38 MAPK activation (Figure 5), which is consistent with others demonstrating that various hyperosmotic stressors impart p38 MAPK activity (34). Despite some activation of p38 MAPK, HTS itself did not induce IL-8 synthesis and release nor ICAM-1 surface expression. These results parallel the findings of acetyltransferase et al. (34) in PMNs demonstrating that HTS alone has little activity on cell functions, but modulates other stimulus signaling.
Though TNFα stimulation under hypertonic conditions further enhances activation of p38 MAPK (Figure 5) in HMVECs, hyperosmolar stress (HTS or sorbitol) interfered with surface expression of ICAM-1 and IL-8 synthesis and release. This indicates that HTS attenuates TNFα stimulated ICAM-1 surface expression in HMVECs, distal to p38 MAPK activity. Indeed, inhibiting p38 MAPK activation did not enhance the ability of HTS to attenuate stimulated ICAM-1 surface expression (Figure 6).
Physiologically relevant HTS attenuated PMN adhesion to activated HMVECs, which is consistent with the decreased IL-8 release and ICAM-1 surface expression. That HTS suppressed TNFα pro-inflammatory activation of HMVECs suggests a protective mechanism involving attenuation of the proximal cytoplasmic signaling events (Figure 7) responsible for activation of NF-kB in the nucleus, leading to decreases in both ICAM-1 surface expression and IL-8 release. Although NF-kB dimers can be diverse (16), the prevalent dimer p65 Rel A - p50 regulates both genes. We measured decreased –kB DNA binding by p65 Rel A in the nuclear fraction by ELISA, that was proportional for both readouts (Figure 8). Thus, HTS joins a large array of NF-kB inhibitors with a translational edge: cost and safety (35). Gratifyingly, our hyperosmolarity values attenuated but did not completely abolish the NF-kB activation, since NF-kB is also required for cell survival. Therefore, about 30 minutes of HTS exposure achieved about 40% immunomodulation, without outright suppression of inflammatory signaling. Our findings may also help resolve the controversies of whether HTS is pro-inflammatory or immunosuppressive. Although HTS may enhance PMN cytotoxic potential in vitro (36), this effect may be countered by decreased pro-inflammatory activation of the vascular endothelium.
The described studies demonstrates for the first time dramatic in vitro effects of HTS on HMVECs, limiting PMN sequestration, and further support the clinical findings that HTS is a beneficial anti-inflammatory therapy after injury. The beneficial effects of HTS in vivo to reduce PMN sequestration in lung parenchyma following hemorrhagic shock may be the result of decreased NF-kB signaling, lowering IL-8 release and ICAM-1 surface expression on pulmonary ECs as demonstrated in vitro. This immunosuppressive effect on vascular EC may clinically prevent syndromes of excessive PMN activity in both ARDS and post injury MOF as our in vivo models demonstrated.
Acknowledgements
The authors thank members of the SHOCK society for valuable discussions. This work was supported by NIGMS P50 49222 and T32GM008315 – 21.
Supported in part by National Institutes of Health Grants P50GM49222, T32GM09315, and a Clinical Associate Physician Award (M0l-RR--69)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
BIBLIOGRAPHY
- 1.Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury. 2009;40(9):912–8. doi: 10.1016/j.injury.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 2.Matthay MA, Zimmerman GA, Esmon C, Bhattacharya J, Coller B, Doerschuk CM, Floros J, Gimbrone MA, Jr., Hoffman E, Hubmayr RD, Leppert M, Matalon S, Munford R, Parsons P, Slutsky AS, Tracey KJ, Ward P, Gail DB, Harabin AL. Future research directions in acute lung injury: summary of a National Heart, Lung, and Blood Institute working group. Am J Respir Crit Care Med. 2003;167(7):1027–35. doi: 10.1164/rccm.200208-966WS. [DOI] [PubMed] [Google Scholar]
- 3.Bulger EM, Jurkovich GJ, Nathens AB, Copass MK, Hanson S, Cooper C, Liu PY, Neff M, Awan AB, Warner K, Maier RV. Hypertonic resuscitation of hypovolemic shock after blunt trauma: a randomized controlled trial. Arch Surg. 2008;143(2):139–48. doi: 10.1001/archsurg.2007.41. discussion 149. [DOI] [PubMed] [Google Scholar]
- 4.Angle N, Hoyt DB, Coimbra R, Liu F, Herdon-Remelius C, Loomis W, Junger WG. Hypertonic saline resuscitation diminishes lung injury by suppressing neutrophil activation after hemorrhagic shock. Shock. 1998;9(3):164–70. doi: 10.1097/00024382-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Yada-Langui MM, Anjos-Valotta EA, Sannomiya P, Rocha e Silva M, Coimbra R. Resuscitation affects microcirculatory polymorphonuclear leukocyte behavior after hemorrhagic shock: role of hypertonic saline and pentoxifylline. Exp Biol Med (Maywood) 2004;229(7):684–93. doi: 10.1177/153537020422900713. [DOI] [PubMed] [Google Scholar]
- 6.Pascual JL, Khwaja KA, Ferri LE, Giannias B, Evans DC, Razek T, Michel RP, Christou NV. Hypertonic saline resuscitation attenuates neutrophil lung sequestration and transmigration by diminishing leukocyte-endothelial interactions in a two-hit model of hemorrhagic shock and infection. J Trauma. 2003;54(1):121–30. doi: 10.1097/00005373-200301000-00015. discussion 130-2. [DOI] [PubMed] [Google Scholar]
- 7.Rizoli SB, Rhind SG, Shek PN, Inaba K, Filips D, Tien H, Brenneman F, Rotstein O. The immunomodulatory effects of hypertonic saline resuscitation in patients sustaining traumatic hemorrhagic shock: a randomized, controlled, double-blinded trial. Ann Surg. 2006;243(1):47–57. doi: 10.1097/01.sla.0000193608.93127.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nick JA, Avdi NJ, Young SK, Knall C, Gerwins P, Johnson GL, Worthen GS. Common and distinct intracellular signaling pathways in human neutrophils utilized by platelet activating factor and FMLP. J Clin Invest. 1997;99(5):975–86. doi: 10.1172/JCI119263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciesla DJ, Moore EE, Gonzalez RJ, Biffl WL, Silliman CC. Hypertonic saline inhibits neutrophil (PMN) priming via attenuation of p38 MAPK signaling. Shock. 2000;14(3):265–9. doi: 10.1097/00024382-200014030-00004. discussion 269-70. [DOI] [PubMed] [Google Scholar]
- 10.McLaughlin NJ, Banerjee A, Kelher MR, Gamboni-Robertson F, Hamiel C, Sheppard FR, Moore EE, Silliman CC. Platelet-activating factor-induced clathrin-mediated endocytosis requires beta-arrestin-1 recruitment and activation of the p38 MAPK signalosome at the plasma membrane for actin bundle formation. J Immunol. 2006;176(11):7039–50. doi: 10.4049/jimmunol.176.11.7039. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Pfeiffer GR, 2nd, Stevens T, Doerschuk CM. Lung microvascular and arterial endothelial cells differ in their responses to intercellular adhesion molecule-1 ligation. Am J Respir Crit Care Med. 2002;166(6):872–7. doi: 10.1164/rccm.2201007. [DOI] [PubMed] [Google Scholar]
- 12.Tamura DY, Moore EE, Johnson JL, Zallen G, Aiboshi J, Silliman CC. p38 mitogen-activated protein kinase inhibition attenuates intercellular adhesion molecule-1 up-regulation on human pulmonary microvascular endothelial cells. Surgery. 1998;124(2):403–7. discussion 408. [PubMed] [Google Scholar]
- 13.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha in pulmonary pathophysiology. Respir Res. 2006;7:125. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70(3):317–25. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 15.Song Y, Shi Y, Harken AH, Meng X, Raeburn CD. [A low level of TNF-mediates hemorrhage-induced acute lung injury via p55 TNF receptor]. Zhonghua Yi Xue Za Zhi. 2003;83(8):691–4. [PubMed] [Google Scholar]
- 16.Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5(5):392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 17.Ciesla DJ, Moore EE, Biffl WL, Gonzalez RJ, Moore HB, Silliman CC. Hypertonic saline activation of p38 MAPK primes the PMN respiratory burst. Shock. 2001;16(4):285–9. doi: 10.1097/00024382-200116040-00009. [DOI] [PubMed] [Google Scholar]
- 18.Partrick DA, Moore EE, Moore FA, Barnett CC, Silliman CC. Lipid mediators up-regulate CD11b and prime for concordant superoxide and elastase release in human neutrophils. J Trauma. 1997;43(2):297–302. doi: 10.1097/00005373-199708000-00015. discussion 302-3. [DOI] [PubMed] [Google Scholar]
- 19.McClay DR, Wessel GM, Marchase RB. Intercellular recognition: quantitation of initial binding events. Proc Natl Acad Sci U S A. 1981;78(8):4975–9. doi: 10.1073/pnas.78.8.4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnett CC, Jr., Moore EE, Mierau GW, Partrick DA, Biffl WL, Elzi DJ, Silliman CC. ICAM-1-CD18 interaction mediates neutrophil cytotoxicity through protease release. Am J Physiol. 1998;274(6 Pt 1):C1634–44. doi: 10.1152/ajpcell.1998.274.6.C1634. [DOI] [PubMed] [Google Scholar]
- 21.Wyman TH, Bjornsen AJ, Elzi DJ, Smith CW, England KM, Kelher M, Silliman CC. A two-insult in vitro model of PMN-mediated pulmonary endothelial damage: requirements for adherence and chemokine release. Am J Physiol Cell Physiol. 2002;283(6):C1592–603. doi: 10.1152/ajpcell.00540.2001. [DOI] [PubMed] [Google Scholar]
- 22.Noel J, Pouyssegur J. Hormonal regulation, pharmacology, and membrane sorting of vertebrate Na+/H+ exchanger isoforms. Am J Physiol. 1995;268(2 Pt 1):C283–96. doi: 10.1152/ajpcell.1995.268.2.C283. [DOI] [PubMed] [Google Scholar]
- 23.Koliakos G, Paletas K, Kaloyianni M. NHE-1: a molecular target for signalling and cell matrix interactions. Connect Tissue Res. 2008;49(3):157–61. doi: 10.1080/03008200802151581. [DOI] [PubMed] [Google Scholar]
- 24.Vedder NB, Harlan JM. Increased surface expression of CD11b/CD18 (Mac-1) is not required for stimulated neutrophil adherence to cultured endothelium. J Clin Invest. 1988;81(3):676–82. doi: 10.1172/JCI113372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontes B, Moore EE, Moore FA, Koike K, Carl V, Banerjee A. PMNs primed for superoxide release and increased CD11b expression do not sequester in normal lung. J Surg Res. 1995;58(6):599–604. doi: 10.1006/jsre.1995.1094. [DOI] [PubMed] [Google Scholar]
- 26.Grau GE, Mili N, Lou JN, Morel DR, Ricou B, Lucas R, Suter PM. Phenotypic and functional analysis of pulmonary microvascular endothelial cells from patients with acute respiratory distress syndrome. Lab Invest. 1996;74(4):761–70. [PubMed] [Google Scholar]
- 27.Farhood A, McGuire GM, Manning AM, Miyasaka M, Smith CW, Jaeschke H. Intercellular adhesion molecule 1 (ICAM-1) expression and its role in neutrophil-induced ischemia-reperfusion injury in rat liver. J Leukoc Biol. 1995;57(3):368–74. [PubMed] [Google Scholar]
- 28.Kelly KJ, Williams WW, Jr., Colvin RB, Bonventre JV. Antibody to intercellular adhesion molecule 1 protects the kidney against ischemic injury. Proc Natl Acad Sci U S A. 1994;91(2):812–6. doi: 10.1073/pnas.91.2.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Issekutz AC, Issekutz TB. The contribution of LFA-1 (CD11a/CD18) and MAC-1 (CD11b/CD18) to the in vivo migration of polymorphonuclear leucocytes to inflammatory reactions in the rat. Immunology. 1992;76(4):655–61. [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez RJ, Moore EE, Ciesla DJ, Neto JR, Biffl WL, Silliman CC. Hyperosmolarity abrogates neutrophil cytotoxicity provoked by post-shock mesenteric lymph. Shock. 2002;18(1):29–32. doi: 10.1097/00024382-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 31.Sheppard FR, Moore EE, McLaughlin N, Kelher M, Johnson JL, Silliman CC. Clinically relevant osmolar stress inhibits priming-induced PMN NADPH oxidase subunit translocation. J Trauma. 2005;58(4):752–7. doi: 10.1097/01.ta.0000159246.33364.72. discussion 757. [DOI] [PubMed] [Google Scholar]
- 32.Krump E, Nikitas K, Grinstein S. Induction of tyrosine phosphorylation and Na+/H+ exchanger activation during shrinkage of human neutrophils. J Biol Chem. 1997;272(28):17303–11. doi: 10.1074/jbc.272.28.17303. [DOI] [PubMed] [Google Scholar]
- 33.Bulger EM, May S, Kerby JD, Emerson S, Stiell IG, Schreiber MA, Brasel KJ, Tisherman SA, Coimbra R, Rizoli S, Minei JP, Hata JS, Sopko G, Evans DC, Hoyt DB. Out-of-hospital hypertonic resuscitation after traumatic hypovolemic shock: a randomized, placebo controlled trial. Ann Surg. 2011;253(3):431–41. doi: 10.1097/SLA.0b013e3181fcdb22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Junger WG, Hoyt DB, Davis RE, Herdon-Remelius C, Namiki S, Junger H, Loomis W, Altman A. Hypertonicity regulates the function of human neutrophils by modulating chemoattractant receptor signaling and activating mitogen-activated protein kinase p38. J Clin Invest. 1998;101(12):2768–79. doi: 10.1172/JCI1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25(51):6887–99. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 36.Partrick DA, Moore EE, Offner PJ, Johnson JL, Tamura DY, Silliman CC. Hypertonic saline activates lipid-primed human neutrophils for enhanced elastase release. J Trauma. 1998;44(4):592–7. doi: 10.1097/00005373-199804000-00006. discussion 598. [DOI] [PubMed] [Google Scholar]