

Abstract
Inhibition of receptor tyrosine kinase (RTK) signaling pathways is an important area for the development of novel anticancer agents. Numerous multikinase inhibitors (MKIs) have been recently approved for the treatment of cancer. Vascular endothelial growth factor receptor-2 (VEGFR-2) is the principal mediator of tumor angiogenesis. In an effort to develop ATP-competitive VEGFR-2 selective inhibitors the 5-chloro-N4-substituted phenyl-9H-pyrimido[4,5-b]indole-2,4-diamine scaffold was designed. The synthesis of the target compounds involved N-(4,5-dichloro-9H-pyrimido[4,5-b]indol-2-yl)-2,2-dimethylpropanamide) as a common intermediate. A nucleophilic displacement of the 4-chloro group of the common intermediate by appropriately substituted anilines afforded the target compounds. Biological evaluation indicated that compound 5 is a potent and selective VEGFR-2 inhibitor comparable to sunitinib and semaxinib.
Keywords: Receptor tryosine kinase inhibitors, Pyrimido[4, 5-b]indol synthesis, Cytotoxicity, VEGFR-2 inhibitors
1. Introduction
Protein tyrosine kinases are a key means of signal transduction in eukaryotic cells and control processes such as cell proliferation, differentiation, migration, survival, and cell cycle progression. Dysregulation of these tightly regulated processes through overexpression of kinases is implicated in numerous disease states including cancer.2,3
Angiogenesis is the process by which new blood vessels are formed from pre-existing vasculature and this process is crucial for a solid tumor to grow beyond 1–2 mm. As a tumor grows in size, it becomes increasingly hypoxic, leading to induction of growth factors including vascular endothelial growth factor (VEGF), and other important proangiogenic regulators like epidermal growth factor (EGF), platelet derived growth factor (PDGF), fibroblast growth factor (FGF), insulin-like growth factor-1 (IGF-1), transforming growth factors (TGFα and β), and tumor necrosis factor-α (TNF-α) among others.2, 4 The growth factors function by binding to receptor tyrosine kinases (RTKs), a super family of transmembrane proteins. Subsequent to binding of the growth factor, RTKs dimerize and undergo autophosphorylation, initiating angiogenesis.5 Anti-angiogenic therapy targets non-tumor cells (endothelial cells) which may be less prone to mutations and hence to resistance compared to tumor cells.6
One of the approaches to circumvent angiogensis is the inhibition of the key RTKs involved in angiogenesis. Several reports on multikinase inhibitors (MKIs) have appeared in the recent literature, some of which have afforded clinically approved agents. Imatinib (Figure 1) was the first RTK inhibitor approved in 2001 for chronic myelogenous leukemia (CML).7,8 Imatinib inhibits Abelson Tyrosine Kinase (Abl), c-kit protein (CD117), PDGFR-α and PDGFR-β.
Figure 1.
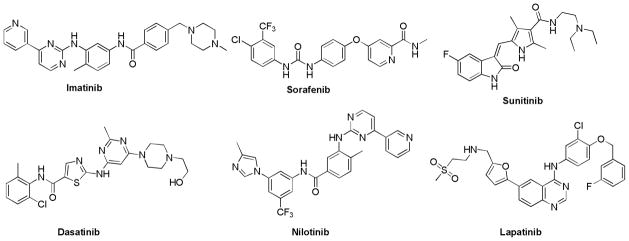
Various RTK inhibitors
Sorafenib, approved in 2005 for the treatment of hepatocellular carcinoma and renal cell carcinoma, inhibits Raf kinase, VEGFR-2, VEGFR-3, PDGFR-β, and c-kit.9 Sunitinib, approved in 2006 for the treatment of renal cell carcinoma and imatinib-resistant gastrointestinal stromal tumors, inhibits VEGFR, PDGFR, c-kit, and FMS-like tyrosine kinase 3 (FLT3).10
Two drugs – dasatinib11 and nilotinib12 were approved in 2006 and 2007 respectively, specifically for imatinib-resistant or unresponsive tumors in CML. Dasatinib inhibits Abl, c-Kit, PDGFR, and Src. Nilotinib inihibits Abl, c-Kit, PDGFR-β, Src, and Ephrin. Lapatinib, approved in 2007 for advanced metastatic breast cancer in conjunction with chemotherapy, inhibits EGFR and erythroblastic leukemia viral oncogene homolog-2 (ErbB-2).13
Although the approval of numerous MKIs for cancer therapy attests to the importance of this approach in cancer chemotherapy, gaining selectivity for a limited subset of kinases for MKIs is a widely recognized challenge facing medicinal chemists in the kinase area.14 The risk of a MKI approach is that such compounds are by their very nature more likely to hit a diverse spectrum of kinases. A recent report studied the selectivity of approved kinase inhibitors and candidates across 317 different kinases.15 The most selective of the currently approved MKIs was lapatinib, and the least selective was sunitinib; the latter bound > 15% of kinases tested with Kd < 100 nM. Bamborough et al.16 screened 577 diverse compounds versus 203 protein kinases and found that two-thirds of the compounds bound to more than 10 kinases, hence clearly demonstrating the extent of the selectivity challenge in the kinase area.
Where a large number of closely related kinase isozymes exist, some of which may be crucial for normal cellular function, it is critical to avoid the off-target activity of MKIs as it might translate into undesirable biology. This has been exemplified by the approved MKIs sorafenib and sunitinib which have been reported to be cardiotoxic.17,18 In addition, imatinib has been reported to have mechanism-based cardiotoxic effects, and nilotinib carries a black box warning for possible heart complications.19 Hence, specific inhibitors of kinases critical in tumor survival like VEGFR-2 or other RTKs, with minimal off-target activity, are also of considerable interest.
In 1996, Traxler et al.20 reported a series of N4-substituted phenyl-9H-pyrimido[4,5-b]indoles as inhibitors of EGFR. From this series, compound 1 (Figure 2) emerged as a single digit nanomolar inhibitors of EGFR (IC50 = 6 nM). Showalter et al.21 reported several elaborations of similar anilino pyrimidine inhibitors of which compound 2 emerged as another potent inhibitor of EGFR (IC50 = 147 nM). Compounds 1 and 2 were not tested in VEGFR-2, which has been implicated as the principal mediator of angiogenesis22,23–25 and VEGFR-2 stimulation alone is enough to initiate tumor growth and metastases.25 It was of interest to structurally design a scaffold to afford either dual EGFR/VEGFR-2 inhibition or perhaps a selective VEGFR-2 inhibition.
Figure 2.

Design of scaffold (I)
On the basis of 1 and 2, we designed general scaffold A – 5-chloro-N4-substituted phenyl-9H-pyrimido[4,5-b]indole-2,4-diamine (Figure 2). A systematic conformational search (5° increments) performed using Sybyl X 1.326 indicated a lower number of conformations (24) about the C4–N bond with the pyrimidine ring for 5-chloro-N4-phenyl-9H-pyrimido[4,5-b]indole-2,4-diamine 3 (Figure 5) compared to its corresponding 5-deschloro analog, 2 (41). Hence, the addition of a 5-Cl substitution was expected to provide conformational restriction through steric hindrance to rotation and this may allow for specificity against some kinases over others and perhaps allow potent inhibition of a different spectrum of RTKs.
Figure 5.

5-Chloro-N4-substituted phenyl-9H-pyrimido[4,5-b]indole-2,4-diamines as potential multiple RTK inhibitors with lead compounds
Molecular modeling was carried out to gain some insight to the binding mode of 3 in the VEGFR-2 active site. The best scored pose on docking compound 3 using Lead IT 2.1.027 is depicted in Figures 3 and 4. The binding site of ATP competitive inhibitors in RTKs consists of a Hinge region, and a hydrophobic binding site (Hydrophobic Region I) among others (Figure 3).28–31 The pyrimido[4,5-b]indole ring of 3 occupies the adenine binding portion of the ATP binding site. The 1-N and the 9-NH are involved in hydrogen bonds with Cys917 in the Hinge region. Hydrophobic interactions of the indole ring with Val914, Phe916, Cys917, Leu1033, Cys1043, Leu838 and Val846 can stabilize the docked pose. In addition, the aniline phenyl extends towards Hydrophobic region I and is involved in interactions with Val846, Ala864, Lys866, Val897, Val914 and Leu1033. The 2-amino group could potentially form a hydrogen bond with Glu915 in the hinge region.
Figure 3.
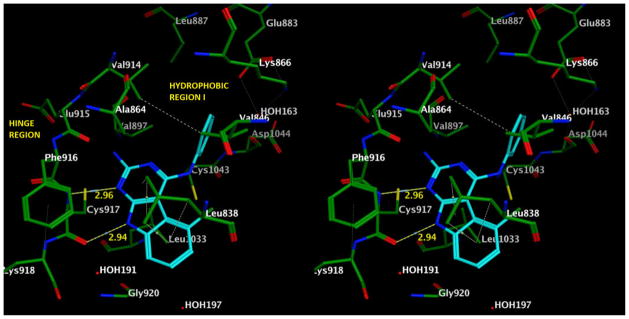
Stereoview of docked pose of compound 3 in VEGFR-2 (PDB code: 1YWN) showing the key interactions.
Figure 4.

Ligand interaction plot of the docked pose of compound 3 in VEGFR-2 (PDB code: 1YWN) showing the key interactions.
A series of six compounds 3–8 (Figure 5) was designed to study scaffold A with respect to inhibition of receptor tyrosine kinases. The selection of the substituents on the 4-position are rationalized below. The 4′-isopropyl group in 4 was included to obtain information about bulk tolerance in this position in the RTK active site. Compound 15 (bearing a 4-chloroaniline) reported by Bold et al.32 demonstrated potent VEGFR-2 and VEGFR-1 inhibition in 15 (Figure 5). Thus compound 5 with the 4′-Cl aniline was proposed with the purpose of obtaining potent inhibition of VEGFR-2 and/or VEGFR-1. The 3′-OMe aniline substitution in 8 is similar to compounds 16 and 17 (Figure 5) which are potent inhibitors of EGFR.33 In order to obtain further SAR information at this position, the 3′-OMe group (8) was replaced with the 3′-F (7) which would afford information regarding electron withdrawing groups at the 3′-position. Compound 6 with the 2′-F, 4′-Cl aniline substituted ring is similar to that in 1834 and was included with the purpose of obtaining potent inhibition of VEGFR-2 and VEGFR-1 as reported for 18. The unsubstituted phenyl ring (3) served as a comparison with the substituted compounds.
The synthesis of 3–8 was designed from the corresponding 2-amino pivaloyl (2,2-dimethylpropan-1-one) protected precursors 9–14. It was decided to biologically evaluate intermediates 9–14 to study the effect of the 2-amino pivaloyl groups on binding to RTKs. It was anticipated that the pivaloyl moieties of 9–14 could interact at hydrophobic region II 35 and perhaps afford increased activity against some RTK.
2. Chemistry
Compounds 3–14 were synthesized as described in Scheme 1. N-(4,5-dichloro-9H-pyrimido[4,5-b]indol-2-yl)pivalamide 19 was synthesized by a method developed by Gangjee et al.35 Compound 19 was condensed with appropriately substituted anilines in isopropanol at reflux, in the presence of three drops of conc. HCl, to afford regiospecifically 9–14. Deprotection of the 2-pivaloyl group in 11 by reaction with 15% KOH in 1,4-dioxane at reflux afforded 5. Compounds 9, 10 and 12 were more soluble in a 1:1 mixture of methanol to methylene chloride than in 1,4-dioxane. However, when 1 N NaOH and the 1:1 methanol/methylene chloride solvent system were used for the deprotection reaction of 9, 10 and 12 to obtain 3, 4 and 6 respectively, the result was an increased reaction time (1–3 days) and lower yields for 3 (52%) and 4 (49%) (This method afforded 6 in 74% yield). Reaction time was significantly decreased when 1 N NaOH in isopropanol was used as the reaction system; 13 and 14 were converted to 7 (70%) and 8 (92%) respectively in 2–14 h. Isopropanol was more efficient, probably because of the enhanced solubility of the starting material (13, 14) as well as the nucleophile (hydroxide ions).
Scheme 1.

Reagents and conditions: (a) ArNH2, Isopropanol, 3 drops of conc. HCl, reflux, 1.5–6 h (48%–95%); (b) 15% KOH, 1,4-dioxane, reflux, 14 h (95%); (c) 1 N NaOH, methanol/CH2Cl2, reflux, 1–3 d (49%–74%); (d) 1 N NaOH, Isopropanol, reflux, 2–14 h (70%–92%).
3. Biological evaluation and discussion
Compounds 3–14 were evaluated as RTK inhibitors using human tumor cells known to express high levels of VEGFR-2, EGFR and PDFGR-β using a phosphotyrosine ELISA cytoblot (Table 1).36 Whole cell assays were used for RTK inhibitory activity since these assays afford more meaningful results for translation to in vivo studies. The effect of compounds on cell proliferation was measured using A431 cancer cells, known to overexpress EGFR. EGFR is known to play a role in the overall survival of A431 cells.36
Table 1.
IC50 values (μM) of kinase inhibiton and A431 cytotoxicity for compounds 3–14
| Compound | VEGFR-2 kinase inhibition | EGFR Kinase Inhibition | PDGFR-β kinase inhibition | A431 cytotoxicity |
|---|---|---|---|---|
| 3 | 123.8 ± 29.0 | >200 | >200 | 193.1 ±29.7 |
| 4 | 30.3 ± 5.0 | 42 ± 10.5 | >200 | 45.1 ±10.1 |
| 5 | 13.2 ± 1.7 | 63.6 ± 8.1 | 196.2 ± 22.4 | 44.1 |
| 6 | 38.9 ± 6.0 | 38.9 ± 6.0 | >200 | 128.3 ± 20.2 |
| 7 | 86.1 ± 7.8 | >200 | 172.2 ± 30.1 | 184.1 ± 22.1 |
| 8 | 70.1 ± 10.1 | 82.9 ± 19.2 | 50.5 ± 7.2 | 167.0 ± 38.6 |
| 9 | 192.0 ± 16.2 | 114.7 ± 30.2 | 70.6 ± 10.7 | 185.2 ± 20.1 |
| 10 | 47.8 ± 10.1 | 39.2 ± 7.8 | >200 | >250 |
| 11 | 21.3 ± 4.0 | 58.7 ± 7.2 | >200 | 55.0 |
| 12 | 89.1 ± 10.9 | >200 | 170.8 ± 19.6 | >250 |
| 13 | 31.2 ± 4.5 | 30.8 ± 7.0 | >200 | >250 |
| 14 | 41.0 ± 7.2 | 130.5 ± 32.8 | 196.2 ± 30.0 | >250 |
| Semaxanib(20) | 12.0 ± 2.7 | |||
| PD153035 (21) | 0.2 ± 0.004 | |||
| DMBI (22) | 3.7 ± 0.06 | |||
| Sunitinib | 18.9 ± 2.7 | 172.1 ± 19.4 | 83.1 ± 10.1 | |
| Erlotinib | 124.7 ± 18.2 | 1.2 ± 0.2 | 12.2 ± 1.9 | |
| Cisplatin (23) | 10.6 ± 2.9 |
Since the IC50 values of compounds vary under different assay conditions (e.g., ATP concentrations), standard compounds (Figure 6) were used as controls in each of the evaluations. The standard compounds used were semaxanib (SU5416), 20 for VEGFR-2;37 4-[(3-bromophenyl)amino]-6,7-dimethoxyquinazoline (PD153035), 21 for EGFR;38 3-(4-dimethylamino-benzylidenyl)-2-indolinone (DMBI), 22 for PDGFR-β.39 and cisplatin, 23 for A431 cytotoxicity. Approved agents sunitinib (a multitargeted RTK inhibitor including VEGFR-2, PDGFRs) and erlotinib40 (a specific EGFR inhibitor approved for non small cell lung cancer) were also used as standard agents.
Figure 6.

In the VEGFR-2 kinase inhibition assay, of the target compounds 3–8, the 4-chlorophenyl substituted compound 5 was the most active and equipotent to the standard 20. Compound 5 was also 1.4- and 9.4-fold more potent than sunitinib and erlotinib respectively in the VEGFR-2 inhibition assay. The next most potent compounds were the 4-isopropylphenyl substituted 4, and the 2-fluoro-4-chlorophenyl substituted 6 which were 2.5-fold less active and 3-fold less active respectively, than 20. VEGFR-2 inhibition decreased on moving the phenyl substitution from the 4- to the 3-position, as exemplified by the 3-fluorophenyl substituted 7 and the 3-methoxy substituted 8, which were 6- and 7-fold less potent then 20 respectively.
For the 2-amino pivaloyl substituted compounds 9–14, the 4-chlorophenyl substituted compound 11 was the most active and was 2-fold less potent than the standard 20 in the VEGFR-2 inhibition assay. The next most potent compounds included the 3-fluorophenyl substituted 13, 3-methoxyphenyl substituted 14, and the 4-isopropylphenyl substituted 10, which were about 2.5-, 3.5- and 4-fold less active than 20, respectively. The increased activity of the 2-pivaloyl analogs 13–14 in the VEGFR-2 assay over the corresponding despivaloyl analogs 7 and 8 respectively indicates a possible interaction with hydrophobic site II of the pivaloyl moiety in VEGFR-2.
A trend in activity was observed for 3–5, however in that these compounds were about 1.5-fold more active than their 2-amino pivaloyl derivatives. However, a similar trend was not observed for 7 and 8 which were about 2.8- and 1.7-fold less active than the corresponding 2-amino pivaloyl compounds 13 and 14. Thus subtle differences in binding orientation of 3–5 compared with 7–8 perhaps accounts for the differences in their respective pivaloyl derivatives.
The most potent compound against EGFR was the 2-amino pivaloyl, 3-fluorophenyl substituted 13 which was 150-fold less potent than the standard 21. However compound 13 was 5.5-fold more active then sunitinib, and 25-fold less active than erlotinib in the EGFR inhibition assay. The presence of a 5-chloro group is detrimental to cell-based EGFR inhibition relative to the 5-deschloro lead compound 2 (Figure 2) which is a reported nanomolar EGFR inhibitor in an isolated enzyme assay. The most potent PDGFR-β inhibitor in this study was the 3-methoxyphenyl substituted 8, which is about 13.5-fold less active than the standard 22. Compound 8 was 1.6-fold more potent than sunitinib and 4-fold less active than erlotinib in the PDGFR-β inhibition assay.
In the A431 cytotoxicity assay, the most potent compounds were the 4-isopropylphenyl substituted 4, and the 4-chlorophenyl substituted 5 which were 4-fold less potent than the standard 23. Since substitution at any other position produced a further decrease in activity, the 4-position is optimum for activity, and tolerates both electron withdrawing and electron donating as well as bulky groups. Of the 2-amino pivaloyl derivatives 9–14, only the 2-amino pivaloyl, 4-chlorophenyl substituted 11 produced potent inibition, which was 5-fold less than the standard 23.
In summary, compounds 3–14 were synthesized to develop a novel scaffold with either selective VEGFR-2 and/or VEGFR-1 inhibition or EGFR/VEGFR-2 dual inhibition. The cellular inhibition assays demonstrated that the compounds were indeed selective for VEGFR-2, relative to EGFR and PDGFR-β. The 4-chlorophenyl substituted 5 was the most potent inhibitor in the VEGFR-2 inhibition assay and was equipotent with the standard compound 20, and was 1.4- and 9.4-fold more potent than sunitinib and erlotinib respectively. The results of this study indicate that the 5-chloro-N4-substituted phenyl-9H-pyrimido[4,5-b]indole-2,4-diamine scaffold is highly selective for VEGFR-2 inihibition with minimal off-target kinase inhibition in the limited number of RTKs these compounds were evaluated against and serves as a new lead analog that is equipotent to sunitinib and almost 10x more potent that erlotinib in the VEGFR-2 inhibition whole cell assay.
4. Experimental section
Analytical samples were dried in vacuo (0.2 mm Hg) in a CHEM-DRY drying apparatus over P2O5 at 80 °C. Melting points were determined on a MEL-TEMP II melting point apparatus with FLUKE 51 K/J electronic thermometer and are uncorrected. Nuclear magnetic resonance spectra for proton (1H NMR) were recorded on a Bruker WH-300 (300 MHz) or Bruker WH-400 (400 MHz) spectrometer. The chemical shift values are expressed in ppm (parts per million): s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad singlet. Thin-layer chromatography (TLC) was performed on Whatman Sil G/UV254 silica gel plates with a fluorescent indicator, and the spots were visualized under 254 and 366 nm illumination. Proportions of solvents used for TLC are by volume. Column chromatography was performed on a 230–400 mesh silica gel (Fisher, Somerville, NJ) column. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Element compositions are within ±0.4% of the calculated values. Fractional moles of water or organic solvents frequently found in some analytical samples of antifolates could not be prevented in spite of 24–48 h of drying in vacuo and were confirmed where possible by their presence in the 1H NMR spectra. All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and were used as received.
General procedure for the synthesis of 9–14
To a 50 mL round bottom flask were added N-(4,5-dichloro-9H-pyrimido[4,5-b]indol-2-yl)-2,2-dimethyl propanamide 19, various substituted anilines, isopropanol (50 mL) followed by 3 drops of concd HCl. The mixture was heated at reflux for 1.5 to 6 h. The solvent was removed by evaporation in vacuo. The residue was dissolved in methanol and silica gel (2 g) was added followed by evaporation in vacuo to afford a dry silica gel plug which was loaded on top of a wet (hexane) silica gel column sequentially eluting with 0%, 1% and 2% methanol in chloroform. Fractions containing desired product (TLC) were pooled and evaporated to dryness to afford the product.
N-(4-anilino-5-chloro-9H-pyrimido[4,5-b]indol-2-yl)-2,2-dimethylpropanamide (9)
Reaction of 19 (150 mg, 0.44 mmol) and aniline (310 mg, 3.33 mmol) using the general procedure described above, gave 9: yield 86%, TLC Rf 0.6 (CHCl3/MeOH, 10:1 with 2 drops concentrated NH4OH); mp 249–250 °C; 1H NMR (DMSO-d6) δ 1.27 (s, 9 H, C(CH3)3), 7.03–7.08 (m, 1 H, Ar), 7.33–7.38 (m, 4 H, Ar), 7.46–7.49 (m, 1 H, Ar), 8.13–8.16 (d, 2 H, Ar), 9.36 (s, 1 H, 2-NH, exch), 9.67 (s, 1 H, 4-NH, exch), 12.25 (s, 1 H, 9-NH, exch). Anal. (C21H20ClN5O) C, H, N, Cl.
N-{5-chloro-4-[(4-isopropylphenyl)amino]-9H-pyrimido[4,5-b]indol-2-yl}-2,2-dimethylpropanamide (10)
Reaction of 19 (250 mg, 0.74 mmol) and 4-isopropyl aniline (750 mg, 5.56 mmol) using the general procedure described above, gave 10: yield 75%, TLC Rf 0.62 (CHCl3/MeOH, 10:1 with 2 drops concentrated NH4OH); mp 265 °C; 1H NMR (DMSO-d6) δ 1.20 (s, 3 H, CH3), 1.23 (s, 3 H, CH3), 1.27 (s, 9 H, C(CH3)3), 7.20–7.23 (m, 1 H, Ar), 7.35–7.40 (m, 2 H, Ar), 7.46–7.49 (m, 1 H, Ar), 8.03–8.06 (d, 2 H, Ar), 9.31 (s, 1 H, 2-NH, exch), 9.63 (s, 1 H, 4-NH, exch), 12.24 (s, 1 H, 9-NH, exch). HRMS (ESI) [M+H]+ calcd. for C24H26ClN5O m/z = 436.1904, found m/z = 436.1868.
N-{5-chloro-4-[(4-chlorophenyl)amino]-9H-pyrimido[4,5-b]indol-2-yl}-2,2-dimethylpropanamide (11)
Reaction of 19 (150 mg, 0.44 mmol) and 4-chloro aniline (188 mg, 1.46 mmol) using the general procedure described above, gave 11: yield 48%, TLC Rf 0.5 (CHCl3/MeOH, 10:1 with 2 drops concentrated NH4OH); mp 281.8–282 °C; 1H NMR (DMSO-d6) δ 1.26 (s, 9 H, C(CH3)3), 7.31–7.40 (m, 4 H, Ar), 7.45–7.48 (m, 1 H, Ar), 8.19–8.22 (d, 2 H, Ar), 9.37 (s, 1 H, 2-NH, exch), 9.70 (s, 1 H, 4-NH, exch), 12.25 (s, 1 H, 9-NH, exch). Anal. (C21H19Cl2N5O) C, H, N, Cl.
N-{5-chloro-4-[(4-chloro, 2-fluorophenyl)amino]-9H-pyrimido[4,5-b]indol-2-yl}-2,2-dimethylpropanamide (12)
Reaction of 19 (150 mg, 0.44 mmol) and 2-fluoro-4-chloroaniline (485 mg, 3.33 mmol) using the general procedure described above, gave 12: yield 74%, TLC Rf 0.6 (CHCl3/MeOH, 10:1 with 2 drops concentrated NH4OH); mp 284–285 °C; 1H NMR (DMSO-d6) δ 1.28 (s, 9 H, C(CH3)3), 7.23–7.26 (m, 1 H, Ar), 7.33–7.40 (m, 2 H, Ar), 7.49–7.57 (m, 2 H, Ar), 9.47–9.53 (m, 1 H, Ar), 9.50 (s, 1 H, 2-NH, exch), 9.82 (s, 1 H, 4-NH, exch), 12.34 (s, 1 H, 9-NH, exch). Anal. (C21H18Cl2FN5O. 0.025 CHCl3) C, H, N, Cl, F.
N-{5-chloro-4-[(3-fluorophenyl)amino]-9H-pyrimido[4,5-b]indol-2-yl}-2,2-dimethylpropanamide (13)
Reaction of 19 (100 mg, 0.29 mmol) and 3-fluoroaniline (242 mg, 2.17 mmol) using the general procedure described above, gave 13: yield 95%, TLC Rf 0.6 (CHCl3/MeOH, 10:1 with 2 drops concentrated NH4OH); mp 273–274 °C; 1H NMR (DMSO-d6) δ 1.28 (s, 9 H, C(CH3)3), 6.82–6.89 (m, 1 H, Ar), 7.33–7.39 (m, 3 H, Ar), 7.48–7.54 (s, 2 H, Ar), 8.61–8.66 (d, 1 H, Ar), 9.46 (s, 1 H, 2-NH, exch), 9.79 (s, 1 H, 4-NH, exch), 12.27 (s, 1 H, 9-NH, exch). Anal. (C21H19ClFN5O) C, H, N, Cl, F.
N-{5-chloro-4-[(3-methoxyphenyl)amino]-9H-pyrimido[4,5-b]indol-2-yl}-2,2-dimethylpropanamide (14)
Reaction of 19 (100 mg, 0.29 mmol) and 3-methoxyaniline (268 mg, 2.17 mmol) using the general procedure described above, gave 14: yield 91%, TLC Rf 0.6 (CHCl3/MeOH, 10:1 with 2 drops concentrated NH4OH); mp 196–197 °C; 1H NMR (DMSO-d6) δ 1.26 (s, 9 H, C(CH3)3), 3.86 (s, 3 H, CH3), 6.61–6.63 (m, 1 H, Ar), 7.19–7.28 (m, 2 H, Ar), 7.31–7.44 (m, 2 H, Ar), 7.48–7.50 (m, 1 H, Ar), 8.18 (s, 1 H, Ar), 9.37 (s, 1 H, 2-NH, exch), 9.79 (s, 1 H, 4-NH, exch), 12.26 (s, 1 H, 9-NH, exch). Anal. (C22H22ClN5O2. 0.09 CHCl3) C, H, N, Cl.
5-chloro-N4-(4-chlorophenyl)-9H-pyrimido[4,5-b]indole-2,4-diamine (5)
In a 50 mL flask 11 (70 mg, 0.16 mmol) was dissolved in 1,4-dioxane 20 mL, and 2 mL of 15% KOH aqueous solution was added. The reaction mixture was heated to reflux for 14 h. The solvent was evaporated to obtain a residue. Water was added and the mixture was extracted with chloroform. Sodium sulfate was used to dry the extract. Silica gel was added and solvent evaporated to make a plug. The plug was loaded on top of a wet (CHCl3) silica gel column (25 cm × 3 cm) and eluted with 60:1 (CHCl3: CH3OH). Fractions containing desired product were pooled and evaporated to afford 5 in 95% yield. TLC Rf 0.35 (CHCl3/MeOH, 10:1 with 3 drops concentrated NH4OH); mp 306.1–307 °C; 1H NMR (DMSO-d6) δ 6.54 (bs, 2 H, NH2, exch), 7.19–7.21 (m, 2 H, Ar), 7.29–7.37 (m, 4 H, Ar), 7.95–7.98 (m, 2 H, Ar), 9.12 (s, 1 H, 4-NH, exch), 11.75 (s, 1 H, 9-NH, exch). HRMS (ESI) [M+H]+ calcd. for C16H11Cl2N5 m/z = 344.0470, found m/z = 344.0449.
5-chloro-N4-(4-isopropylphenyl)-9H-pyrimido[4,5-b]indole-2,4-diamine (4)
In a 50 mL flask 10 (20 mg, 0.044 mmol) was dissolved in CH2Cl2/CH3OH, 1:1 (10 mL), and 2 mL of 1 N NaOH solution was added. The reaction mixture was heated to reflux for 44 h. The solvent was evaporated to obtain a residue. Water was added and the mixture was filtered and dried. The solid was dissolved in MeOH, silica gel was added and solvent evaporated to make a plug. The plug was loaded on top of a wet (CHCl3) silica gel column and eluted with 20:1 (CHCl3: MeOH). Fractions containing desired product were pooled and evaporated to afford the 4 in 52% yield. TLC Rf 0.40 (CHCl3/MeOH, 10:1 with 3 drops concentrated NH4OH); mp 235–236 °C; 1H NMR (DMSO-d6) δ 1.19–1.21 (m, 6 H, CH3 x 2), 2.85–2.90 (m, 1 H, CH), 6.42 (bs, 2 H, NH2, exch), 7.17–7.20 (m, 4 H, Ar), 7.27–7.30 (m, 2 H, Ar), 7.75–7.77 (m, 2 H, Ar), 9.00 (s, 1 H, 4-NH, exch), 11.68 (s, 1 H, 9-NH, exch). Anal. (C19H18ClN5) C, H, N, Cl.
5-chloro-N4-phenyl-9H-pyrimido[4,5-b]indole-2,4-diamine (3)
Compound 3 (synthesized from 9 (130 mg, 0.33 mmol) as described for 4; reaction time = 15 h): yield 49%; TLC Rf 0.40 (CHCl3/MeOH, 10:1 with 3 drops concentrated NH4OH); mp 271–272 °C; 1H NMR (DMSO-d6) δ 6.48 (bs, 2 H, NH2, exch), 7.01–7.04 (m, 1 H, Ar), 7.18–7.19 (m, 2 H, Ar), 7.29–7.34 (m, 3 H, Ar), 7.89–7.91 (d, 2 H, Ar), 9.08 (s, 1 H, 4-NH, exch), 11.71 (s, 1 H, 9-NH, exch). Anal. (C16H12ClN5. 0.04 CHCl3) C, H, N, Cl.
5-chloro-N4-(4-chloro-2-fluorophenyl)-9H-pyrimido[4,5-b]indole-2,4-diamine (6)
Compound 6 (synthesized from 12 (100 mg, 0.22 mmol) as described for 4; reaction time = 48 h): yield 74%; TLC Rf 0.40 (CHCl3/MeOH, 10:1 with 3 drops concentrated NH4OH); mp 296 °C; 1H NMR (DMSO-d6) δ 6.61 (s, 2 H, NH2, exch), 7.21–7.33 (m, 4 H, Ar), 7.52–7.55 (m, 1 H, Ar), 8.91–8.96 (m, 1 H, Ar), 9.14 (s, 1 H, 4-NH, exch), 11.82 (s, 1 H, 9-NH, exch). HRMS (ESI) [M+H]+ calcd. for C16H11Cl2FN5 m/z = 362.0376, found m/z = 362.0381.
5-chloro-N4-(3-fluorophenyl)-9H-pyrimido[4,5-b]indole-2,4-diamine (7)
In a 50 mL flask 13 (84 mg, 0.20 mmol) was dissolved in isopropanol (50 mL), and 2 mL of 1 N NaOH solution was added. The reaction mixture was heated to reflux for 14 h. The solvent was evaporated to obtain a residue. Water was added and the mixture was filtered and dried. The solid was dissolved in CH3OH, silica gel was added and solvent evaporated to make a plug. The plug was loaded on top of a wet (CHCl3) silica gel column and eluted with 30:1 (CHCl3: MeOH). Fractions containing desired product were pooled and evaporated to afford the 7 in 70% yield. TLC Rf 0.47 (CHCl3/MeOH, 10:1 with 3 drops concentrated NH4OH); mp 257–258 °C; 1H NMR (DMSO-d6) δ 6.64 (bs, 2 H, NH2, exch), 6.81–6.86 (m, 1 H, Ar), 7.20–7.22 (m, 2 H, Ar), 7.30–7.37 (m, 2 H, Ar), 7.43–7.46 (m, 1 H, Ar), 8.13–8.16 (m, 1 H, Ar), 9.21 (s, 1 H, 4-NH, exch), 11.79 (s, 1 H, 9-NH, exch). Anal. (C16H11ClFN5. 0.5 H2O) C, H, N, Cl, F.
5-chloro-N4-(3-methoxyphenyl)-9H-pyrimido[4,5-b]indole-2,4-diamine (8)
Compound 8 (synthesized from 14 (85 mg, 0.20 mmol) as described for 7; reaction time = 2 h): yield 92%; TLC Rf 0.42 (CHCl3/MeOH, 10:1 with 3 drops concentrated NH4OH); mp 242–243 °C; 1H NMR (DMSO-d6) δ 3.78 (s, 3 H, CH3), 6.48 (bs, 2 H, NH2, exch), 6.59–6.62 (m, 1 H, Ar), 7.18–7.23 (m, 3 H, Ar), 7.27–7.31 (m, 2 H, Ar), 7.66 (m, 1 H, Ar), 9.06 (s, 1 H, 4-NH, exch), 11.72 (s, 1 H, 9-NH, exch). Anal. (C17H14ClN5O) C, H, N, Cl.
Cells
All cells were maintained at 37 °C in a humidified environment containing 5% CO2 using media from Mediatech (Hemden, NJ). A-431 cells were from the American Type Tissue Collection (Manassas, VA).
Chemicals
All growth factors (bFGF, VEGF, EGF, and PDGF-β) were purchased from Peprotech (Rocky Hill, NJ). PD153035, SU5416, AG1295, and CB676475 (4-[(4′-chloro-2′-fluoro)phenylamino]-6,7-dimethoxyquinazoline) were purchased from Calbiochem (San Diego, CA). The CYQUANT cell proliferation assay was from Molecular Probes (Eugene, OR). All other chemicals were from Sigma Chemical unless otherwise noted.
Antibodies
The PY-HRP antibody was from BD Transduction Laboratories (Franklin Lakes, NJ). Antibodies against EGFR, PDGFR-β, FGFR-1, Flk-1, and Flt-1 were purchased from Upstate Biotech (Framingham, MA).
Phosphotyrosine ELISA
Cells used were tumor cell lines naturally expressing high levels of EGFR (A431), Flk-1 (U251), Flt-1 (A498), PDGFR-β (SF-539), and FGFR-1 (NIH OVCAR-8). Expression levels at the RNA level were derived from the NCI Developmental Therapeutics Program (NCI-DTP) web site public molecular target information (http://www.dtp.nci.nih.gov/mtargets/mt_index.html). Briefly, cells at 60–75% confluence were placed in serum-free medium for 18 h to reduce the background of phosphorylation. Cells were always >98% viable by Trypan blue exclusion. Cells were then pretreated for 60 min with 10, 3.33, 1.11, 0.37, and 0.12 μM compounds followed by 100 ng/ml EGF, VEGF, or PDGF-BB for 10 min. The reaction was stopped and cells permeabilized by quickly removing the media from the cells and adding ice-cold Tris-buffered saline (TBS) containing 0.05% Triton X-100, protease inhibitor cocktail, and tyrosine phosphatase inhibitor cocktail. The TBS solution was then removed and cells fixed to the plate for 30 min at 60 °C and further incubation in 70% ethanol for an additional 30 min. Cells were further exposed to block (TBS with 1% BSA) for 1 h, washed, and then a horseradish peroxidase (HRP)-conjugated phosphotyrosine (PY) antibody added overnight. The antibody was removed, cells were washed again in TBS, exposed to an enhanced luminal ELISA substrate (Pierce Chemical, Rockford, IL), and light emission measured using a UV product (Upland, CA) BioChemi digital darkroom. The known RTK-specific kinase inhibitor, PD153035, was used as a positive control compound for EGFR kinase inhibition; SU5416 for Flk1 kinase inhibition; AG1295 for PDGFR-β kinase inhibition; and CB676475 (4-[(4′-chloro-2′-fluoro)phenylamino]-6,7-dimethoxyquinazoline) was used as a positive control for both Flt1 and Flk1 kinase inhibition. Data were graphed as a percent of cells receiving growth factor alone and IC50 values were determined from two to three separate experiments (n = 8–24) using non-linear regression dose-response analysis with Prism 5.0 software (GraphPad, San Diego, CA). In each case, the activity of a positive control inhibitor did not deviate more than 10% from the IC50 values listed in the text.
CYQUANT cell proliferation assay
As a measure of cell proliferation, the CYQUANT cell counting/proliferation assay was used as previously described.36 Briefly, cells are first treated with compounds for 12 h and then allowed to grow for an additional 36 h. The cells are then lysed and the CYQUANT dye, which intercalates into the DNA of cells, is added and after 5 min the fluorescence of each well measured using a BioTek plate reader (Biotek, Winooski, VT). A positive control used for cytotoxicity in each experiment was cisplatin, 23. Data are graphed as a percent of cells receiving growth factor alone and IC50 values determined from two to three separate experiments (n = 6–15) using non-linear regression dose-response analysis with Prism 5.0 software (GraphPad, San Diego, CA).
Statistics
All analysis was done using Prism 5.0. (GraphPad Software, San Diego, CA).
Molecular Modeling and Computational Studies
The X-ray crystal structure of VEGFR2 at 1.71Å resolution was obtained from the protein database (PDB ID 1YWN)41. This crystal structure contains VEGFR2 in complex with a novel 4-amino-furo[2,3-d]pyrimidine. Docking studies were performed using Lead IT.27 The protein was prepared in Lead IT using default settings. Docking with Lead IT was carried out in by defining the active site as a sphere of ~6.5 Å from the X-ray crystal structure ligand. The protonation states utilized for the proteins and the ligands were calculated using the default settings. Water molecules in the active site were permitted to rotate freely. Ligands for docking were prepared using MOE 2010.1042 and energy minimized using the MMFF94X forcefield to a constant of 0.05 kcal/mol. Triangle matching was used as the placement method and the docked poses were scored using default settings. The docked poses were exported and visualized in MOE. The docking protocol was validated by re-docking the X-ray crystal structure ligand. The best docked pose in the re-docking study had an RMSD of 0.7686 Å. Docking studies were performed for 3 in Lead IT using a similar procedure. Poses from the docking experiment performed in Lead IT were exported and visualized in MOE.
Supplementary Material
Acknowledgments
This work was supported, in part, by the National Institutes of Health and National Cancer Institute Grant CA98850 (A.G.) and the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (AG).
Abbreviations
- ATP
adenosine 5′triphosphate
- VEGF
vascular endothelial growth factor
- EGF
epidermal growth factor
- PDGF
platelet derived growth factor
- RTK
receptor tyrosine kinase
- VEGFR-2
vascular endothelial growth factor receptor-2
- VEGFR-1
vascular endothelial growth factor receptor-1
- PDGFR-β
platelet derived growth factor receptor-β
- FGF
fibroblast growth factor
- IGF-1
insulin-like growth factor-1
- TGFα and β
transforming growth factors
- TNF-α
tumor necrosis factor-α
- MKIs
multikinase inhibitors
- CML
chronic myelogenous leukemia
- Abl
Abelson Tyrosine Kinase
- CD117
c-kit protein
- FLT3
FMS-like tyrosine kinase 3
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Taken in part from the dissertation submitted by N.Z. to the Graduate School of Pharmaceutical Sciences, Duquesne University, in partial fulfillment of the requirements for the degree of Doctor of Philosophy, July 2006 Presented in part at the 237th American Chemical Society National Meeting; Salt Lake City, UT, United States. March 22–26, 2009; p. MEDI-226.
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The Protein Kinase Complement of the Human Genome. Science (Washington, DC, U S) 2002;298:1912–1916. 1933–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 4.Tabernero J. The Role of VEGF and EGFR Inhibition: Implications for Combining Anti-VEGF and Anti-EGFR Agents. Mol Cancer Res. 2007;5:203–220. doi: 10.1158/1541-7786.MCR-06-0404. [DOI] [PubMed] [Google Scholar]
- 5.Gschwind A, Fischer OM, Ullrich A. Timeline: The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 6.Quesada AR, Munoz-Chapuli R, Medina MA. Anti-angiogenic drugs: from bench to clinical trials. Med Res Rev. 2006;26:483–530. doi: 10.1002/med.20059. [DOI] [PubMed] [Google Scholar]
- 7.Marcucci G, Perrotti D, Caligiuri MA. Understanding the Molecular Basis of Imatinib Mesylate Therapy in Chronic Myelogenous Leukemia and the Related Mechanisms of Resistance: Commentary re: A. N. Mohamed et al., The Effect of Imatinib Mesylate on Patients with Philadelphia Chromosome-positive Chronic Myeloid Leukemia with Secondary Chromosomal Aberrations. Clin. Cancer Res., 9: 1333–1337 2003. Clin Cancer Res. 2003;9:1248–1252. [PubMed] [Google Scholar]
- 8.Mohamed AN, Pemberton P, Zonder J, Schiffer CA. The Effect of Imatinib Mesylate on Patients with Philadelphia Chromosome-positive Chronic Myeloid Leukemia with Secondary Chromosomal Aberrations. Clin Cancer Res. 2003;9:1333–1337. [PubMed] [Google Scholar]
- 9.Gridelli C, Maione P, Del Gaizo F, Colantuoni G, Guerriero C, Ferrara C, Nicolella D, Comunale D, De Vita A, Rossi A. Sorafenib and sunitinib in the treatment of advanced non-small cell lung cancer. Oncologist. 2007;12:191–200. doi: 10.1634/theoncologist.12-2-191. [DOI] [PubMed] [Google Scholar]
- 10.Chow LQM, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 11.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC. 2-Aminothiazole as a Novel Kinase Inhibitor Template. Structure-Activity Relationship Studies toward the Discovery of N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl)]-2-methyl-4-pyrimidinyl]amino)]-1,3-thiazole-5-carboxamide (Dasatinib, BMS-354825) as a Potent pan-Src Kinase Inhibitor. J Med Chem. 2006;49:6819–6832. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- 12.Deininger MW. Nilotinib. Clin Cancer Res. 2008;14:4027–4031. doi: 10.1158/1078-0432.CCR-07-5015. [DOI] [PubMed] [Google Scholar]
- 13.Johnston SRD, Leary A. Lapatinib: a novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs of Today. 2006;42:441–453. doi: 10.1358/dot.2006.42.7.985637. [DOI] [PubMed] [Google Scholar]
- 14.Morphy R. Selectively Nonselective Kinase Inhibition: Striking the Right Balance. J Med Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- 15.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nature Biotechnology. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 16.Bamborough P, Drewry D, Harper G, Smith GK, Schneider K. Assessment of Chemical Coverage of Kinome Space and Its Implications for Kinase Drug Discovery. J Med Chem. 2008;51:7898–7914. doi: 10.1021/jm8011036. [DOI] [PubMed] [Google Scholar]
- 17.Force T, Krause DS, Van Etten RA. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat Rev Cancer. 2007;7:332–344. doi: 10.1038/nrc2106. [DOI] [PubMed] [Google Scholar]
- 18.Crespo A, Zhang X, Fernandez A. Redesigning Kinase Inhibitors to Enhance Specificity. J Med Chem. 2008;51:4890–4898. doi: 10.1021/jm800453a. [DOI] [PubMed] [Google Scholar]
- 19.Kerkelae R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand J-B, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med (N Y, NY, U S) 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 20.Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T, Lydon N. 4-(Phenylamino)pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase. J Med Chem. 1996;39:2285–2292. doi: 10.1021/jm960118j. [DOI] [PubMed] [Google Scholar]
- 21.Showalter HDH, Bridges AJ, Zhou H, Sercel AD, McMichael A, Fry DW. Tyrosine Kinase Inhibitors. 16. 6,5,6-Tricyclic Benzothieno[3,2-d]pyrimidines and Pyrimido[5,4-b]- and -[4,5-b]indoles as Potent Inhibitors of the Epidermal Growth Factor Receptor Tyrosine Kinase. J Med Chem. 1999;42:5464–5474. doi: 10.1021/jm9903949. [DOI] [PubMed] [Google Scholar]
- 22.Han L, Lorincz A, Sukumar S. In: In Antiangiogenic Agents in Cancer Therapy. Teicher B, Ellis L, editors. Vol. 2. Humana Press; New Jersey: 2008. pp. 331–352. [Google Scholar]
- 23.Ferrara N. Vascular endothelial growth factor as a target for anticancer therapy. Oncologist. 2004;9:2–10. doi: 10.1634/theoncologist.9-suppl_1-2. [DOI] [PubMed] [Google Scholar]
- 24.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nature Medicine (New York, NY, United States) 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 25.Shinkaruk S, Bayle M, Lain G, Deleris G. Vascular endothelial cell growth factor (VEGF), an emerging target for cancer chemotherapy. Current Medicinal Chemistry: Anti-Cancer Agents. 2003;3:95–117. doi: 10.2174/1568011033353452. [DOI] [PubMed] [Google Scholar]
- 26.SYBYL X. 1.3. Tripos Inc; St. Louis, MO: [Google Scholar]
- 27.Lead IT. 2.1.0. Biosolve IT GmbH; St. Augustin, Germany: [Google Scholar]
- 28.Gangjee A, Yang J, Ihnat MA, Kamat S. Antiangiogenic and antitumor agents. Design, synthesis, and evaluation of novel 2-amino-4-(3-bromoanilino)-6-benzylsubstituted pyrrolo[2,3-d]pyrimidines as inhibitors of receptor tyrosine kinases. Bioorg Med Chem. 2003;11:5155–5170. doi: 10.1016/j.bmc.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 29.Traxler PM, Furet P, Mett H, Buchdunger E, Meyer T, Lydon N. 4-(Phenylamino)pyrrolopyrimidines: Potent and Selective, ATP Site Directed Inhibitors of the EGF-Receptor Protein Tyrosine Kinase. J Med Chem. 1996;39:2285–2292. doi: 10.1021/jm960118j. [DOI] [PubMed] [Google Scholar]
- 30.Laufer SA, Domeyer DM, Scior TRF, Albrecht W, Hauser DRJ. Synthesis and Biological Testing of Purine Derivatives as Potential ATP-Competitive Kinase Inhibitors. J Med Chem. 2005;48:710–722. doi: 10.1021/jm0408767. [DOI] [PubMed] [Google Scholar]
- 31.Choowongkomon K, Sawatdichaikul O, Songtawee N, Limtrakul J. Receptor-based virtual screening of EGFR kinase inhibitors from the NCI diversity database. Molecules. 2010;15:4041–4054. doi: 10.3390/molecules15064041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bold G, Altmann KH, Frei J, Lang M, Manley PW, Traxler P, Wietfeld B, Brueggen J, Buchdunger E, Cozens R, Ferrari S, Furet P, Hofmann F, Martiny-Baron G, Mestan J, Roesel J, Sills M, Stover D, Acemoglu F, Boss E, Emmenegger R, Laesser L, Masso E, Roth R, Schlachter C, Vetterli W, Wyss D, Wood JM. New Anilinophthalazines as Potent and Orally Well Absorbed Inhibitors of the VEGF Receptor Tyrosine Kinases Useful as Antagonists of Tumor-Driven Angiogenesis. J Med Chem. 2000;43:2310–2323. doi: 10.1021/jm9909443. [DOI] [PubMed] [Google Scholar]
- 33.Traxler P, Bold G, Frei J, Lang M, Lydon N, Mett H, Buchdunger E, Meyer T, Mueller M, Furet P. Use of a Pharmacophore Model for the Design of EGF-R Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4-d]pyrimidines. J Med Chem. 1997;40:3601–3616. doi: 10.1021/jm970124v. [DOI] [PubMed] [Google Scholar]
- 34.Hennequin LF, Thomas AP, Johnstone C, Stokes ES, Ple PA, Lohmann JJ, Ogilvie DJ, Dukes M, Wedge SR, Curwen JO, Kendrew J, Lambert-van der Brempt C. Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors. J Med Chem. 1999;42:5369–89. doi: 10.1021/jm990345w. [DOI] [PubMed] [Google Scholar]
- 35.Gangjee A, Zaware N, Raghavan S, Ihnat M, Shenoy S, Kisliuk RL. Single Agents with Designed Combination Chemotherapy Potential: Synthesis and Evaluation of Substituted Pyrimido[4,5-b]indoles as Receptor Tyrosine Kinase and Thymidylate Synthase Inhibitors and as Antitumor Agents. Journal of Medicinal Chemistry. 2010;53:1563–1578. doi: 10.1021/jm9011142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson SM, Barsoum MJ, Wilson BW, Pappone PA. Purine nucleotides modulate proliferation of brown fat preadipocytes. Cell Proliferation. 1999;32:131–140. doi: 10.1046/j.1365-2184.1999.32230131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315:971–979. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 38.Hennequin LF, Thomas AP, Johnstone C, Stokes ESE, Ple PA, Lohmann JJM, Ogilvie DJ, Dukes M, Wedge SR, Curwen JO, Kendrew J, Lambert-van der Brempt C. Design and Structure-Activity Relationship of a New Class of Potent VEGF Receptor Tyrosine Kinase Inhibitors. J Med Chem. 1999;42:5369–5389. doi: 10.1021/jm990345w. [DOI] [PubMed] [Google Scholar]
- 39.Kovalenko M, Gazit A, Bohmer A, Rorsman C, Ronnstrand L, Heldin CH, Waltenberger J, Bohmer FD, Levitzki A. Selective platelet-derived growth factor receptor kinase blockers reverse sis-transformation. Cancer Res. 1994;54:6106–14. [PubMed] [Google Scholar]
- 40.Comis RL. The current situation: Erlotinib (Tarceva) and Gefitinib (Iressa) in non-small cell lung cancer. Oncologist. 2005;10:467–470. doi: 10.1634/theoncologist.10-7-467. [DOI] [PubMed] [Google Scholar]
- 41.Miyazaki Y, Matsunaga S, Tang J, Maeda Y, Nakano M, Philippe RJ, Shibahara M, Liu W, Sato H, Wang L, Nolte RT. Novel 4-aminofuro[2,3-d]pyrimidines as Tie-2 and VEGFR2 dual inhibitors. Bioorg Med Chem Lett. 2005;15:2203–2207. doi: 10.1016/j.bmcl.2005.03.034. [DOI] [PubMed] [Google Scholar]
- 42.Molecular Operating environment (MOE) 2010.10. C. C. G., Inc; 1255 University Street, Suite 1600, Montreal, Quebec, Canada: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.