

Abstract
Purpose
Many tyrosine kinase inhibitors (TKIs) undergo extensive hepatic metabolism, but mechanisms of their hepatocellular uptake remain poorly understood. We hypothesized that liver uptake of TKIs is mediated by the solute carriers OATP1B1 and OATP1B3.
Experimental Design
Transport of crizotinib, dasatinib, gefitinib, imatinib, nilotinib, pazopanib, sorafenib, sunitinib, vandetanib, and vemurafenib was studied in vitro using artificial membranes (PAMPA) and HEK293 cell lines stably transfected with OATP1B1, OATP1B3, or the ortholog mouse transporter, Oatp1b2. Pharmacokinetic studies were performed with Oatp1b2-knockout mice and humanized OATP1B1- or OATP1B3-transgenic mice.
Results
All 10 TKIs were identified as substrates of OATP1B1, OATP1B3, or both. Transport of sorafenib was investigated further, since its diffusion was particularly low in the PAMPA assay (<4%) compared to other TKIs that were transported by both OATP1B1 and OATP1B3. While Oatp1b2 deficiency in vivo had minimal influence on parent and active metabolite N-oxide drug exposure, plasma levels of the glucuronic-acid metabolite of sorafenib (sorafenib-glucuronide) were increased >8-fold in Oatp1b2-knockout mice. This finding was unrelated to possible changes in intrinsic metabolic capacity for sorafenib-glucuronide formation in hepatic or intestinal microsomes ex vivo. Ensuing experiments revealed that sorafenib-glucuronide was itself a transported substrate of Oatp1b2 (17.5-fold vs control), OATP1B1 (10.6-fold), and OATP1B3 (6.4-fold), and introduction of the human transporters in Oatp1b2-knockout mice provided partial restoration of function.
Conclusions
These findings signify a unique role for OATP1B1 and OATP1B3 in the elimination of sorafenib-glucuronide, and suggest a role for these transporters in the in vivo handling of glucuronic acid conjugates of drugs.
Keywords: Sorafenib, OATP1B, pharmacokinetics, mouse model
INTRODUCTION
Over the last decade, human genome sequence data has suggested that many of the abnormalities associated with cancer are due to the abnormal function of protein kinases, and a major thrust of the current oncology drug discovery era has been to identify small molecule tyrosine kinase inhibitors (TKIs). To date, already 18 of such agents have been approved by the U.S. Food and Drug Administration for the treatment of a variety of diseases that were previously essentially resistant to standard chemotherapy. Nonetheless, the recent advances in cancer care have been relatively modest in a number of diseases and many TKIs, like their cytotoxic counterparts, have rather limited efficacy combined with a significant degree of unexpected and unexplained toxicity (1).
In spite of the revolutionary changes in the various stages of drug development employed for TKIs (2), the standard strategy still in use for dose selection is to establish a therapeutic dose in Phase II trials and subsequently, at best, modify it for individual differences in body-surface area in children. However, there is already a wealth of experimental data indicating that both the efficacy and safety of TKIs might be optimized if dosing strategies would take into consideration the unique pharmacokinetic profiles of these agents (3, 4). Indeed, while TKIs offer possibly a number of important theoretical advantages over conventional cytotoxic agents, they are still afflicted by some of the same problems, including an extensive interindividual pharmacokinetic variability and the existence of a rather narrow therapeutic window (5).
It is likely that a critical determinant of pharmacokinetic variability observed with TKIs is associated with differential expression of drug-metabolizing enzymes and/or transporters at sites of absorption and elimination. Although the metabolic pathways of the approved TKIs have been reasonably well established (3, 6), the mechanisms by which these agents are taken up into human liver cells, in advance of metabolism, are still largely unknown. Previously reported in vitro studies have provided preliminary evidence that the hepatocellular uptake of some TKIs may be regulated, at least in part, by OATP1B1 (formerly, OATP-C, LST-1, OATP2) and OATP1B3 (formerly, OATP8, LST-2), polymorphic organic anion transporting polypeptides encoded by the SLCO1B1 (formerly, SLC21A6) and SLCO1B3 (formerly, SLC21A8) genes. For example, axitinib (7) and imatinib (8) were previously identified as substrates of OATP1B1 and OATP1B3, respectively, and a number of other TKIs, including lapatinib (9), pazopanib (10), and vandetanib (Caprelsa package insert; see: www.astrazeneca-us.com), were recently demonstrated to potently inhibit the function of OATP1B1 and/or OATP1B3 using in vitro model systems. However, a systematic approach to evaluate the ability of TKIs to interact with OATP1B-type transporters and subsequently affect drug disposition profiles is still lacking (11). The aims of the current study were to (i) evaluate transport of 10 different TKIs by OATP1B-type transporters in vitro; (ii) determine the pharmacokinetics of a lead TKI substrate, sorafenib, in mice that are knock-out for the ortholog transporter Oatp1b2; and (iii) assess the degree of functional restoration in mice that are knock-in for either OATP1B1 or OATP1B3.
MATERIALS AND METHODS
Chemicals and cell lines
Crizotinib, nilotinib, sorafenib, and vemurafenib were obtained from Chemie Tek, dasatinib, pazopanib, and vandetanib from LC Laboratories, and sunitinib and gefitinib from Toronto Research Chemicals. Imatinib was kindly provided by Novartis Pharmaceuticals. General tritium-labeled crizotinib, dasatinib, gefitinib, imatinib, nilotinib, pazopanib, sorafenib, vandetanib, and vemurafenib (specific activity, >1 Ci/mmol; radiochemical purity, >97.1%) were custom made by Moravek Biochemicals. [3H]Sunitinib (specific activity, 12.5 Ci/mmol; radiochemical purity, 99.0%) and [3H]estradiol-17β-D-glucuronide (specific activity, 50.1 Ci/mmol; radiochemical purity, 99.0%), a positive control substrate for OATP1B1 and OATP1B3 (12), were obtained from American Radiolabeled Chemicals. Dimethyl sulfoxide (Sigma Aldrich) was used as solvent for all TKIs.
The generation of stable, isogenic Flp-in T-Rex293 cells expressing OATP1B1*1a (wildtype), OATP1B1*5 [c.521T>C (V174A); rs4149056], or OATP1B1*15 (N130D, V174A) has been described previously (13). Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen) supplemented with 10% FBS, hygromycin B (25 μg/mL; Invitrogen), and blasticidin (37.5 μg/mL; Biovision). A secondary model of OATP1B1- or OATP1B3-expressing cells was created by transfecting HEK293 cells with the pIRES2-EGFP vector (BD Biosciences) containing SLCO1B1 and SLCO1B3 cDNA, respectively, as described previously (14). Cells were maintained in DMEM supplemented with 10% FBS and G418 sulfate (1000 μg/mL; A.G. Scientific). Similarly, HEK293 cells were transfected with the pDream2.1/MCS vector (GenScript) containing SLCO1b2 cDNA (14), and a stable cell line was selected and maintained in DMEM supplemented with 10% FBS and G418 sulfate (500 μg/mL; A.G. Scientific). All cell lines were cultured and maintained at 37°C under 5% CO2.
In vitro transport in cellular models
Cells were seeded in 6-well plates in phenol red-free DMEM media containing 10% FBS, hygromycin (25 μg/mL), blasticidin (37.5 μg/mL) and doxycycline (1 μg/mL) and were incubated at 37°C for 24 hours. Cells were then washed with warm PBS and incubated with the TKI of interest in phenol-free DMEM media (without FBS and supplements) at 37°C. In the initial TKI screen, a concentration of 0.1 μM was used in order to evaluate OATP1B1-mediated transport under non-saturating conditions. In select experiments for sorafenib, uptake experiments were performed also at a concentration of 10 μM, which is a concentration achievable in mice and humans receiving the drug orally. The experiment was terminated by placing cells on ice and washing twice with ice-cold PBS. Cells were collected and centrifuged at 1050 RPM for 5 min at 4°C. The cell pellet was lysed in 1N NaOH by vortex-mixing, incubated at 4°C overnight, and then the solution was neutralized with 2M HCl. Total protein was measured using a Pierce BCA Protein Assay Kit (Thermo Scientific) and total protein content was quantified using a Biotek μQuant microplate spectrophotometer. Intracellular drug concentrations were determined in the remaining cell lysate by liquid scintillation counting using a LS 6500 Multipurpose Scintillation Counter (Beckman). Intracellular concentrations of sorafenib-glucuronide were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS), as described previously (15), with modifications. Briefly, 100 μl of cell lysate was mixed with 100 μl MeOH in a 1.5 ml microcentrifuge tube. The tube was vortex-mixed for 45 sec, followed by centrifugation at 13,000 rpm for 8 min at 4°C, and 1 μL supernatant was injected for analysis. Sorafenib-glucuronide stock solution (0.5 mg/mL in MeOH) was diluted with MeOH to prepare calibration standards at concentrations ranging from 1 to 2,000 ng/mL. Quality control (QC) samples were prepared independently at three different concentrations (low, medium and high concentrations).
In vitro transport in a parallel artificial membrane permeability assay (PAMPA)
Diffusion was assessed using Millipore 96-well multi-screen filter plates with polyvinylidine fluoride membranes and transport receiver plates (Millipore). The artificial membrane was prepared by adding 5 μl of 5% lecithin in dodecane (Avanti Polar Lipids Inc.) to the donor well. Then 150 μl of PBS solution containing the radiolabeled TKI of interest (1 μM), [3H]midazolam (1 μM), or [3H]methotrexate (500 μM), each containing 5% DMSO, at pH 7.4 was added to the lipid-treated donor wells. The donor plate was placed onto the acceptor plate, and incubation was carried out over 16 hours as directed in the Millipore assay protocol at room temperature with the wells covered, to prevent evaporation. After incubation, 75-μl samples from each donor well and 150-μl samples from each acceptor well were analyzed using liquid scintillation alongside theoretical equilibrium drug solutions containing 150 μl of each stock drug solution in 300 μl of the buffer. Membrane integrity was confirmed by determining the equilibrium transport of Brilliant Cresyl Blue and Lucifer Yellow. Results from the PAMPA analysis were expressed as a percentage of the drug accumulating in the acceptor wells, which values can vary between 0%, showing no diffusion, and 50%, showing free diffusion.
Pharmacokinetic studies
Female mice knockout for Oatp1b2 [Oatp1b2(-/-)] and age-matched wildtype mice on a DBA1/lacJ background were bred in-house (16). Female mice knockout for the entire Oatp1a and Oatp1b locus [Oatp1a/b(-/-)] (17), age-matched transgenic mice with liver-specific expression of OATP1B1 (OATP1B1tg) or OATP1B3 (OATP1B3tg) (18), and age-matched wildtype mice, all on an FVB background, were obtained from Taconic . Mice were housed in a temperature-controlled environment with a 12-hour light cycle and given a standard diet and water ad libitum. Experiments were approved by the Institutional Animal Care and Use Committee (St. Jude Children's Research Hospital).
Sorafenib was formulated in 50% Cremophor EL (Sigma Aldrich) and 50% ethanol, and diluted 1:4 (vol/vol) with deionized water immediately before administration by oral gavage. Mice were fasted for 3 h before and during the study, with unrestricted access to drinking water. At select time points after sorafenib administration, blood samples (30 μL each) were taken from individual mice at 0.25, 0.5, and 1.5 h from the submandibular vein using a lancet, and at 3, and 4.5 h from the retro-orbital venous plexus using a capillary. A final blood draw was obtained at 7.5 h by a cardiac puncture using a syringe and needle. The total blood volume collected during the procedure from each mouse was 150 μL. All blood samples were centrifuged at 3000 × g for 5 min, and plasma was separated and stored at -80°C until analysis. A separate group of mice was euthanized by CO2 asphyxiation and livers were immediately collected and flash-frozen on dry ice. Liver specimens were stored at -80°C until further processing.
Plasma and liver concentrations of sorafenib, sorafenib N-oxide, and sorafenib glucuronide were determined by LC-MS/MS, as described previously (15), with modifications. Briefly, 10 μL of plasma was extracted with 60 μL acetonitrile containing internal standards. Liver was homogenized with 10 times volume of water, and 20 μL homogenate was extracted with 80 μL acetonitrile containing internal standards. Samples were centrifuged as above, and 2 μL supernatant was injected for analysis. Calibrators and QCs were made using blank plasma or liver homogenate from the same mouse strains the pharmacokinetic studies were conducted in. Sorafenib was quantitated over the concentration range of 10 to 10,000 ng/mL and sorafenib N-oxide and sorafenib-glucuronide were quantitated over the range of 10 to 5,000 ng/mL. Pharmacokinetic parameters were calculated using non-compartmental methods in WinNonlin 6.2 software (Pharsight).
Ex vivo microsomal incubations
Mouse liver and intestinal microsomes were prepared as described previously (19). Activity assays for UDP-glucuronosyltransferases (UGT) and cytochrome P450s (CYP) were performed as described by the manufacturer (BD Biosciences). Briefly, sorafenib (10 μM) was incubated with liver or intestinal microsomes (1 mg/mL) for 60 min at 37°C, and the reaction was terminated by adding 100-μL of ice-cold acetonitrile. Concentrations of sorafenib, sorafenib N-oxide and sorafenib-glucuronide were determined in reaction mixtures by LC-MS/MS as described previously (15).
Statistical analysis
All data are presented as mean ± SE. Statistical analysis was performed using GraphPad Prism 5.0 (GraphPad Software Inc.). All t-tests were two-tailed, and P<0.05 was considered statistically significant.
RESULTS
In vitro transport of TKIs by OATP1B-type transporters
We initially determined whether OATP1B1 can transport TKIs by measuring cellular uptake in HEK293 cells that overexpress OATP1B1. Estradiol-17β-D-glucuronide was used as a prototypical model substrate for OATP1B-mediated transport (12), and was evaluated in each experiment as a positive control (Supplementary Fig. S1). As shown in Fig. 1A, each TKI, with the exception of vandetanib and vemurafenib, had statistically significantly enhanced cellular uptake compared to that observed in vector control (VC) cells. Among the tested TKIs, the highest uptake by OATP1B1 was observed for sorafenib, about 1.5-fold greater than that observed in VC cells. This is in the same order of magnitude as the contribution of OATP1B1 to the uptake of the irinotecan metabolite, SN-38, using similar in vitro experiments (20).
Figure 1. Transport of sorafenib by OATP1B transporters.
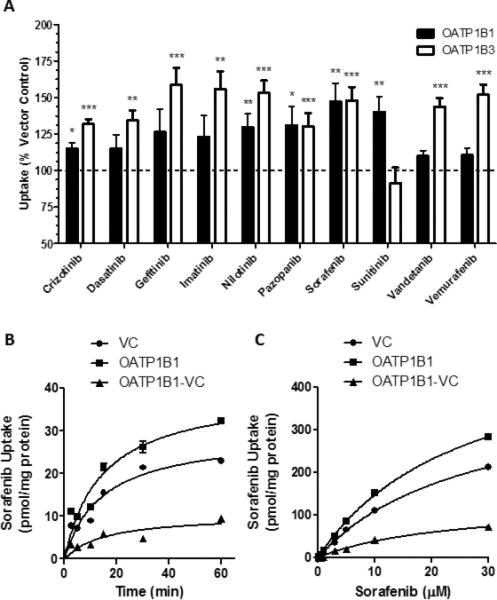
A) [3H]-Labeled TKIs (0.1 μM) were incubated with HEK293 cells expressing OATP1B1, OATP1B3, or vector control (VC) for 30 min. Intracellular concentrations of drug were determined by liquid scintillation counting. Data represent the mean ± SE of two experiments with triplicate samples (n=6) (*, P<0.05; **, P<0.01; ***, P<0.001). B, C) T-Rex293 cells expressing OATP1B1*1A or VC were incubated with 0.1 μM for the indicated time (B) or the indicated concentration of [3H]sorafenib for 15 min (C). Data represent the mean sorafenib uptake (symbols) ± SE (error bars, shown when larger than symbol) of triplicate samples (n=3). VC represents the cells transfected with an empty vector, representing background transport. The OATP1B1-VC data points, representing the specific contribution of OATP1B1 to the observed transport of sorafenib, were determined by averaging the triplicate values for each group, then subtracting the uptake values obtained in cells transfected with VC from the uptake values obtained in cells expressing OATP1B1.
Since the OATP1B3 isoform has 80% amino acid identity with OATP1B1 (12) and shares multiple overlapping substrates, including the anticancer agent docetaxel (14), we next measured TKI uptake using HEK293 cells that overexpress OATP1B3. These cells displayed uptake of a broad set of TKIs, similar to the OATP1B1-expressing cells, with the exception of sunitinib (Fig. 1A), which was not recognized as a transported substrate under the experimental conditions applied.
To assess the extent of TKI diffusion across cell membranes, a PAMPA assay was performed under steady-state conditions. The lowest percent transfer was observed for sorafenib (<4%) and vemurafenib (<1%), compared with 0.56% for methotrexate and about 50% for midazolam, agents representative of those known to diffuse poorly or diffuse freely across cell membranes, respectively (Supplementary Fig. S2). The low permeability observed for sorafenib and vemurafenib suggests that membrane transport of these agents is likely to be particularly dependent on solute carriers. Since of these TKIs only sorafenib was a confirmed substrate for both OATP1B1 and OATP1B3, further studies focused specifically on sorafenib. More comprehensive profiling showed that transport of sorafenib (0.1 μM) by OATP1B1 was time-dependent (Fig. 1B) as well as saturable (15-min incubations) (Fig. 1C), with a Michaelis-Menten constant (Km) of 23.5 μM and a maximum velocity (Vmax) of 128 pmol/mg/min.
Sorafenib pharmacokinetics in Oatp1b2-knockout mice
To test whether sorafenib is transported by OATP1B-type carriers in vivo, we determined the pharmacokinetic profile of sorafenib in DBA mice deficient in the ortholog transporter Oatp1b2 [Oatp1b2(-/-) mice]. Unexpectedly, minimal differences in peak plasma concentration and plasma area under the curve (AUC) for sorafenib, sorafenib N-oxide, and total active compounds (sorafenib + sorafenib N-oxide) were observed between Oatp1b2(-/-) and wildtype mice after a single oral sorafenib dose of 10 mg/kg (Fig. 2A; Table 1; Supplementary Fig. S3). Interestingly, a dramatic increase in the plasma levels of sorafenib-glucuronide was observed in the Oatp1b2(-/-) mice at each time point, resulting in an 5.5-fold increase in AUC compared to wildtype mice (Fig. 2B; Table 1). Furthermore, the metabolic ratio for sorafenib-glucuronide to sorafenib was substantially increased in the Oatp1b2(-/-) mice compared to wildtype mice (Table 1), whereas the metabolic ratio for sorafenib N-oxide was unchanged by Oatp1b2-deficiency (Supplementary Fig. S4). Importantly, the metabolic ratio of sorafenib-glucuronide in wildtype mice (0.17±0.02) was within the same order of magnitude as that observed in humans (0.30±0.19) (15). As anticipated, the liver-to-plasma concentration ratio of sorafenib-glucuronide was substantially reduced in Oatp1b2(-/-) mice compared with wildtype mice (mean, 5.6-fold), but this was not observed for sorafenib or sorafenib N-oxide (Supplementary Fig. S5).
Figure 2. Influence of Oatp1b2-deficiency on sorafenib pharmacokinetics.
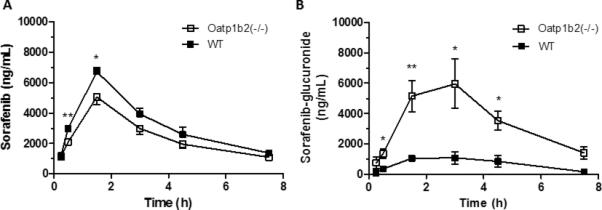
A, B) Wildtype (WT) and Oatp1b2(-/-) female mice (n = 4/group) were given 10 mg/kg sorafenib via oral gavage. Plasma sorafenib (A) and sorafenib-glucuronide (B) concentrations were determined by LC-MS/MS. Data represent the mean ± SE (*, P<0.05; **, P<0.01).
Table 1.
Sorafenib pharmacokinetic parameters in mice after a single oral administration of 10 mg/kg.
| Sorafenib | Sorafenib-glucuronide | |||||
|---|---|---|---|---|---|---|
| Strain | Genotype | Cmax (μg/mL) | AUC (μg×h/mL) | Cmax (μg/mL) | AUC (μg×h/mL) | S-glu:Sorafenib AUC Ratio |
| DBA | WT | 6.70±0.27 | 24.0±2.20 | 1.10±0.40 | 4.20±1.00 | 0.17±0.02 |
| DBA | Oatp1b2(-/-) | 5.10±0.50* | 18.0±2.00 | 6.00±1.60* | 23.0±5.60* | 1.36±0.39* |
| FVB | WT | 5.10±0.75 | 23.0±4.40 | 0.19±0.12 | 0.83±0.42 | 0.03±0.01 |
| FVB | Oatp1a/1b(-/-) | 5.60±0.53 | 18.0±2.00 | 4.70±0.63*** | 24.0±2.30*** | 1.35±0.15*** |
| FVB | OATP1B1tg | 5.60±0.59 | 16.0±1.40 | 3.30±0.40*** | 14.0±1.30*** | 0.90±0.12*** |
| FVB | OATP1B3tg | 5.70±0.32 | 19.0±1.60 | 2.70±0.12*** | 12.0±1.40*** | 0.61±0.08*** |
Values are mean ± SEM. Abbreviations: AUC, Area under the curve; Cmax, maximum plasma concentration; S-glu, sorafenib-glucuronide; WT, wildtype
P<0.05
P<0.001.
To test whether differences in sorafenib pharmacokinetics between wildtype mice and Oatp1b2(-/-) mice were due to alteration in sorafenib glucuronidation or oxidation, ex vivo assays were performed using intestinal and liver microsomes. As shown in Fig. 3A, the rate of sorafenib glucuronidation in liver microsomes was not substantially different between mouse genotypes, and reaction velocity was negligible in intestinal microsomes. Although sorafenib N-oxide formation in liver microsomes was slightly increased in samples from Oatp1b2(-/-) mice (Supplementary Fig. S6), the reaction velocity for this metabolic pathway was insignificant in these mice and about 10-fold lower than that observed for sorafenib glucuronidation; intestinal sorafenib oxidation was undetectable (Supplementary Fig. S6). This suggests that the altered systemic levels of sorafenib-glucuronide in Oatp1b2(-/-) mice cannot be explained by an intrinsically altered ability to metabolize sorafenib.
Figure 3. Transport of sorafenib-glucuronide by OATP1B transporters.
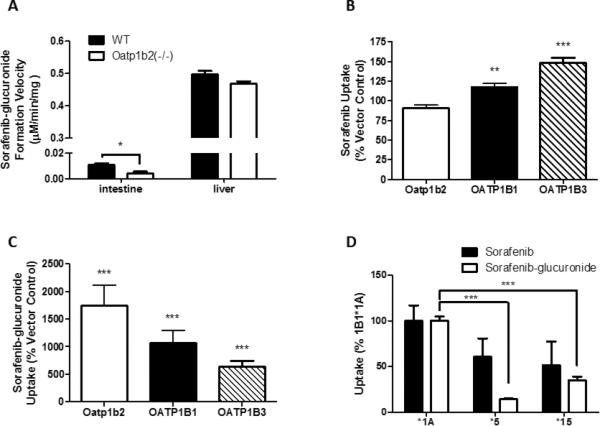
A) Ex vivo sorafenib-glucuronide formation was determined in intestine and liver microsomes from wildtype (WT) and Oatp1b2(-/-) mice. Microsomes (1 mg/mL) were incubated with 10 μM sorafenib for 60 min and sorafenib-glucuronide formation velocity was determined. Data represent the mean ± SE of 4 (intestine) or 16 (liver) samples. B, C) HEK293 cells expressing Oatp1b2, OATP1B1, or OATP1B3 were incubated with 10 μM [3H]sorafenib (B) or 10 μM sorafenib-glucuronide (C) for 15 min and intracellular concentrations were determined by liquid scintillation or LC-MS/MS, respectively. Data represent the mean ± SE drug uptake relative to vector control cells from 2 experiments performed with triplicate samples (n = 6). D) T-Rex293 cells expressing OATP1B1*1A (*1A), OATP1B1*5 (*5), or OATP1B1*15 (*15) were incubated with 0.1 μM [3H]sorafenib or 1 μM sorafenib-glucuronide for 15 min. Data represent the mean ± SE drug uptake of 6-15 replicates (*, P<0.05; **, P<0.01; ***, P<0.001).
In vitro transport of sorafenib and sorafenib-glucuronide by OATP1B-type transporters
The mouse Oatp1b2 transporter shares 65% amino acid homology with the human OATP1B1 transporter (21), and has overlapping substrate specificity (22). To determine whether Oatp1b2 transports sorafenib, we measured sorafenib uptake in HEK293 cells transfected with Oatp1b2. In line with the in vivo data, sorafenib uptake was not increased compared to VC cells, whereas sorafenib uptake was facilitated by OATP1B1 and OATP1B3 under the same experimental conditions (Fig. 3B). In contrast, cells expressing Oatp1b2 had a 17.5-fold greater cellular uptake of sorafenib-glucuronide compared to VC cells (Fig. 3C). Similarly, sorafenib-glucuronide was identified as a transported substrate for OATP1B1 and OATP1B3, with a 10.6- and 6.4-fold increase in cellular incorporation over VC cells, respectively (Fig. 3C).
Sorafenib and sorafenib-glucuronide uptake was also determined using cell lines expressing clinically-relevant protein variants of OATP1B1 associated with single-nucleotide substitutions. Compared to the wildtype OATP1B1 (OATP1B1*1A), the cells transfected with reduced function variant haplotypes containing a c.521C substitution (OATP1B1*5 and OATP1B1*15) incorporated less sorafenib (Fig. 3D). Similar observations were made in these cells when testing the uptake of estradiol-17β-D-glucuronide (Supplementary Fig. S7).
Sorafenib pharmacokinetics in humanized OATP1B1tg and OATP1B3tg mice
Because sorafenib was not transported by Oatp1b2 in vitro, we next determined the pharmacokinetics of sorafenib in humanized transgenic mice with liver-specific expression of OATP1B1 (OATP1B1tg) or OATP1B3 (OATP1B3tg) on an FVB strain deficient in the entire Oatp1a and Oatp1b gene locus [Oatp1a/b(-/-) mice]. Despite the ability of OATP1B1 and OATP1B3 to transport sorafenib in vitro, sorafenib maximum plasma concentration and AUC were not significantly different between OATP1B1tg or OATP1B3tg mice and Oatp1a/b/(-/-) mice, as well as between WT and Oatp1a/b/(-/-) mice (Fig. 4A; Table 1). Similar to the observed pharmacokinetic parameters in Oatp1b2(-/-) mice, Oatp1a/b(-/-) mice displayed a 28.9-fold increase in the AUC of sorafenib-glucuronide compared to the corresponding wildtype (Fig. 4A-B; Table 1). Notably, the introduction of OATP1B1 or OATP1B3 into livers of Oatp1a/b/(-/-) mice was associated with a sorafenib-glucuronide AUC that was 16.9- and 14.5-fold higher, respectively, compared to wildtype mice (Fig. 4B; Table 1), thereby providing partial restoration of function. Evaluation of sorafenib metabolism ex vivo in liver and intestinal microsomes from wildtype and Oatp1b2(-/-) mice showed no differences between mouse genotypes (Supplementary Fig. S8). Interestingly, the sorafenib-glucuronide to sorafenib AUC ratio was about 4.5-fold higher in DBA wildtype mice in comparison with FVB wildtype mice (Table 1). This finding is consistent with the notion that the sorafenib-glucuronide formation rate was faster in liver microsomes from DBA wildtype mice (Fig. 3A) compared with the FVB strain (Supplementary Fig. 8A). It is possible, however, that additional mechanisms exist beyond the scope of this investigation that may have contributed to these phenotypic differences, including mouse strain-dependent differences in the expression of sorafenib transporters in the liver.
Figure 4. Influence of Oatp1a/b-deficiency on sorafenib pharmacokinetics.
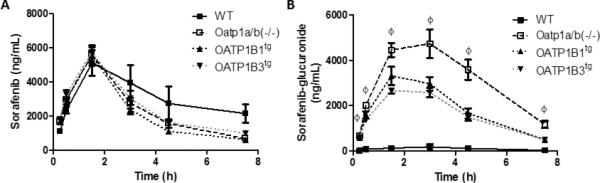
A, B) Female wildtype (WT), Oatp1a/b(-/-), OATP1B1tg, and OATP1B3tg mice (n =4/group) were given 10 mg/kg sorafenib via oral gavage. Plasma (A) sorafenib and (B) sorafenib-glucuronide concentrations were determined by LC-MS/MS. Data represent the mean ± SE (Ø, P<0.001 for all groups compared to WT).
DISCUSSION
In this study, we demonstrate that several TKIs, including sorafenib, are substrates for the human OATP1B1 and OATP1B3 transporters. Using a HEK293 cell model over-expressing the OATP1B-type proteins, we determined that sorafenib is incorporated into cells in both a time- and concentration-dependent manner. These data, however, are in contrast to our previous study to identify sorafenib carriers using a Xenopus laevis oocyte model transfected with OATP1B1 cRNA (23). This discrepancy suggests that cell context may affect OATP-mediated transport of xenobiotics, and similar cell context-dependent observations have been made with the OATP1B1 substrate docetaxel (14). Furthermore, using the T-Rex293 cell model, sorafenib transport was decreased in cells expressing naturally-occurring OATP1B variants (OATP1B1*5, OATP1B1*15) exhibiting reduced function.
Our in vivo studies using the Oatp1b2(-/-) mouse model demonstrated minimal differences in the pharmacokinetic profile of sorafenib compared to wildtype mice. We previously demonstrated by microarray analysis that Oatp1b2-deficiency in mice is not associated with any pronounced compensatory alterations in metabolic enzyme or transporter expression in the liver (24). Moreover, there were no changes in the functional expression of the key cytochrome P450 enzymes associated with sorafenib metabolism, including Cyp3a isoforms, in these transporter knockout mice (24). Furthermore, our in vivo data was supported by a lack of sorafenib transport in HEK293 cells transfected with the Oatp1b2 transporter. It should be pointed out that one possible limitation of the Oatp1b2(-/-) mouse model is the fact that, unlike in humans, mouse hepatocytes express multiple members of Oatp1a, a related subfamily of transporters that can potentially provide compensatory restoration of function when Oatp1b2 is lost (25). However, in our current study, we found no substantial differences in sorafenib pharmacokinetic parameters when comparing results in Oatp1b2(-/-) mice to those in sex and age-matched Oatp1a/b(-/-) mice, which are additionally deficient in all members of the Oatp1a subfamily. This finding eliminates the possibility that the lack of a change in sorafenib plasma levels in Oatp1b2(-/-) mice, compared to wildtype animals, was due to compensatory alteration by Oatp1a transporters.
Although the rodent and human OATP1B-type transporters share a high degree of sequence homology, similarity in basolateral membrane localization, and have largely overlapping substrate specificity (26), our current in vitro data suggest that sorafenib is a substrate for both the human OATP1B1 and OATP1B3 transporters, but not for mouse Oatp1b2. Such demonstration of inherent interspecies differences in the affinity for OATP1B-type transporters is not unprecedented, as prior findings showed that the OATP1B3 substrate digoxin is not transported by the rodent Oatp1b2 ortholog (27). Considering the relatively low intrinsic permeability of sorafenib observed here in a PAMPA assay, and its apparent lack of interaction with the mouse Oatp1a or Oatp1b transporters, additional uptake transporters likely exist that contribute to the hepatic uptake of sorafenib in mice. The low permeability observed for sorafenib also supports the existence of uptake transporters with a role in the drug's intestinal absorption after oral administration, and such transport component may have additional contribution to the overall pharmacokinetic variability.
In contrast to sorafenib, the systemic exposure to sorafenib-glucuronide was dramatically increased in Oatp1b2(-/-) mice compared to their wildtype counterparts. No apparent differences in the ex vivo sorafenib-glucuronide formation rate were observed between Oatp1b2(-/-) and wildtype mice, suggesting that the observed kinetic differences were due to aberrant transport. Moreover, compared to the corresponding wildtype mice, an increase in the plasma concentrations of sorafenib-glucuronide was also observed in Oatp1a/b(-/-) mice. A similar phenomenon has recently been reported to occur for conjugated bilirubin (18), whereby Oatp1a/b-deficiency leads to excessive buildup of bilirubin-glucuronide in the systemic circulation, which can ultimately result in jaundice. Indeed, a substantial fraction of bilirubin that is being conjugated in hepatocytes can be secreted back into the circulation (by ABCC3), and be subsequently taken up again in downstream hepatocytes by OATP1B-type transporters. This unusual mechanism, dubbed “hepatocyte hopping” (25), facilitates efficient detoxification by circumventing saturation of alternate detoxification pathways, including terminal excretion of Phase II conjugated metabolites into the bile. It is likely that the same hepatocyte-hopping principle demonstrated for bilirubin-glucuronide applies also to sorafenib-glucuronide. Among the class of TKIs, this pharmacokinetic process may be particularly important for sorafenib since it is one of only few currently FDA-approved TKIs for which glucuronidation contributes substantially to drug elimination in addition to CYP3A4-mediated biotransformation (15).
The potential clinical ramification of the hepatocyte-hopping phenomenon of sorafenib-glucuronide requires additional investigation. For example, sorafenib undergoes enterohepatic recirculation (28) following bacterial β-glucuronidase-mediated de-conjugation of sorafenib-glucuronide within the intestinal lumen (29), and interference of this de-conjugation by neomycin treatment decreases the systemic exposure to sorafenib by more than 50% (Nexavar package insert: see: berlex.bayerhealthcare.com). It can be envisaged that interference of the biliary excretion of sorafenib-glucuronide by inhibition of OATP1B-mediated uptake into hepatocytes could potentially lead to diminished enterohepatic recycling of sorafenib and reduced sorafenib systemic exposure. In this context, it is noteworthy that there was a trend for a lower sorafenib AUC by 25% in the Oatp1b2(-/-) mice and by 22% in the Oatp1a/b(-/-) mice compared with their respective wildtype controls. A similar phenomenon has been recently reported for mycophenolate mofetil, an immune-suppressive drug that undergoes extensive glucuronidation. In this case, a cohort of renal transplant patients with a genotype associated with decreased OATP1B function had reduced circulating levels of the active moiety, mycophenolic acid, and a concomitant increase in the levels of its glucuronide metabolite, presumably due to a disturbance in enterohepatic cycling (30).
Based on in vitro uptake studies, multiple functionally different haplotypes, including OATP1B1*5 and OATP1B1*15, were found to have a detrimental impact on sorafenib transport. This finding is consistent with previous studies showing substantially diminished transport activity of several OATP1B1 substrates by these particular variants when transfected into mammalian cells (31). Moreover, in humans, these variants have been associated with altered systemic exposure and toxicity in response to multiple substrates (32). In our current study, introduction of OATP1B1 or OATP1B3 into the liver of Oatp1a/b(-/-) mice resulted only in partially restored function to eliminate sorafenib-glucuronide. It is possible that this partial restoration is due to differences in affinity of sorafenib-glucuronide for Oatp1b2 relative to OATP1B1 and OATP1B3, and/or that the hepatic expression of OATP1B1 or OATP1B3 in the transgenic mice is lower than that of Oatp1b2 in the parental strain. Regardless, these data suggests the possibility that even partial deficiency of either OATP1B1 or OATP1B3 function, for example as a result of an inherited genetic defect, can lead to a buildup of sorafenib-glucuronide in the circulation that leads to altered recycling of sorafenib.
Overall, our findings signify an unusual role for OATP1B1 and OATP1B3 in the elimination of sorafenib, whereby sorafenib-glucuronide can enter a sinusoidal liver-blood shuttling loop that becomes defective when OATP1B-function is compromised, leading to excessive systemic accumulation. Future study of the association between sorafenib-glucuronide levels and sorafenib-associated toxicity and efficacy, and the connection with polymorphic OATP1B-type transport, is warranted.
Supplementary Material
Translational Relevance.
The interindividual pharmacokinetic variability seen with most approved tyrosine kinase inhibitors (TKIs) remains high, and this phenomenon may have important ramification for the clinical activity and toxicity of these agents. We hypothesized that differential expression of polymorphic transporters involved in the hepatic elimination of TKIs plays a crucial role in explaining this pharmacologic variability. Here, we investigated the contribution of organic anion transporting polypeptides of the OATP1B family to the transport of 10 FDA approved TKIs using an array of experimental model systems. Our results indicate the existence of at least two uptake transporters in the human liver (OATP1B1 and OATP1B3) with similar affinity for most of the tested TKIs that likely regulate the initial step in hepatic elimination. Deficiency of these transporters in vivo was associated with a particularly striking change in disposition phenotype for the glucuronic acid-conjugated metabolite of sorafenib. These findings signify a unique role for OATP1B-type transporters in the handling of drug glucuronides, and have implication for the design of future clinical studies exploring the drug-drug interaction potential of TKIs, as well as for pharmacogenomic association analyses involving this class of agents.
Acknowledgments
We would like to thank Richard Kim and Jeffrey Stock for providing the Oatp1b2(-/-) mice.
Acknowledgment of research support: This study was supported in part by the American Lebanese Syrian Associated Charities (ALSAC), USPHS Cancer Center Support Grant 3P30CA021765 (to SDB), and NCI Grants 5R01CA138744-03 (to SDB), 5R01CA151633-03 (to AS), and 5R25CA023944.
Footnotes
Conflicts of Interest: All authors declare no potential conflict of interest.
Disclosure of Potential Conflict of Interest
The authors declared no conflict of interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
References
- 1.Caraglia M, Santini D, Bronte G, Rizzo S, Sortino G, Rini GB, et al. Predicting efficacy and toxicity in the era of targeted therapy: focus on anti-EGFR and anti-VEGF molecules. Curr Drug Metab. 2011;12:944–55. doi: 10.2174/138920011798062346. [DOI] [PubMed] [Google Scholar]
- 2.Humphrey RW, Brockway-Lunardi LM, Bonk DT, Dohoney KM, Doroshow JH, Meech SJ, et al. Opportunities and challenges in the development of experimental drug combinations for cancer. J Natl Cancer Inst. 2011;103:1222–6. doi: 10.1093/jnci/djr246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Gion P, Kanefendt F, Lindauer A, Scheffler M, Doroshyenko O, Fuhr U, et al. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on pyrimidines, pyridines and pyrroles. Clin Pharmacokinet. 2011;50:551–603. doi: 10.2165/11593320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 4.Scheffler M, Di Gion P, Doroshyenko O, Wolf J, Fuhr U. Clinical pharmacokinetics of tyrosine kinase inhibitors: focus on 4-anilinoquinazolines. Clin Pharmacokinet. 2011;50:371–403. doi: 10.2165/11587020-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Baker SD, Hu S. Pharmacokinetic considerations for new targeted therapies. Clin Pharmacol Ther. 2009;85:208–11. doi: 10.1038/clpt.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35:692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Brennan M, Williams JA, Chen Y, Tortorici M, Pithavala Y, Liu YC. Meta-analysis of contribution of genetic polymorphisms in drug-metabolizing enzymes or transporters to axitinib pharmacokinetics. Eur J Clin Pharmacol. 2012;68:645–55. doi: 10.1007/s00228-011-1171-8. [DOI] [PubMed] [Google Scholar]
- 8.Hu S, Franke RM, Filipski KK, Hu C, Orwick SJ, de Bruijn EA, et al. Interaction of imatinib with human organic ion carriers. Clin Cancer Res. 2008;14:3141–8. doi: 10.1158/1078-0432.CCR-07-4913. [DOI] [PubMed] [Google Scholar]
- 9.Polli JW, Humphreys JE, Harmon KA, Castellino S, O'Mara MJ, Olson KL, et al. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- 10.Keisner SV, Shah SR. Pazopanib: the newest tyrosine kinase inhibitor for the treatment of advanced or metastatic renal cell carcinoma. Drugs. 2011;71:443–54. doi: 10.2165/11588960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Mandery K, Glaeser H, Fromm MF. Interaction of innovative small molecule drugs used for cancer therapy with drug transporters. Br J Pharmacol. 2012;165:345–62. doi: 10.1111/j.1476-5381.2011.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konig J, Cui Y, Nies AT, Keppler D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J Biol Chem. 2000;275:23161–8. doi: 10.1074/jbc.M001448200. [DOI] [PubMed] [Google Scholar]
- 13.Ramsey LB, Bruun GH, Yang W, Trevino LR, Vattathil S, Scheet P, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22:1–8. doi: 10.1101/gr.129668.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Graan AJ, Lancaster CS, Obaidat A, Hagenbuch B, Elens L, Friberg LE, et al. Influence of polymorphic OATP1B-type carriers on the disposition of docetaxel. Clin Cancer Res. 2012;18:4433–4440. doi: 10.1158/1078-0432.CCR-12-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zimmerman EI, Roberts JL, Li L, Finkelstein D, Gibson AA, Chaudhry AS, et al. Ontogeny and sorafenib metabolism. Clin Cancer Res. 2012;18:5788–5795. doi: 10.1158/1078-0432.CCR-12-1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaher H, Meyer zu Schwabedissen HE, Tirona RG, Cox ML, Obert LA, Agrawal N, et al. Targeted disruption of murine organic anion-transporting polypeptide 1b2 (Oatp1b2/Slco1b2) significantly alters disposition of prototypical drug substrates pravastatin and rifampin. Mol Pharmacol. 2008;74:320–9. doi: 10.1124/mol.108.046458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van de Steeg E, Wagenaar E, van der Kruijssen CM, Burggraaff JE, de Waart DR, Elferink RP, et al. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest. 2010;120:2942–52. doi: 10.1172/JCI42168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van de Steeg E, Stranecky V, Hartmannova H, Noskova L, Hrebicek M, Wagenaar E, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122:519–28. doi: 10.1172/JCI59526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emoto C, Yamazaki H, Yamasaki S, Shimada N, Nakajima M, Yokoi T. Characterization of cytochrome P450 enzymes involved in drug oxidations in mouse intestinal microsomes. Xenobiotica. 2000;30:943–53. doi: 10.1080/00498250050200104. [DOI] [PubMed] [Google Scholar]
- 20.Nozawa T, Minami H, Sugiura S, Tsuji A, Tamai I. Role of organic anion transporter OATP1B1 (OATP-C) in hepatic uptake of irinotecan and its active metabolite, 7-ethyl-10-hydroxycamptothecin: in vitro evidence and effect of single nucleotide polymorphisms. Drug Metab Dispos. 2005;33:434–9. doi: 10.1124/dmd.104.001909. [DOI] [PubMed] [Google Scholar]
- 21.Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, et al. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J Biol Chem. 1999;274:17159–63. doi: 10.1074/jbc.274.24.17159. [DOI] [PubMed] [Google Scholar]
- 22.Evers R, Chu XY. Role of the murine organic anion-transporting polypeptide 1b2 (Oatp1b2) in drug disposition and hepatotoxicity. Mol Pharmacol. 2008;74:309–11. doi: 10.1124/mol.108.048991. [DOI] [PubMed] [Google Scholar]
- 23.Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, et al. Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res. 2009;15:6062–9. doi: 10.1158/1078-0432.CCR-09-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lancaster CS, Bruun GH, Peer CJ, Mikkelsen TS, Corydon TJ, Gibson AA, et al. OATP1B1 pPolymorphism as a determinant of erythromycin disposition. Clin Pharmacol Ther. 2012;92:642–650. doi: 10.1038/clpt.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iusuf D, van de Steeg E, Schinkel AH. Functions of OATP1A and 1B transporters in vivo: insights from mouse models. Trends Pharmacol Sci. 2012;33:100–8. doi: 10.1016/j.tips.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–87. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hagenbuch B, Adler ID, Schmid TE. Molecular cloning and functional characterization of the mouse organic-anion-transporting polypeptide 1 (Oatp1) and mapping of the gene to chromosome X. Biochem J. 2000;345(Pt 1):115–20. [PMC free article] [PubMed] [Google Scholar]
- 28.Jain L, Woo S, Gardner ER, Dahut WL, Kohn EC, Kummar S, et al. Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br J Clin Pharmacol. 2011;72:294–305. doi: 10.1111/j.1365-2125.2011.03963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hilger RA, Richly H, Grubert M, Kredtke S, Thyssen D, Eberhardt W, et al. Pharmacokinetics of sorafenib in patients with renal impairment undergoing hemodialysis. Int J Clin Pharmacol Ther. 2009;47:61–4. doi: 10.5414/cpp47061. [DOI] [PubMed] [Google Scholar]
- 30.Picard N, Yee SW, Woillard JB, Lebranchu Y, Le Meur Y, Giacomini KM, et al. The role of organic anion-transporting polypeptides and their common genetic variants in mycophenolic acid pharmacokinetics. Clin Pharmacol Ther. 2010;87:100–8. doi: 10.1038/clpt.2009.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15:513–22. doi: 10.1097/01.fpc.0000170913.73780.5f. [DOI] [PubMed] [Google Scholar]
- 32.Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–81. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.