

Abstract
Insulin and contraction-mediated glucose transporter 4 (GLUT4) trafficking have different kinetics in mature skeletal muscle. Intravital imaging indicates that insulin-stimulated GLUT4 trafficking differs between t-tubules and sarcolemma. In contrast, contraction-induced GLUT4 trafficking does not differ between membrane surfaces. This distinction is likely due to differences in the underlying signaling pathways regulating GLUT4 vesicle depletion, GLUT4 membrane fusion and GLUT4 re-internalization.
Keywords: Living mice, Intravital, In-Vivo, PI3-Kinase, AMP-Kinase, LKB1
INTRODUCTION
Glucose transport in skeletal muscle is regulated by glucose transporter protein 4 (GLUT4) transporter movement (translocation) to the membrane surfaces. During the last 30 years of research on GLUT4 translocation several biochemical techniques have established the key steps in the muscle GLUT4 translocation and its regulation by insulin and muscle contractions. Specifically, it has been shown that GLUT4 translocation and in turn glucose uptake is stimulated by insulin and muscle contractions through different signaling cascades and these stimuli act in an additive fashion (30). Furthermore, it has been established that GLUT4 moves from intracellular vesicular depots to the two distinct plasma membrane domains, the t-tubules (a specialized plasma membrane network), and the sarcolemma (the plasma membrane proper) (28;30;38). At maximal translocation a significant part of GLUT4 is inserted into the plasma membranes with the majority of GLUT4 at the t-tubules membranes (90%) and a minor fraction (10%) at the sarcolemma (30;38). Once inserted into the plasma membranes GLUT4 can transport glucose molecules across the trans-membrane barrier into the muscle fiber (38). After the stimulation ceases GLUT4 re-internalizes from the membrane surfaces back into the basal storage depots. Despite the significant advancement in the understanding of the key steps in the GLUT4 translocation process, the previously used biochemical techniques have not been able to characterize the complete process from basal to stimulated state in high spatial-temporal detail. Thus, the intermediate trafficking steps in GLUT4 translocation have to a large extent been missed (for a in-depth review see (22)). However, recently developments in intravital imaging have provided new spatial and temporal details about GLUT4 trafficking kinetics previously not obtainable. We provided the first evidence to document the spatial-temporal kinetics of insulin-induced GLUT4 trafficking steps in mature muscle: the GLUT4 vesicle recruitment/depletion, -membrane appearance, -recycling, and re-internalization (18;20;21). Interestingly, we found that the intermediate GLUT4 trafficking steps differed significantly between t-tubules and sarcolemma (16–18;21;22). In contrast to our intravital studies of insulin stimulation in mature muscle, our recent intravital studies of contraction-induced GLUT4 trafficking in muscle did not reveal any kinetic differences between the membrane surfaces (19). Furthermore, intravital imaging after contraction stimulation showed different re-internalization kinetics compared to insulin stimulation (18;19). Thus, it appears that the two major stimuli of GLUT4 translocation in muscle, insulin and contractions, differ in regard to compartmentalization of trafficking kinetics. This review will discuss some of the underlying factors that may contribute to the stimulus-dependent difference in GLUT4 trafficking kinetics in mature muscle.
PREVIOUS TECHNIQUES TO STUDY GLUT4 TRANSLOCATION IN MATURE MUSCLE
The established knowledge on GLUT4 translocation in mature muscle has emerged from the use of biochemical techniques primarily such as muscle-membrane fractionation in combination with either antibody or photo-labeling based detection of glucose transporters (27;28). Furthermore, techniques such as GLUT4 immuno-staining of muscle fibers or sections after fixation of the entire isolated muscle have added important spatial information about the translocation process (20;38). However, these techniques have, due to methodological limitations, not been able to characterize GLUT4 translocation with sufficient spatial-temporal resolution to uncover the intermediate steps between basal and stimulated state. One of the main reasons for technical limitations can be attributed to the complexity of this tissue. Fully differentiated muscle consists of thousands of long, large muscle fibers containing thousands of nuclei, and large amounts of contractile proteins are embedded together into a complex three dimensional structure (Fig 1A). The muscle fibers contain complex membrane architecture with deep narrow plasma membrane invaginations, denoted the t-tubules, stretching from the sarcolemma into the muscle fiber core (Fig 1B). Furthermore, specialized membrane compartments like the calcium releasing sarcoplamic reticulum and mitochondria are packed in between the high content of contractile proteins (myofibrils) that makes up the majority of the muscle cell interior (Fig 1B). This tissue complexity has made previously applied techniques such as precise muscle cell-sorting or isolation, in vitro cultivation, cell staining and sub-cellular fractionation complex and challenging (see (22)). Previous studies have applied a high degree of invasive methodological steps such as excision and isolation of the muscle from its natural environment in situ, disruption of muscle architecture during homogenization (as in membrane fractionation based techniques) and muscle fixation and sectioning. These methodological steps all risk severe interference with the biological processes thereby adding the risk of artifacts. Furthermore, muscle membrane fractionation based techniques severely reduce the spatial resolution as the cellular architecture is broken up completely. In addition, it has not been practically feasible to isolate or fix muscles with very small time intervals (e.g. every 2–15 sec) in order to capture intermediate steps of the dynamic GLUT4 translocation process with higher time-resolution. Thus, the understanding of the combined spatial and temporal details of GLUT4 translocation kinetics in mature muscle remains incomplete.
Fig 1. The structure of fully differentiated skeletal muscle.
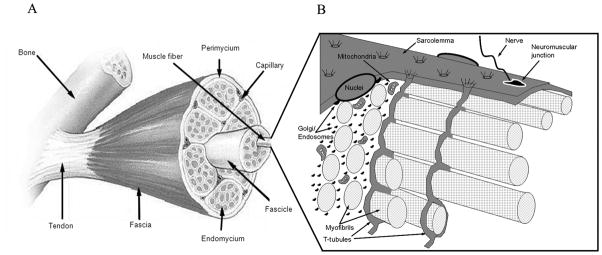
(A) A macroscopic view of skeletal muscle consisting of the tendon attached muscle belly embedded within the Fascia, a layer of connective tissue that envelops the muscle. The Perimysium is inward projections of the fascia that segregate bundles of fibers (fasciles). The Endomysium is another layer of connective tissue that surrounds each muscle fiber within fascicles. (B) The surface membrane of each multinucleated muscle fiber consists of the sarcolemma (the plasma membrane proper) with neuromuscular junctions mediating the nerve impulses for muscle contraction.
In addition to the sarcolemma, a deep network of membrane invaginations, the t-tubules, runs from the sarcolemma surface into the muscle fiber. Further adding structural complexity is a high content of myofibrils (contractile proteins) filling up the majority of the intracellular space. In addition, cellular compartments like the Golgi complex and the endosomal compartment are fragmentized and distributed throughout the muscle cell in unconnected elements. (Reprinted from (22). Copyright © The American Physiological Society 2010. Used with permission.)
INTRAVITAL MEASUREMENTS OF GLUT4 TRAFFICKING IN MATURE MUSCLE WITH HIGH SPATIAL-TEMPORAL RESOLUTION
One way to reduce the invasiveness and improve the spatial-temporal resolution of GLUT4 translocation analysis has been the development of intravital imaging. Recent advances in intravital microscopy imaging have permitted us to directly assess GLUT4 vesicle localization and movement inside intact living muscle fibers in situ in anesthetized mice (16;21). By tagging the protein of interest with enhanced green fluorescent protein (EGFP) i.e. GLUT4-EGFP, GLUT4 traffic can be followed in living cells by fluorescent microscopy (Fig 2). Fluorescent protein tagging has during the last 20 years been proven to be a non-interfering way of tracking the localization and movement of intracellular proteins in living cells. We and others have previously found GLUT4-EGFP to co-localize with endogenous GLUT4 when expressed in muscle fibers (12;18). Intravital imaging of GLUT4-EGFP is minimally invasive, as intact muscle fiber architecture is preserved in its natural environment (Fig 2A, B &C). There is no need for muscle isolation, fixation, the adding of co-factors or artificial buffers since the GLUT4-EGFP is visualized by blue light excitation in situ (Fig 2D). Specifically, by gene gun mediated GLUT4-EGFP transfection of superficial quadriceps muscle fibers in situ we have achieved transient expression of GLUT4-EGFP in situ in the superficial muscle fibers (Fig 2C) (16;17). After transfection the skin is closed and the animal left to recover (16;17). Five days later, the animal is anesthetized and mounted on the confocal microscope stage (Fig 2A). Unless muscle fibers are obviously damaged, gene gun transfection mediated GLUT4-EGFP expression does not appear to interfere with the muscle fiber morphology, the ability to contract or the basal GLUT4 localization (17–19). Previous studies of electroporation mediated gene transfer have shown that muscle torque is severely reduced the first 3 days after gene transfer but back to 90–100 % after 5 days recovery (31). However, the muscle damage was shown to primarily be associated with the eletroporation current in combination with the saline solution injected and not a result of the cDNA transfer itself (31). No current is used with gene gun transfection, only a short low pulse of helium gas that propel gold beads containing GLUT4-EGFP into the superficial muscle fibers. By keeping helium pressure and gold particle size low damage is minimal (17). Regarding the effect of expression a EGFP plasmid, the EGFP over-expression that follows did not induce more muscle damage or macrophage infiltration than electroporation treatment alone or FLAG tag cDNA (31). This indicates that the EGFP expression does not induce inflammation. The gene gun technique only produces 2–3 fold GLUT4-EGFP over expression compared to endogenous GLUT4 in a few superficial located muscle fibers (Fig 2C) (18). This is ideal as only fibers in the muscle surface can be imaged with appropriate resolution (Fig 2B). Furthermore, as neighboring fibers rarely are transfected, the GLUT4-EGFP positive fibers therefore have dark surroundings making the GLUT4-EGFP signal at the sarcolemma easier to define (Fig 2D, horizontal arrow in the 15 min post contraction image). The time-resolution of image sampling can be chosen in order to fit the pace of the cellular event analyzed. Thus, the time resolution used to study insulin stimulated GLUT4-EGFP trafficking has primarily been imaging every 15 sec (16;18;21) although higher resolution (sampling every 1–2 sec) has been used without adding additional information (18;20). In contrast, a higher time resolution (sampling every 3.3 sec or 2 sec) was needed to fluidly capture the rapid dynamics of the progressing PI3-K activation during insulin stimulation (20;21).
Fig 2. The set-up for intravital microscopy analysis of GLUT4-GFP trafficking kinetics in quadriceps muscle fibers in situ in anaesthetized mice.

(A) An anesthetized mouse is shown imbedded onto the mounting platform on the stage of a microscope using visible light (confocal detection) or multiphoton laser light excitation (for further details see (17)). (B) Images (emission fluorescence light) of the superficial muscle fibers are obtained in situ through the cover glass on top of the muscle surface. (C) A low magnification image shows the quadriceps muscle surfaces with some transfected muscle fibers expressing GLUT4-EGFP appearing bright against the majority of non-transfected fibers (bar = 500 μm). (D) A part of a GLUT4-EGFP positive muscle fiber from (C) is shown in higher magnification (bar = 20 μm). (D) In the basal high magnification image, GLUT4-EGFP is localized to larger and smaller vesicular depots (white fluorescent) throughout the muscle fiber and around the lateralized nuclei (diagonal arrows). After 15 min of in situ muscle contraction (for further details see (19)), the basal GLUT4-EGFP depots have been extensively depleted. Furthermore, GLUT4-EGFP has now been inserted into the sarcolemma indicated by staining along the plasma membrane (horizontal arrow) and to the t-tubules indicated by a striated pattern (vertical arrow). Forty minutes after the contraction bout stopped, GLUT4-EGFP re-internalization is only partially complete. GLUT4-EGFP appearance at the membrane surfaces is reduced but still present and the basal GLUT4-EGFP depots have gradually re-emerged but are not back to basal levels yet. Notice the unique position of the nuclei in all three images (diagonal arrows) that also ensures that images throughout the time-lapse series are obtained in the same x,y,z position of the muscle fiber within the living mouse.
Due to the diffraction limit of light the GLUT4-EGFP inserted into the membranes surfaces (sarcolemma and t-tubules) cannot be differentiated from GLUT4-EGFP close (within a 0.2 μm distance) to the membrane surfaces. However, a recent study analyzed freshly isolated cardiomyocytes from transgenic mice with muscle tissue expressing GFP tagged GLUT4 with a exofacial Human influenza hemagglutinin (HA) tag (5). In response to cardiomyocyte stimulation with insulin, contractions or hypoxia the study showed parallel increases in both GLUT4-GFP fluorescence at the membranes and membrane inserted GLUT4-GFP using antibody detection of the exofacial HA-tag (5). The overlap between GFP signal and HA-tag detection was present at both sarcolemma and t-tubules membranes (5). Thus, despite the diffraction limit of light there appears to be a good correlation between GLUT4-EGFP membrane staining and the GLUT4-EGFP inserted into the membranes. When GLUT4-EGFP transgene expression is optimal five days after transfection, the imaging of GLUT4 trafficking kinetics can be performed in situ with a high degree of time resolution (seconds to min), before, during and after stimulation (Fig 2D). Specifically, in the quadriceps muscle of each mouse, one superficial fiber with GLUT4-EGFP expression is normally imaged during a high resolution imaging experiment (Fig 2C to 2D). Therefore only the fiber type predominant in the muscle surface can be analyzed. Thus, by imaging the upper white part of the quadriceps muscle, information about the type IIB glycolytic fibers can be obtained (17). Alternatively, imaging GLUT4-EGFP transfected fibers in the surface of a red muscle such as soleus can be performed to highlight fiber type specific differences in GLUT4 trafficking. Intravital imaging requires that the animal is anesthetized in order to minimize movement during imaging. Finally, since muscle fibers are imaged in situ in the living animals, trafficking kinetics are measured under in vivo conditions of the animal such as normal healthy, obese, insulin resistant, or a trained state.
The intravital technique has by direct imaging confirmed several of the established GLUT4 translocation findings such as; insulin and muscle contractions induce GLUT4 translocation to both the sarcolemma and t-tubules (19;21), that GLUT4 re-internalizes after stimulation (18;19) and that GLUT4 translocation is reduced during states of insulin resistance (20). However, due to the vastly improved spatial and temporal resolution of intravital imaging, the technique has provided new dynamic details of GLUT4 trafficking kinetics during vesicle depletion, movement, fusion and re-internalization in living intact tissue.
SPATIAL-TEMPORAL KINETICS OF INSULIN MEDIATED GLUT4 TRAFFICKING
It has been extensively documented that the signaling steps leading to insulin and contraction-mediated GLUT4 build-up at the plasma membranes are independent pathways as reflected by their additivity. Thus, the insulin signaling leading to GLUT4 trafficking is mediated as a unidirectional pathway from the insulin receptor downstream through the insulin receptor/insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3 kinase (PI3-K)/Protein kinase B (Akt) pathway. Therefore, the insulin mediated GLUT4 translocation and glucose uptake can be completely inhibited by blocking a single step in the signaling pathway by wortmannin induced PI3-K inhibition. Our intravital imaging studies have showed that the signaling cascade at the level of PI3-K activation is initiated locally in both the sarcolemma and t-tubules membrane regions in white quadriceps muscle after insulin administration i.v. (20;21). The total time of events we recorded confirmed previous studies showing that PI3-K activation takes place within a 20 min period after insulin administration to white muscle (36). However, due to the high spatial-temporal resolution, we could add new details and show that local PI3-K signaling is initiated as insulin binds to resident insulin receptors in the respective membranes, an effect shown to correlate with local insulin arrival (21). Specifically, we showed that insulin first binds to receptors and initiates local PI3-K signaling in the sarcolemma within 1–2 min after i.v. administration (Fig 3 A & B, t=2 min), correlating with insulin’s arrival to this surface (20;21). Subsequently, PI3-K signaling ceases locally in the sarcolemma and insulin slowly diffuses into the t-tubule network. As insulin diffuses into the lumen of the t-tubules, PI3-K is progressively activated, resulting in maximal PI3-K activation approximately 10 min (Fig 3 A & B, t=13 min) later in these membranes compared to sarcolemma (20;21). The diffusion delay has been proposed to be a result of several factors such as the t-tubules having a narrow diameter (100 nm), a high degree of network tortuosity and a viscose lumen content (21;35). In addition, insulin binding to local receptors during diffusion into the t-tubules could limit the progressing lumen concentration of insulin (21;35). The diffusion-dependent delay in insulin mediated PI3-K signaling found between sarcolemma and t-tubules (Fig 3), correlates with a similar 10 minute delay downstream at the level of GLUT4 vesicle depletion and appearance at the plasma membranes (21). Together, our intravital data showed that the insulin signal moves from the surface into the muscle cell interior and in turn induces GLUT4 translocation locally. In regard to GLUT4 vesicle trafficking during the insulin induced translocation process, we did not see any high degree of GLUT4 vesicle movements over larger distances (>1 μm) (18). The lack of vesicle movements is in contrast to abundant GLUT4 vesicles movements previously described in in vitro cultured cell systems (26;29;33). In contrast, we found that the majority of GLUT4 vesicles remains static and “melt away” as they deplete and fuse with the local membrane (sarcolemma or t-tubules) during insulin stimulation (18). Recently, our findings were confirmed and extended to red muscle with a different approach using confocal total internal reflection fluorescence (TIRF) microscopy that increases the resolution of light microscopy so even vesicles below 0.2 μm can be followed precisely (25). In this study TIRF imaging was used to analyze isolated GLUT4-GFP positive muscle fibers from soleus and flexor digitorum brevis muscles obtained from GLUT4-GFP transgenic mice (25). Thus, when single freshly isolated GLUT4-GFP transgenic fibers were imaged in vitro before and during insulin stimulation using the higher resolution TIRF microscopy, only 10% of the total GLUT4 vesicles showed any significant movement (>1 μm) (25). The majority of GLUT4 vesicles remained static during insulin stimulation as local membrane fusion was induced (25). Together, this indicates that in both white and red mature muscle the amount of GLUT4 vesicle movement is minimal during insulin stimulation. This difference between GLUT4 vesicle mobility in cultured cells compared to mature muscle may also be reflected by the different effects found when manipulating Akt substrate 160 (AS160). AS160 has been proposed to be a distal step in insulin signaling, acting as a direct brake and retaining GLUT4 vesicles inside the cell via its guanosine triphosphatase (GTPase) activating domain that interacts with Rab proteins on the GLUT4 vesicles (1). Thus, when AS160 is phosphorylated by Akt during insulin stimulation, the AS160 GTPase brake is inactivated and GLUT4 vesicles released to move to the membrane surface (1). In adipocyte based cultured cell systems, inactivation of AS160 GTPase activity by over-expression of a GTPase inactive mutant leads to build-up of GLUT4 at the plasma membrane in the basal cell surface and in turn an increased basal glucose uptake (4). Furthermore, in cultured muscle the expression of a AS160 mutant that cannot be phosphorylated and thereby not inactivated results in reduced GLUT4 at the plasma membrane at basal conditions (37). In contrast, in tibialis anterior muscle the basal in vivo glucose uptake is not affected by over-expression of any the above AS160 mutants, suggesting that the release of GLUT4 vesicles for movement towards the plasma membranes is not sufficient for inducing increased basal glucose uptake in muscle (15). Together, these findings provide evidence that the traditional understanding of insulin induced GLUT4 translocation as process consisting of 1) a large degree of GLUT4 vesicle movement towards the membranes; 2) GLUT4 vesicle docking at the membranes; and 3) GLUT4 vesicle fusion into the membranes may not accurately reflect GLUT4 trafficking in mature muscle. Instead, recent GLUT4 vesicle analysis from mature muscle indicates that the majority of GLUT4 vesicles are already docked at the membranes, and GLUT4 vesicle fusion into the membranes may be the key step that is regulated in muscle in vivo (18;25).
Fig 3. Compartmentalization and the signaling of insulin mediated GLUT4 trafficking in mature muscle.
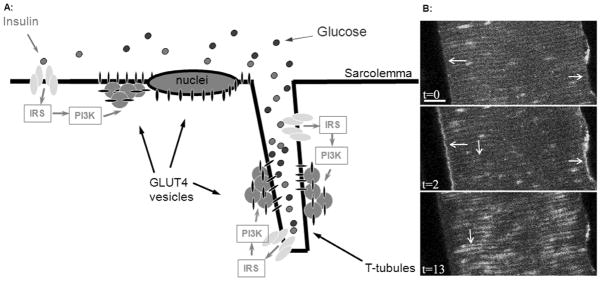
(A) Intravital imaging of insulin mediated GLUT4 translocation has shown that insulin (green dots) arrives at the sarcolemma and first act on local insulin receptors, thereby activating the insulin receptor/insulin receptor substrate-1 (IRS-1)/phosphatidylinositol 3 kinase (PI3-K)/Protein kinase B (Akt) signaling axis at the sarcolemma and initiating local GLUT4 recruitment from perinuclear and subsarcolemmal GLUT4 vesicular depots to fuse into the sarcolemma. As insulin slowly diffuses into the t-tubules, local t-tubular PI3-K activation and local GLUT4 trafficking are initiated. (B) Intravital imaging of PI3-K activation in a part of a muscle fiber in situ in a living ICR mouse (visualized by translocation of the phosphatidylinositol 3,4,5 P3 (PIP3) receptor GFP- ARNO, for further details see (17;21)). At t=0 no localized PIP3 production is present indicated by GFP fluorescence that are evenly distributed with no continuous staining of the sarcolemma (t=0, horizontal arrows) or t-tubules (t=0, vertical arrows). Two minutes after insulin injection i.v., PI3-K and, in turn, PIP3 production areis maximally activated at the sarcolemma (t=2 min, horizontal arrows). In contrast, no insulin mediated PI3-kinase activation is initiated in the t-tubule region at this time point (t=2 min). Subsequently, as insulin slowly diffuses into the t-tubule network PI3-K and, in turn, PIP3 are progressively activated in this membrane compartment (t=13 min, vertical arrow). Bar = 10 μm. This time difference in insulin mediated activation at the sarcolemma and t-tubules was also found further downstream at the level of GLUT4 vesicle depletion and membrane appearance (21).
SPATIAL-TEMPORAL KINETICS OF MUSCLE CONTRACTION MEDIATED GLUT4 TRAFFICKING
In contrast to insulin stimulation, our intravital imaging of contraction stimulated GLUT4 trafficking showed no delay of GLUT4 vesicle depletion and appearance at the plasma membranes (19;21). This indicates contraction induced GLUT4 trafficking is not compartmentalized. This could be because muscle contractions activate several parallel pathways with different temporal kinetics of activation compared insulin’s unidirectional pathway (Fig 4A). Several signaling pathways involved in contraction-mediated glucose transport have so far been identified; the calcium activated calmodulin dependent kinase CAMKII activation; the Serine/threonine kinase 11 (LKB1) that phosphorylates downstream targets adenosine monophosphate (AMP) activated kinase (AMPK) and the AMPK related kinase sucrose non-fermenting AMPK related kinase (SNARK) (9;14;39). Potentially, these pathways might differ not only in their temporal activation characteristics but also in their spatial interaction with GLUT4 trafficking in the muscle fiber, e.g., only in one membrane domain such as the t-tubules. During the early part of a contraction bout, calcium is released instantly and phosphorylation of CAMKII has been shown to be an early transient signaling event in isolated contracting epitrochlearis muscle (7). Studies using inhibitor peptides against CAMKII thereby reducing CAMKII activity have shown reduced basal and contraction mediated glucose transport in tibialis anterior muscle (39). How the GLUT4 trafficking kinetics were affected was not determined (39). However, during muscle contractions, t-tubules are known to play an early and central role in distributing the depolarization signal as calcium is released from sarcoplasmic reticulum during the excitation-contraction coupling process leading to contractions. It could therefore be anticipated that CAMKII-induced GLUT4 trafficking could have different kinetics in the t-tubules compared to the sarcolemma at least during the early period of a contraction bout. However, our recent intravital GLUT4 analysis did not detect any difference in GLUT4 vesicle depletion or appearance at the membranes when comparing the sarcolemma and t-tubules at the 5, 10 or 15 min time-points during in situ contraction bouts (19). This indicates that CAMKII activation during the early phase is not compartmentalized (Fig 4). This observation could however be a result of the lower time resolution of the study, which was limited to an image every 5 min during the breaks between the 3×5 min contraction periods (19). Higher image resolution sampling did not reveal additional information due to out of focus movement of the imaged muscle fibers during the contraction bouts (19). Thus, very early differences in GLUT4 trafficking kinetics during the first 5 min of contractions may have been overlooked. Another signaling pathway shown to modulate contraction-induced glucose transport is LKB1. This constitutively active kinase affects several downstream signaling steps such as AMPK and SNARK thereby regulating glucose transport (13;14;32) Recently, in vivo glucose transport stimulated by in situ contraction was reported to be greatly reduced (40%–100%) in extensor digitorum longus, soleus and tibialis anterior muscles from LKB1 knock-out mice (14;32). Thus, due to this large effect on glucose transport, LKB1 activity must have a significant impact on GLUT4 trafficking kinetics. So far, the spatial and temporal changes in GLUT4 trafficking kinetics in LKB1 knock-out muscle have not yet been investigated (Fig 4). However, since LKB1 was not completely knocked down in the previous LKB1 models studied (70–95% depending of model) achieving accurate information about spatial effects of LKB1 on GLUT4 trafficking could be challenging without further analysis about the cellular location of LKB1 (13;32). Thus, immuno-staining of muscle fibers for LKB1 in the knockout model and its littermate control is needed. Thereby it can be determined how LKB1 is distributed both normally and how the remaining LKB1 is localized in the knockout. Thus, if the remaining LKB1 protein resides in a specific cellular region in the knockout, then GLUT4 trafficking could still be normal in that part of the muscle fiber because LKB1 has not been eliminated from the functionally important cellular location.
Fig 4. Contraction mediated GLUT4 trafficking in mature muscle.
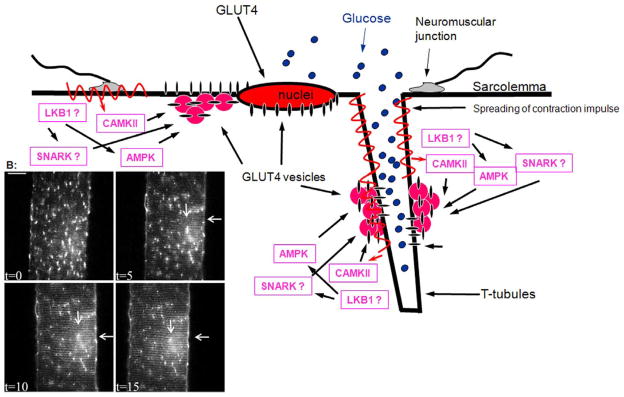
(A) Several parallel signalling pathways lead to contraction mediated GLUT4 translocation. Intravital imaging has not found any indication of differential compartmentalization in regard to calcium activated calmodulin dependent kinase (CAMKII) - or AMP activated kinase (AMPK) - activation induced GLUT4 translocation. However, any local differences in Serine/threonine kinase 11 (LKB1) or the AMPK related kinase sucrose non-fermenting AMPK related kinase (SNARK) signalling have not yet been studied. Therefore potential compartmentalization of these signals remains to be elucidated. (B) Intravital imaging of contraction mediated GLUT4-EGFP translocation in a part of a muscle fiber in situ in a living mouse (for further details see (19)). At t=0, GLUT4-EGFP is localized to larger and smaller vesicular depots throughout the muscle fiber and around the lateralized nuclei. Immediately after t=0 in situ contractions were initiated for 3×5 min periods (for further details see (19)). After each 5 min period, an image was taken at t=5, t=10 and t=15. It is seen that GLUT4-EGFP is gradually being inserted into the sarcolemma indicated by staining along the plasma membrane (horizontal arrow). Simultaneously a striated pattern is gradually emerging indicating GLUT4-EGFP translocation to t-tubules (vertical arrow). This indicates that translocation to both membrane surfaces takes place with similar kinetics (for quantification see (19). In addition, the basal GLUT4-EGFP depots have gradually been depleted with contractions. Bar = 20 μm.
One of the downstream targets of the constitutively activated LKB1 is AMPK and the activity of this kinase is greatly reduced in LKB1 deficient muscles (13;32). Phosphorylated AMPK has been shown to correlate with AMPK activity and increased glucose transport (6). Specifically, the increasing AMP/ATP ratio accompanying energy depletion with muscle contractions stabilizes the LKB1 phosphorylated AMPK against phosphatase mediated de-phosphorylation, leading to increased level of phosphorylated AMPK and in turn AMPK activity. Furthermore, the decrease in glycogen concentration associated with prolonged periods of muscle contraction also regulates AMPK activity. Thus, not surprisingly studies have shown that AMPK, in contrast to CAMKII, is maximally phosphorylated during the later stages of a contraction bout associated with energy depletion in epitrochlearis (7). In regard to GLUT4 trafficking kinetics, AMPK stimulation by the pharmacological agent 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) has in a previous study using muscle membrane fractionation of extensor digitorum longus suggested that only GLUT4 translocation to sarcolemma and not the t-tubules is regulated by AMPK activation (24). This study suggested therefore that AMPK-activated GLUT4 translocation is compartmentalized. In contrast, our intravital studies of white quadriceps clearly showed that AICAR-induced a specific AMPK mediated depletion of GLUT4 vesicles in the entire muscle fiber and an appearance of GLUT4 at both the sarcolemma and t-tubules membranes (19). It has been proposed that AICAR is a nonspecific drug activating several other signaling pathways, thereby potentially also affecting GLUT4 translocation through AMPK. Thus, more specific new AMPK stimulators such as A769662 have been developed. However, we imaged AICAR induced GLUT4 translocation in superficial quadriceps muscle fibers in AMPKα2i transgenic mice where no AMPKα2 activity is present and with only insignificant levels of AMPKα1 (6). In the AMPKα2i mice the majority of AICAR induced GLUT4-EGFP translocation was inhibited indicating that the AICAR effect on GLUT4 translocation in white muscle is AMPK specific (19). Furthermore, the AMPKα2i transgenic mice did not have altered contraction induced glucose transport in extensor digitorum longus or GLUT4 vesicle depletion and membrane appearance kinetics compared to controls in white quadriceps muscle fibers (6;19). This indicates that AMPK-activated GLUT4 trafficking kinetics are not spatial-temporal compartmentalized within the muscle fiber in the AMPKα2i transgenic model (Fig 4). This could potentially also indicate that LKB1 mediated AMPK phosphorylation is also not compartmentalized, since LKB1 is upstream of AMPK (32). Future intravital imaging studies of GLUT4 trafficking will be needed in the LKB1 transgenic models to analyze this further.
SNARK is another newly identified substrate downstream of LBK1 involved in contraction-induced glucose transport (14). SNARK activation could be a potential parallel pathway to AMPK and could compensate for GLUT4 trafficking in AMPKα2i transgenic mice during contraction stimulation. Muscle glucose transport stimulated by contractions in vivo is reduced in tibialis anterior muscle from mice transiently over expressing a mutant SNARK form and in soleus muscle from hetero-SNARK knockout mice with a 30 % reduction in SNARK protein (14). However, this study did not investigate how GLUT4 trafficking kinetics was affected. Both LKB1 and SNARK activity should potentially affect the spatial-temporal kinetics of contraction stimulated GLUT4 trafficking significantly but the details remain to be determined in the transgenic models (Fig 4).
KINETICS OF GLUT4 RE-INTERNALIZATION
Our intravital imaging of GLUT4 re-internalization kinetics following maximal insulin stimulation has shown that declining plasma insulin concentration correlates with gradual GLUT4 re-internalization (18). Specifically, GLUT4 appearance at the membrane surfaces gradually declined and GLUT4 vesicles gradually re-emerged as plasma insulin concentration was reduced (18). Insulin degradation in vivo is primarily dependent on central clearance by the liver and kidneys and to a minor degree by insulin receptor mediated binding and degradation in muscle tissues. This indicates that plasma insulin clearance is the primary regulating factor of GLUT4 re-internalization (18). Interestingly and corresponding with events during the initiation of insulin stimulation, a 10 min delay in full GLUT4 re-internalization at the t-tubules was observed compared to sarcolemma (18). Thus, the insulin concentration is probably cleared less rapidly in the t-tubules due to a slow insulin diffusion out (in parallel to a slow diffusion in) of the network as the plasma insulin concentration and in turn the interstitial insulin concentration wanes (18). The regulating role of insulin on GLUT4 re-internalization has also been found using pulse-chase surface photo-labeling of GLUT4 in isolated epitrochlearis muscle (11). Here GLUT4 re-internalization from homogenized plasma membranes (both sarcolemma and t-tubules combined) was found to be completed within 10 min of insulin removal (11). Similarly, a study in isolated epitrochlearis muscles also found that insulin signaling at the level of phosphorylation of Akt or AS160 reversed within 10–20 min upon insulin removal (34). However, in contrast to our observations on GLUT4-EGFP re-internalization in mice, the glucose transport reversed more slowly in the in vitro isolated muscle study, indicating a delayed GLUT4 internalization after insulin stimulation (34). The reason for this contrast could be difference in the preparation. In human clamp studies, a significant delay in the offset of proximal insulin signaling and glucose infusion rate and in turn glucose uptake across the limb was found after withdrawal of insulin infusion (40). However, this delay was likely due to a slower decrease in interstitial insulin concentration compared to plasma insulin (40). We did not see a delay in GLUT4-EGFP re-internalization in quadriceps muscle of mice as plasma insulin was cleared. Since the half-life of insulin in humans and mice is comparable (7–10 min), the lack of delay in mice could potentially be due to the significantly higher tissue perfusion rate (~5x the perfusion in humans) and thereby faster tissue insulin washout to the circulation for central clearance (3).
In contrast to the progressively declining insulin concentration in vivo, the direct stimulatory effect of contractions stops almost instantly once muscle contractions end. Upon cessation of muscle contraction, free calcium is pumped back to the sarcoplasmic reticulum within milliseconds. It could therefore be expected that GLUT4 would re-internalize at least as rapidly as after insulin removal. Surprisingly, intravital imaging showed that GLUT4 re-internalization in mouse quadriceps required 100–130 min after cessation of contractions to be completed depending on mouse strain (19). Interestingly, previous studies have shown similarly increased rates of glucose transport well into the post-exercise period in isolated white extensor digitorum longus mouse muscle measured at both 30 and 85 min time-points post-exercise (8). In contrast, no increase in glucose transport was found in epitrochlearis or soleus muscles 85 min after exercise (8). The reason for the difference in glucose transport reversal between extensor digitorum longus and epitrochlearis muscles could be due to that extensor digitorum longus muscle has a higher contraction-induced glucose transport and thereby a possible higher increase in GLUT4 translocation compared to epitrochlearis. The slow reversal in glucose transport in extensor digitorum longus muscle compared with epitrochlearis could therefore be due to more GLUT4 needs to re-internalize in this muscle.
The mechanism behind the slow GLUT4 re-internalization might be reduced energy levels after exercise increasing AMPK activity, e.g., reduced levels of glycogen after contractions. Specifically, glycogen levels have been shown to be reduced for up to several hours post exercise and correlate with increased AMPK activity. Furthermore, glycogen has been shown to directly associate with the AMPK beta-subunit thereby inactivating AMPK. Muscle contractions associated with treadmill exercise have also been shown to cause sustained AMPK phosphorylation at least 10 min after exercise in mice (8). Furthermore, in favor of a role of AMPK are findings using pulse-chase surface photo-labeling of GLUT4 in isolated epitrochlearis muscle (11). Here it was shown that GLUT4 re-internalization from unspecified membrane surfaces after stimulation with AMPK activator AICAR was not complete before 30–60 min after AICAR removal, compared to 10 min after insulin removal in white epitrochlearis muscles (11). These findings could indicate that AMPK regulates the GLUT4 re-internalization. However, this delay could also just reflect a delayed AICAR monophosphate (ZMP) degradation in the pulse-chase study. In contrast, our intravital studies in white quadriceps fibers in AMPKα2i mice with muscle specific deletion of AMPK activity showed that GLUT4 re-internalization was not altered compared to controls, indicating no role for AMPK in regulating the slow GLUT4 re-internalization from either sarcolemma or t-tubules after contractions (19). However, in the AMPKα2i transgenic mice it is possible that parallel pathways are compensating thereby masking the effect of the AMPK activity loss.
How the LKB1/SNARK activation pathway may affect GLUT4 re-internalization after exercise has not yet been addressed using intravital imaging or other techniques assaying GLUT4 trafficking. However, muscles from both LKB1 and SNARK knock-out mice have reduced contraction-mediated glucose uptake measured during a 15 min contraction period and a subsequent 30 min post contraction period (14). This reduced glucose transport could potentially be a result of altered (e.g. higher endocytosis rate) GLUT4 re-internalization kinetics and not only an impaired GLUT4 exocytosis. Thus, a faster re-internalization due to the lack of either LKB1 or SNARK activity would reduce the GLUT4 “dwell time” in plasma membrane surfaces, thereby reducing the time available for GLUT4 to transport glucose across the membrane barrier. Taken together, modulation of GLUT4 re-internalization after muscle contractions is likely an important step in the regulation of glucose transport. However, several signaling steps potentially responsible for the slow GLUT4 re-internalization remain to be elucidated. Furthermore, studies of how the GLUT4 re-internalization kinetics correlate with the well described increased effect of a sub-maximal insulin dose on glucose transport (increased insulin sensitivity) after an exercise bout would also be interesting to investigate in relation to these signaling steps.
STIMULUS SPECIFIC POOL OF GLUT4?
In addition to the differences in signaling pathways and GLUT4 exocytosis/re-internalization kinetics, insulin and contraction stimulation have previously been proposed to recruit GLUT4 from different vesicle pools. In muscle fractionation studies several groups have shown that GLUT4 vesicles can be divided into two pools (30). Approximately half of the GLUT4 vesicles are transferrin receptor-positive and have been characterized as part of the endosomal compartment (30). The other half is a non-endosomal transferrin receptor-negative GLUT4 pool, hypothesized to be part of a specialized GLUT4 compartment (30). The biochemical studies are in accordance with immuno-staining studies showing an ~50 % co-localization overlap between the transferrin receptor and GLUT4 present in both larger and smaller vesicle structures throughout the muscle fiber core and surface area under basal conditions in soleus and flexor digitorum brevis muscle fibers (10;30). It has furthermore been proposed that recruitment of the two GLUT4 vesicle pools is stimulus specific. Specifically, muscle contractions were shown to recruit GLUT4 and transferrin receptor from primarily the transferrin positive membrane fractions in hind limb muscle membranes (23;30). In contrast, insulin was shown to recruit GLUT4 from primarily the transferrin receptor-negative membrane fractions (30). Furthermore, in the fractionation study with spatial differentiation of membranes, muscle contractions were reported to also recruit transferrin receptor to the sarcolemma but not the t-tubules (23). Insulin stimulation did not recruit transferrin receptors to any of the membrane surfaces (23). The stimulus specific recruitment of GLUT4 pools was also found using light microscopy in intact soleus fibers immuno-stained for transferrin receptor and GLUT4 (30). However, the stimuli specific co-localization were only determined in the area around the nuclei at the sarcolemma (30). Thus, stimuli specific co-localization between GLUT4 and the transferrin receptor was not determined in the rest of the fiber, including the central t-tubules region (30).
In contrast, intravital imaging of GLUT4 vesicle recruitment in white quadriceps has not revealed regional differences in the GLUT4 vesicle depletion in white muscle fibers during and after stimulation with insulin or contractions (19;21). Specifically, stimulation with either insulin or contraction reduced basal GLUT4 vesicle depots with 60–70% throughout the muscle fibers (19;21). Thus, if 60–70% of the basal GLUT4 vesicles are depleted by either stimulus and approximately half the muscle fiber GLUT4 vesicles are transferrin-positive and half transferrin-negative, recruitment must at least partially take place from both GLUT4 pools (19;21). Therefore, the present intravital imaging data in intact muscle do not support a strict stimulus specific GLUT4 pools based on the previously proposed criteria at least in white muscle. Since the previous immuno-staining work was performed in red soleus or flexor digitorum brevis muscle, the difference could also reflect fiber type differences. However, further co-localization analysis with fluorescent tagged transferrin receptor combined with GLUT4-EGFP is needed to investigate the dynamic degree of overlap at the sarcolemma and t-tubules during stimulation.
PERSPECTIVES
Intravital imaging studies in muscle have so far provided new information about stimulus dependent differences in GLUT4 trafficking kinetics with an improved spatial-temporal resolution. However, how the contraction stimulated signaling pathways interact with local GLUT4 trafficking kinetics in the muscle fiber needs to be further clarified. This will be of particular interest to further investigate during conditions such as obesity and insulin resistance as the contraction mediated GLUT4 translocation is still intact. Interestingly, our intravital studies have shown that both insulin signaling and GLUT4 trafficking defects associated with some forms of insulin resistance are compartmentalized (20). Specifically, ten days of denervation or 12 weeks of high fat feeding primarily affected insulin signaling and GLUT4 translocation to the t-tubules and less to the sarcolemma (20). In addition, recent studies have described that it is primarily reductions in the intramyofibrillar mitochondria pool that correlate with mitochondrial defects seen in insulin resistance (2). Interestingly, this pool of mitochondria is located in close vicinity the t-tubule region. Although muscle contraction during normal physiological conditions appears to lack compartmentalization, contractions may modulate GLUT4 trafficking or related factors locally during insulin resistant conditions. Potentially this could be in an unexpected way either by a compensatory increase in GLUT4 trafficking to sarcolemma or by a normalization of GLUT4 trafficking in parts of the t-tubule system. Therefore intravital imaging of contraction induced GLUT4 trafficking kinetics during insulin resistant conditions is warranted.
SUMMARY
Insulin and contraction-mediated GLUT4 trafficking to the sarcolemma and the t-tubules in fully differentiated skeletal muscle have different spatial and temporal kinetics. This compartmentalization difference appears primarily to be attributed to differences in signaling pathways. Insulin mediated GLUT4 trafficking is dependent on the local arrival of the hormone and the subsequent unilateral downstream pathway leading to GLUT4 trafficking. In contrast, muscle contraction induces GLUT4 trafficking through several parallel pathways that also differ in their temporal activation kinetics.
Summary.
Intravital imaging studies in mature muscle indicate that insulin and contraction-mediated GLUT4 trafficking kinetics have signal pathway mediated differences.
Acknowledgments
The author wishes to acknowledge the work of researchers who could not be cited due to reference limitations. I wish to thank Professor Henrik Galbo, Dr. Ho-Jin Koh and Mike Hirshman for critical suggestions.
Disclosure of funding: This project was supported by funding from the Weimann Foundation and the DERC P30DK036836 at the Joslin Diabetes Center.
Footnotes
Disclosure of interest: There is no conflict of interest.
Reference List
- 1.Cartee GD, Funai K. Exercise and insulin: Convergence or divergence at AS160 and TBC1D1? Exerc Sport Sci Rev. 2009 Oct;37(4):188–95. doi: 10.1097/JES.0b013e3181b7b7c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chomentowski P, Coen PM, Radikova Z, Goodpaster BH, Toledo FG. Skeletal muscle mitochondria in insulin resistance: differences in intermyofibrillar versus subsarcolemmal subpopulations and relationship to metabolic flexibility. J Clin Endocrinol Metab. 2011 Feb;96(2):494–503. doi: 10.1210/jc.2010-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993 Jul;10(7):1093–5. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 4.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, McGraw TE. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005 Oct;2(4):263–72. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Fazakerley DJ, Lawrence SP, Lizunov VA, Cushman SW, Holman GD. A common trafficking route for GLUT4 in cardiomyocytes in response to insulin, contraction and energy-status signalling. J Cell Sci. 2009 Mar 1;122(Pt 5):727–34. doi: 10.1242/jcs.041178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, Goodyear LJ. AMP-activated protein kinase alpha2 activity is not essential for contraction- and hyperosmolarity-induced glucose transport in skeletal muscle. J Biol Chem. 2005 Nov 25;280(47):39033–41. doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- 7.Funai K, Cartee GD. Contraction-stimulated glucose transport in rat skeletal muscle is sustained despite reversal of increased PAS-phosphorylation of AS160 and TBC1D1. J Appl Physiol. 2008 Dec;105(6):1788–95. doi: 10.1152/japplphysiol.90838.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamada T, Arias EB, Cartee GD. Increased submaximal insulin-stimulated glucose uptake in mouse skeletal muscle after treadmill exercise. J Appl Physiol. 2006 Nov;101(5):1368–76. doi: 10.1152/japplphysiol.00416.2006. [DOI] [PubMed] [Google Scholar]
- 9.Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998 Aug;47(8):1369–73. doi: 10.2337/diab.47.8.1369. [DOI] [PubMed] [Google Scholar]
- 10.Kaisto T, Rahkila P, Marjomaki V, Parton RG, Metsikko K. Endocytosis in skeletal muscle fibers. Exp Cell Res. 1999 Dec 15;253(2):551–60. doi: 10.1006/excr.1999.4659. [DOI] [PubMed] [Google Scholar]
- 11.Karlsson HK, Chibalin AV, Koistinen HA, Yang J, Koumanov F, Wallberg-Henriksson H, Zierath JR, Holman GD. Kinetics of GLUT4 Trafficking in Rat and Human Skeletal Muscle. Diabetes. 2009 Feb 2; doi: 10.2337/db08-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan AH, Thurmond DC, Yang C, Ceresa BP, Pessin JE. Munc18c regulates insulin-stimulated GLUT4 translocation to the transverse tubules in skeletal muscle. J Biol Chem. 2000 Oct 27;276(6):4063–9. doi: 10.1074/jbc.M007419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006 Nov;26(22):8217–27. doi: 10.1128/MCB.00979-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koh HJ, Toyoda T, Fujii N, Jung MM, Rathod A, Middelbeek RJ, Lessard SJ, Treebak JT, Tsuchihara K, Esumi H, Richter EA, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci U S A. 2010 Aug 31;107(35):15541–6. doi: 10.1073/pnas.1008131107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem. 2006 Oct 20;281(42):31478–85. doi: 10.1074/jbc.M605461200. [DOI] [PubMed] [Google Scholar]
- 16.Lauritzen HPMM, Reynet C, Schjerling P, Ralston E, Thomas S, Galbo H, Ploug T. Gene gun bombardment-mediated expression and translocation of EGFP-tagged GLUT4 in skeletal muscle fibers in vivo. Pflugers Arch Euro J Physiol. 2002 Jun 27;444(6):710–21. doi: 10.1007/s00424-002-0862-5. [DOI] [PubMed] [Google Scholar]
- 17.Lauritzen HP. Imaging of protein translocation in situ in skeletal muscle of living mice. Methods Mol Biol. 2010;637:231–44. doi: 10.1007/978-1-60761-700-6_12. [DOI] [PubMed] [Google Scholar]
- 18.Lauritzen HP, Galbo H, Brandauer J, Goodyear LJ, Ploug T. Large GLUT4 vesicles are stationary while locally and reversibly depleted during transient insulin stimulation of skeletal muscle of living mice: imaging analysis of GLUT4-enhanced green fluorescent protein vesicle dynamics. Diabetes. 2008 Feb;57(2):315–24. doi: 10.2337/db06-1578. [DOI] [PubMed] [Google Scholar]
- 19.Lauritzen HP, Galbo H, Toyoda T, Goodyear LJ. Kinetics of contraction-induced GLUT4 translocation in skeletal muscle fibers from living mice. Diabetes. 2010 Sep;59(9):2134–44. doi: 10.2337/db10-0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lauritzen HP, Ploug T, Ai H, Donsmark M, Prats C, Galbo H. Denervation and high-fat diet reduce insulin signaling in T-tubules in skeletal muscle of living mice. Diabetes. 2008 Jan;57(1):13–23. doi: 10.2337/db07-0516. [DOI] [PubMed] [Google Scholar]
- 21.Lauritzen HP, Ploug T, Prats C, Tavare JM, Galbo H. Imaging of insulin signaling in skeletal muscle of living mice shows major role of T-tubules. Diabetes. 2006 May;55(5):1300–6. doi: 10.2337/db05-1216. [DOI] [PubMed] [Google Scholar]
- 22.Lauritzen HP, Schertzer JD. Measuring GLUT4 translocation in mature muscle fibers. Am J Physiol Endocrinol Metab. 2010;299(2):E169–79. doi: 10.1152/ajpendo.00066.2010. [DOI] [PubMed] [Google Scholar]
- 23.Lemieux K, Han XX, Dombrowski L, Bonen A, Marette A. The transferrin receptor defines two distinct contraction-responsive GLUT4 vesicle populations in skeletal muscle. Diabetes. 2000 Feb;49(2):183–9. doi: 10.2337/diabetes.49.2.183. [DOI] [PubMed] [Google Scholar]
- 24.Lemieux K, Konrad D, Klip A, Marette A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. FASEB J. 2003 Sep;17(12):1658–65. doi: 10.1096/fj.02-1125com. [DOI] [PubMed] [Google Scholar]
- 25.Lizunov V, Stenkula K, Lisinski I, Gavrilova O, Yver DR, Chadt A, Al-Hasani H, Zimmerberg J, Cushman SW. Insulin stimulates fusion, but not tethering, of GLUT4 vesicles in skeletal muscle of transgenic mouse. Am J Physiol Endocrinol Metab. 2012 Jan 31; doi: 10.1152/ajpendo.00466.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lizunov VA, Matsumoto H, Zimmerberg J, Cushman SW, Frolov VA. Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J Cell Biol. 2005 May 9;169(3):481–9. doi: 10.1083/jcb.200412069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lund S, Holman GD, Schmitz O, Pedersen O. Glut 4 content in the plasma membrane of rat skeletal muscle: comparative studies of the subcellular fractionation method and the exofacial photolabelling technique using ATB-BMPA. FEBS Lett. 1993 Sep 20;330(3):312–8. doi: 10.1016/0014-5793(93)80895-2. [DOI] [PubMed] [Google Scholar]
- 28.Marette A, Burdett E, Douen A, Vranic M, Klip A. Insulin induces the translocation of GLUT4 from a unique intracellular organelle to transverse tubules in rat skeletal muscle. Diabetes. 1992 Dec;41(12):1562–9. doi: 10.2337/diab.41.12.1562. [DOI] [PubMed] [Google Scholar]
- 29.Patki V, Buxton J, Chawla A, Lifshitz L, Fogarty K, Carrington W, Tuft R, Corvera S. Insulin action on GLUT4 traffic visualized in single 3T3-l1 adipocytes by using ultra-fast microscopy. Mol Biol Cell. 2001 Jan;12(1):129–41. doi: 10.1091/mbc.12.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ploug T, vanDeurs B, Ai H, Cushman SW, Ralston E. Analysis of GLUT4 distribution in whole skeletal muscle fibers: Identification of distinct storage compartments that are recruited by insulin and muscle contractions. J Cell Biol. 1998 Sep 21;142(6):1429–46. doi: 10.1083/jcb.142.6.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roche JA, Ford-Speelman DL, Ru LW, Densmore AL, Roche R, Reed PW, Bloch RJ. Physiological and histological changes in skeletal muscle following in vivo gene transfer by electroporation. Am J Physiol Cell Physiol. 2011 Nov;301(5):C1239–C1250. doi: 10.1152/ajpcell.00431.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame HD, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J. 2005 May 18;24(10):1810–20. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Semiz S, Park JG, Nicoloro SM, Furcinitti P, Zhang C, Chawla A, Leszyk J, Czech MP. Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. EMBO J. 2003 May 15;22(10):2387–99. doi: 10.1093/emboj/cdg237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma N, Arias EB, Cartee GD. Rapid reversal of insulin-stimulated AS160 phosphorylation in rat skeletal muscle after insulin exposure. Physiol Res. 2010;59(1):71–8. doi: 10.33549/physiolres.931707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shorten PR, McMahon CD, Soboleva TK. Insulin transport within skeletal muscle transverse tubule networks. Biophys J. 2007 Nov 1;93(9):3001–7. doi: 10.1529/biophysj.107.107888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song XM, Ryder JW, Kawano Y, Chibalin AV, Krook A, Zierath JR. Muscle fiber type specificity in insulin signal transduction. Am J Physiol. 1999 Dec;277(6 Pt 2):R1690–R1696. doi: 10.1152/ajpregu.1999.277.6.R1690. [DOI] [PubMed] [Google Scholar]
- 37.Sun Y, Bilan PJ, Liu Z, Klip A. Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci U S A. 2010 Nov 16;107(46):19909–14. doi: 10.1073/pnas.1009523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang W, Hansen PA, Marshall BA, Holloszy JO, Mueckler M. Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J Cell Biol. 1996 Oct;135(2):415–30. doi: 10.1083/jcb.135.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witczak CA, Jessen N, Warro DM, Toyoda T, Fujii N, Anderson ME, Hirshman MF, Goodyear LJ. CaMKII regulates contraction- but not insulin-induced glucose uptake in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2010 Jun;298(6):E1150–E1160. doi: 10.1152/ajpendo.00659.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wojtaszewski JF, Hansen BF, Gade, Kiens B, Markuns JF, Goodyear LJ, Richter EA. Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes. 2000 Mar;49(3):325–31. doi: 10.2337/diabetes.49.3.325. [DOI] [PubMed] [Google Scholar]