

Abstract
An estimated 170 million people worldwide are chronically infected with the Hepatitis C Virus (HCV), which is characterized histologically by a persistent immune and inflammatory response that fails to clear HCV from hepatocytes. This response is recruited to the liver in part by the chemokine CXCL10, the serum and intrahepatic levels of which have been inversely linked to the outcome of interferon (IFN)-based therapies for hepatitis C. Bystander tissue damage from this ineffective response is thought to lead to increased hepatocyte turnover and the development of fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). However, CXCL10 is traditionally viewed as an orchestrator of the angiostatic and anti-tumor immune response. In this review, we will explore this duality and the pathways by which CXCL10 is produced by hepatocytes during HCV infection, its effects on resident and infiltrating immune cells, and how deregulation of these cell populations within the liver may lead to chronic liver inflammation. We will also discuss potential host-directed therapies to slow or reverse HCV-induced inflammation that leads to fibrosis, cirrhosis, and HCC.
BACKGROUND
Chronic hepatitis C virus (HCV) infection affects an estimated 170 million people globally, and is the leading cause of liver transplantation in many countries(1,2). Activation of innate immune pathways in hepatocytes following infection leads to infiltration of pro-inflammatory, anti-viral immune effector cells into the liver(3). Many of these cells are recruited to the liver by the chemokine CXCL10, which binds to and activates the CXCR3 receptor found most commonly on pro-inflammatory CD8+ cytotoxic T (TC) cells, CD4+ type I helper T (TH1) cells, and natural killer (NK) cells(4,5). However, this response is incapable of eliminating the virus in approximately 85% of patients with acute infection and instead contributes to a chronic immune cell presence in the liver(6). Indeed, CXCR3+ CD8+ Tc cells have been identified among intrahepatic immune cells in chronic hepatitis C patients(4,5). Damage to bystander tissue from this persistent yet ineffective inflammatory response has been linked to the development of fibrosis, cirrhosis, and hepatocellular carcinoma (HCC)(7). CXCL10 plasma levels are also negatively correlated with the outcome of interferon (IFN)-based therapy for HCV infection(8). However, as an angiostatic chemkoine that recruits CD8+ Tc and NK cells, CXCL10 could orchestrate an anti-tumor response(9). Herein we will explore this apparent paradox by defining the innate immune signaling pathways that lead to CXCL10 induction in hepatocytes, examining how deregulation of the recruited immune response during HCV infection may lead to inflammatory liver disease, and discussing possible avenues for controlling inflammation and preventing the development of HCC.
Innate Immune Sensing of HCV in Hepatocytes
Activation of cellular innate immune pathways depends upon recognition of foreign DNA, RNA, or protein motifs known as pathogen associated molecular patterns (PAMPs). Specific PAMPs are recognized by innate pattern recognition receptors (PRRs) from one of three families: Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), or Nod-like-receptors (NLRs). The interplay of these receptors and their downstream signaling pathways is what determines the resultant innate immune response. For example, the positive sense HCV RNA genome is separately recognized by two different PRRs within the hepatocyte: retinoic acid inducible gene 1 (RIG-I) and Toll-like receptor 3 (TLR3; Figure 1, Insert)(10,11)
Figure 1. Deregulation of the Inflammatory Response Recruited by CXCL10 Following HCV Infection.

Sensing of viral RNA by the innate immune receptors RIG-I and TLR3 following hepatitis C virus (HCV,
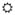 ) infection of the hepatocyte leads to signal transduction through MAVS and TRIF, respectively, activation of transcription factors (NF-κB, IRFs, AP-1, C/EBP-β), and transcription of CXCL10 (Δ) (“CXCL10 Induction”). Secreted CXCL10 forms a chemotactic gradient that recruits immune cells (Natural Killer [NK], CD4+ TH1, and CD8+ Tc cells) and non-parenchymal liver cells (Kupffer cells and Hepatic Stellate Cells [HSCs]) to the site of infection (“Recruitment, Inflammation, and Cell Death”). Upon arriving, these cells produce pro-inflammatory, pro-apoptotic mediators (❖) such as type I interferon (IFN), type III IFN, TNFα, IL-1β, and reactive oxygen species (ROS). This response fails to clear HCV in 80–85% of patients and instead generates persistent inflammation and hepatocyte turnover. It also leads to liver fibrosis through chronic HSC activation, the overproduction of type I collagen, and the inhibition of collagen-degrading matrix metalloproteinases (MMPs) by tissue inhibitor of metalloproteinases (TIMPs) (“Cell Turnover and Fibrosis”). Over several decades, progressive fibrosis can lead to cirrhosis and hepatocellular carcinoma.
) infection of the hepatocyte leads to signal transduction through MAVS and TRIF, respectively, activation of transcription factors (NF-κB, IRFs, AP-1, C/EBP-β), and transcription of CXCL10 (Δ) (“CXCL10 Induction”). Secreted CXCL10 forms a chemotactic gradient that recruits immune cells (Natural Killer [NK], CD4+ TH1, and CD8+ Tc cells) and non-parenchymal liver cells (Kupffer cells and Hepatic Stellate Cells [HSCs]) to the site of infection (“Recruitment, Inflammation, and Cell Death”). Upon arriving, these cells produce pro-inflammatory, pro-apoptotic mediators (❖) such as type I interferon (IFN), type III IFN, TNFα, IL-1β, and reactive oxygen species (ROS). This response fails to clear HCV in 80–85% of patients and instead generates persistent inflammation and hepatocyte turnover. It also leads to liver fibrosis through chronic HSC activation, the overproduction of type I collagen, and the inhibition of collagen-degrading matrix metalloproteinases (MMPs) by tissue inhibitor of metalloproteinases (TIMPs) (“Cell Turnover and Fibrosis”). Over several decades, progressive fibrosis can lead to cirrhosis and hepatocellular carcinoma.
RIG-I is a cytoplasmic sensor of double-stranded, 5′ tri-phosphate RNAs containing poly-U or poly-A motifs(12). Following the binding of this PAMP, RIG-I undergoes a conformational change and binds to the mitochondrial antiviral-signaling protein (MAVS) signaling adaptor(13). In contrast, TLR3 recognizes longer double-stranded RNAs generated during viral replication that have been re-localized to the endosome(11). Activated TLR3 binds the signaling adaptor TIR-domain-containing adapter-inducing IFN-β (TRIF) through its cytoplasmic receptor domain(13).
Induction of CXCL10 in Hepatocytes
MAVS and TRIF signaling activates various transcription factors including nuclear factor (NF)-κB, activator protein (AP)-1, C/EBP-β, and IFN regulatory factors (IRFs), which translocate into the nucleus to induce gene transcription. (13,14). Putative binding sites for these transcription factors have been annotated in the CXCL10 promoter(15). Indeed, HCV can induce NF-κB binding to this site in TLR3-expressing hepatoma cells(11). NF-κB also drives CXCL10 transcription during rhinovirus infection, while AP-1 and C/EBP-β activate transcription of the structurally similar chemokine CXCL8 (i.e. IL-8) (15–17). IRF1, IRF2, IRF3, and IRF7 also reportedly bind the CXCL10 promoter during influenza A infection(18).
Activation of IRF3 and IRF7 can also lead to the induction of anti-viral type I IFNs (IFN-α and IFN-β) and type III IFNs (IL-28A, IL-28B, IL-29) in hepatocytes(14,19). These secreted cytokines can act in a paracrine manner to amplify chemokine and cytokine responses in adjacent liver cells through activation of Janus kinases (JAKs) and various signal transducer and activator of transcription (STAT) proteins(19,20). Activation of JAK-STAT signaling induces IFN-stimulated genes (ISGs) through the binding of STAT dimers to IFN-stimulated response elements (ISREs) or gamma-IFN activation site elements in their promoters(19,20). Type II IFN, a related cytokine produced by infiltrating NK cells, CD8+ Tc cells, and CD4+ TH1 cells, can also induced STAT1-signaling through these elements(20,21). Since the CXCL10 promoter contains both putative IFN-stimulatory regulatory elements (ISREs) and putative STAT-binding sites, it can potentially respond to all three types of IFN(15).
Despite these observations in other systems, we observed that neutralization of type I and type III IFNs had no effect on CXCL10 production during HCV infection in hepatoma cells expressing functional TLR3 and RIG-I (Brownell et. al.; submitted manuscript). These data suggest that CXCL10 induction in hepatocytes during the initial steps of HCV infection occurs predominantly through direct activation of transcription factors following PRR signaling rather than through secondary paracrine signaling of IFNs. Of course, IFNs secreted from immune cells recruited to the HCV-infected liver as well as from non-parenchymal cells likely contribute to CXCL10 induction in vivo. This secondary induction would supplement the initial CXCL10 output by hepatocytes.
Induction of CXCL10 in hepatocytes may also involve non-traditional PRR signaling pathways. Ho et al. reported IFN-independent activation of STAT1 and STAT3 proteins during infection with Dengue Virus, another member of the Flaviviridae(22). STAT1 can also be activated via p38 MAP kinase following TLR7 stimulation in plasmacytoid dendritic cells(23). Since STAT1 can bind to ISREs, it is possible that this alternative pathway contributes to CXCL10 induction in hepatocytes.
CXCL10 Recruits Pro-Inflammatory Effector Cells for the Anti-HCV Response
Once induced, CXCL10 recruits a pro-inflammatory, anti-viral immune response to sites of infection by binding to the CXCR3 receptor on CD4+ TH1 and CD8+ Tc cells (Figure 1)(4,5). CXCR3 was recently reported to be universally expressed and exists in two isoforms: CXCR3A and CXCR3B(24). CXCR3A is the activating isoform highly expressed by leukocytes and is associated with proliferation and chemotactic migration of these cells (24,25). CXCR3 is also expressed by NK cells as well as by minority cell populations within the liver including resident macrophages (i.e. Kupffer cells) and hepatic stellate cells (HSCs) (4,26–28). Thus, CXCL10 induction from hepatocytes could also localize non-parenchymal cells within the liver to specific sites of infection.
Once recruited to the inflamed liver, activated CD8+ Tc and NK cells kill virus-infected cells via Fas/TRAIL-mediated apoptosis, the release of granzymes and perforin, and secretion of type II IFN (26,29). Apoptotic bodies released from dying hepatocytes are then phagocytosed by Kupffer cells, which further promote Fas-mediated hepatocyte apoptosis and release reactive oxygen and nitrogen species (ROS/NOS)(30). Kupffer cells also activate HSCs by releasing TGF-β(30). This causes HSCs to differentiate from quiescent, Vitamin A-storage bodies into proliferative myofibroblasts that secrete type I collagen as part of the general wound healing response to liver injury (31).
Kupffer cells, HSCs, and liver sinusoidal endothelial cells (LSECs) also perpetuate the existing inflammatory state by secreting additional cytokines and chemokines as part of a positive feedback loop. As in hepatocytes, this secretion can be triggered by pro-inflammatory cytokines produced by infiltrating immune cells (TNFα, IFNs, etc) or by innate PRRs. Recognition of HCV non-structural proteins by TLR4 in Kupffer cells during chronic infection can increase secretion of TNFα(32). TNFα and IL-1β activated HSCs show increased secretion of CXCL8 when exposed to ligands for TLR2, which recognizes HCV Core and NS3 proteins(33,34). Supernatants from LSECs treated with TLR3 and TLR4-specific PAMPs were also able to suppress HCV replication in HCV replicon-bearing cells(35). Thus, the primary sensing of HCV RNA by hepatocytes initiates an anti-viral, pro-inflammatory response that involves recruitment of multiple immune cell types to the liver that further amplify the response.
Deregulation of Recruited Cells Leads to Fibrosis, Cirrhosis, and HCC
Despite the robust inflammatory response initiated and recruited by CXCL10, chronic hepatitis C develops in up to 85% of subjects with acute infection(6). Viral evolution plays a considerable role in establishing this persistence, as immune escape variants of the HCV NS3 epitope recognized by CD4+ TH1 cells fail to stimulate proliferation while simultaneously causing these cells to shift to a TH2 response profile(36). This causes induction of anti-inflammatory cytokines (i.e. IL-10) and reduction of CD8+ Tc and NK cell-stimulating cytokines (i.e. type II IFN and IL-2)(37). Direct inactivation of infiltrating effector cells can also lead to ineffective viral clearance. HCV-specific CD8+ Tc cells from patients with chronic hepatitis C display an exhausted phenotype, with decreases in both type II IFN production and epitope-specific degranulation(38). Virus-mediated dendritic cell dysfunction may contribute to the development of anergy through ineffectual co-stimulation or antigen presentation, as could the presence of an antagonistic variant of CXCL10 which may inhibit migration of these CXCR3+ cells from plasma into tissue(39,40). Higher frequencies of both intrahepatic and peripheral CD4+ CD25+ FoxP3+ immunosuppressive regulatory T (Treg) cells have also been reported in HCV-infected patients, further indicating that suppression of effector immune responses maintains viral persistence in chronic hepatitis C(39,41)
HCV proteins also interfere with anti-viral and IFN responses in hepatocytes during chronic infection(42,43). Despite this interference, elevated levels of inflammatory cytokines and chemokines are still found in the liver parenchyma of patients with chronic hepatitis C (see above). Kupffer cells also remain activated and continue to release ROS/NOS and TGF-β, perpetuating HSC activation and type I collagen deposition. Eventually, chronic activation causes HSCs to secrete tissue inhibitor of metalloproteinases (TIMPs), which inhibit collagen-degrading matrix metalloproteinases (MMPs) and leads to an excessive accumulation of fibrotic scar tissue known as fibrosis(31). Progressive disruption of the liver architecture and continued hepatocyte turnover can then lead to cirrhosis, a condition where the liver parenchyma is divided into isolated nodules of regenerative tissue with severely reduced functionality(30). Accumulation of genetic aberrations from repeated rounds of cell death and renewal within these nodules then leads to neoplasm and HCC(7).
The pro-inflammatory and cytotoxic immune responses recruited by CXCL10 can normally eliminate pre-cancerous and cancerous cells through recognition of tumor-specific antigens(7). However, as these responses are already impaired during chronic hepatitis C, it is likely that the ability to identify and eliminate neoplastic cells is also defective. CXCL10 may still inhibit development of HCC through its reported angiostatic activity, but recent literature suggests that CXCL10 may accelerate cancer growth in non-immune cell types(44,45). Neoplastic cells may also exploit chemokine gradients as “roads” during metastasis. Treatment with CXCL10 increases motility of prostate cancer-derived but not normal prostate epithelial cells via reduced CXCR3B expression, which normally inhibits cell growth and migration in non-motile cell types (24,46). CXCR3B expression was also reduced in two breast cancer cell lines, while induction of CXCR3A and repression of CXCR3B have been reported in clear cell ovarian cancers(47,48). It remains to be determined whether down-regulation of growth inhibitory receptor CXCR3B and/or up-regulation of the growth promoting receptor CXCR3A occurs during hepatocyte transformation to HCC and metastasis.
CLINICAL-TRANSLATIONAL ADVANCES
Current therapies for chronic hepatitis C seek to limit the development of persistent inflammation by reducing systemic viral load using a combination treatment of pegylated-IFN-α and the non-specific anti-viral Ribavirin (peg-IFNα/RBV). Unfortunately, this regimen fails to eliminate the infection in roughly 50% of patients(6). While recently developed HCV-specific protease inhibitors improve the likelihood of success for some patients, IFN-containing regimens are still poorly tolerated, require 24–48 weeks of administration, and do not address the underlying inflammatory sequelae that cause liver disease(49,50). For patients that have already progressed to decompensated cirrhosis, liver transplantation represents the only available treatment option(51). However, re-infection of the new liver occurs in nearly all cases of active infection, and anti-HCV therapy is both less efficacious and associated with increased toxicity after transplantation(51). Thus, new treatments that prevent or reverse the onset of these inflammatory sequelae must be pursued. As a master regulator of the infiltrating pro-inflammatory response, the CXCL10/CXCR3 signaling pathway makes an attractive therapeutic target.
Potential Anti-CXCL10 Therapies
Agents that selectively neutralize CXCL10 would theoretically increase patient responsiveness to traditional IFN-based HCV therapy while simultaneously dampening inflammatory immune cell activation. For example, specific inhibitors of the CXCR3A isotype could prevent aberrant activation of CD8+ Tc cells and NK cells that lead to excessive hepatocyte death. This in turn would limit Kupffer cell and HSC activation and delay or prevent development of fibrosis. Such drugs would likely mimic Maraviroc, an antagonist of the chemokine receptor CCR5 that is used clinically to block HIV entry(52). However, it is possible that reducing a patient’s sensitivity to CXCL10 by blocking its receptor may also interfere with the immune system’s ability to respond to other pathogens(53).
Broadly Acting Anti-Inflammatory Therapies
A safer alternative may be to identify new applications for existing anti-inflammatory drugs. One advantage to this approach is the ability to counteract the excessive immune response recruited by CXCL10 through multiple mechanisms. For example, since oxidative stress causes direct cellular damage in addition to activating HSCs, herbal antioxidant compounds have been suggested as both anti-fibrotic and anti-inflammatory therapy for liver diseases of multiple etiologies (30). Vitamin E has successfully reduced inflammation and halted fibrosis progression among those with non-alcoholic steatohepatitis (NASH) in clinical trials(54). The routinely consumed herbal medications Sho-saiko-to and Silymarin also appear to have direct anti-fibrotic activity on HSCs as well as general hepatoprotective properties, although their mechanisms of action remain undefined(55,56). Traditional anti-fibrotic drugs have also had demonstrable effects on oxidative stress within the liver: Long-term treatment with Losartan reduces NADPH oxidase activity in HCV patients (57).
Successful anti-inflammatory therapies may also target pathways other than those involved in generating oxidative stress. Broadly acting corticosteroids remain a standard therapy for autoimmune hepatitis(58). Sorafenib, a chemotherapeutic agent already approved to treat HCC, also inhibits the Raf/ERK pro-inflammatory and pro-fibrotic signaling pathways(59). Finally, TNFα inhibitors have been used reduce serum levels of liver enzymes, IL-6, and TGF-β in animal models, although limited success has been seen in human clinical trials for alcohol-related liver disease or advanced cirrhosis(30).
Targeting multiple pathways simultaneously may also increase the risk of adverse events occurring during treatment. Severe side effects have been reported among patients taking experimental broadly anti-apoptotic drugs such as caspase-3 inhibitors(30). The duration of anti-inflammatory therapy will also likely depend upon the extent of fibrosis or cirrhosis present within the liver, increasing the likelihood of adverse events occurring in patients with severe disease. Additionally, administering anti-inflammatory drugs to patients simultaneously undergoing IFN treatment for hepatitis C may interfere with the anti-viral efficacy of IFN.
Ultimately, a better understanding of immune and inflammatory signaling within the liver is required before the full extent of the efficacy and side effects for these proposed treatments can be known. Since HCV-related cirrhosis and HCC are predicted to rise substantially in the next decade(60), it is imperative that research into this area accelerate. Routine clinical application of hepatoprotective therapies in the near future may help to prevent or reverse the effects of end-stage liver disease in millions of chronically infected HCV patients worldwide. Furthermore, these types of host-directed therapies may be beneficial to other, non-viral forms of liver diseases that include an inflammatory component.
Acknowledgments
Financial Support: National Institutes of Health (NIH U19AI066328 to SJP), University of Washington Pathobiology Training Grant (NIH 2T32AI007509 to JB).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to report.
References
- 1.Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 (Suppl 1):S10–5. doi: 10.1093/cid/cis361. [DOI] [PubMed] [Google Scholar]
- 2.Smith BD, Jorgensen C, Zibbell JE, Beckett GA. Centers for Disease Control and Prevention initiatives to prevent hepatitis C virus infection: a selective update. Clin Infect Dis. 2012;55 (Suppl 1):S49–53. doi: 10.1093/cid/cis363. [DOI] [PubMed] [Google Scholar]
- 3.Heydtmann M, Adams DH. Chemokines in the immunopathogenesis of hepatitis C infection. Hepatology (Baltimore, Md ) 2009;49:676–88. doi: 10.1002/hep.22763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J, Holmes TH, Cheung R, Greenberg HB, He X-S. Expression of chemokine receptors on intrahepatic and peripheral lymphocytes in chronic hepatitis C infection: its relationship to liver inflammation. J Infect Dis. 2004;190:989–97. doi: 10.1086/423283. [DOI] [PubMed] [Google Scholar]
- 5.Shields PL, Morland CM, Salmon M, Qin S, Hubscher SG, Adams DH. Chemokine and chemokine receptor interactions provide a mechanism for selective T cell recruitment to specific liver compartments within hepatitis C-infected liver. J Immunol. 1999;163:6236–43. [PubMed] [Google Scholar]
- 6.Shepherd J, Brodin H, Cave C, Waugh N, Price A, Gabbay J. Pegylated interferon alpha-2a and -2b in combination with ribavirin in the treatment of chronic hepatitis C: a systematic review and economic evaluation. Health Technol Assess. 2004;8:iii–iv. 1–125. doi: 10.3310/hta8390. [DOI] [PubMed] [Google Scholar]
- 7.Flecken T, Spangenberg HC, Thimme R. Immunobiology of hepatocellular carcinoma. Langenbecks Arch Surg. 2012;397:673–80. doi: 10.1007/s00423-011-0783-x. [DOI] [PubMed] [Google Scholar]
- 8.Askarieh G, Alsiö A, Pugnale P, Negro F, Ferrari C, Neumann AU, et al. Systemic and intrahepatic interferon-gamma-inducible protein 10 kDa predicts the first-phase decline in hepatitis C virus RNA and overall viral response to therapy in chronic hepatitis C. Hepatology (Baltimore, Md ) 2010;51:1523–30. doi: 10.1002/hep.23509. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, et al. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immun. 2002;70:3433–42. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sumpter R, Loo Y-M, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79:2689–99. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology (Baltimore, Md ) 2012;55:666–75. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–7. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawai T, Akira S. Toll-like Receptor and RIG-1-like Receptor Signaling. Annals of the New York Academy of Sciences. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 15.Spurrell JCL, Wiehler S, Zaheer RS, Sanders SP, Proud D. Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2005;289:L85–95. doi: 10.1152/ajplung.00397.2004. [DOI] [PubMed] [Google Scholar]
- 16.John AE, Zhu YM, Brightling CE, Pang L, Knox AJ. Human airway smooth muscle cells from asthmatic individuals have CXCL8 hypersecretion due to increased NF-kappa B p65, C/EBP beta, and RNA polymerase II binding to the CXCL8 promoter. J Immunol. 2009;183:4682–92. doi: 10.4049/jimmunol.0803832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haim K, Weitzenfeld P, Meshel T, Ben-Baruch A. Epidermal growth factor and estrogen act by independent pathways to additively promote the release of the angiogenic chemokine CXCL8 by breast tumor cells. Neoplasia. 2011;13:230–43. doi: 10.1593/neo.101340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veckman V, Osterlund P, Fagerlund R, Melén K, Matikainen S, Julkunen I. TNF-alpha and IFN-alpha enhance influenza-A-virus-induced chemokine gene expression in human A549 lung epithelial cells. Virology. 2006;345:96–104. doi: 10.1016/j.virol.2005.09.043. [DOI] [PubMed] [Google Scholar]
- 19.Ank N, West H, Paludan SR. IFN-lambda: novel antiviral cytokines. J Interferon Cytokine Res. 2006;26:373–9. doi: 10.1089/jir.2006.26.373. [DOI] [PubMed] [Google Scholar]
- 20.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–5. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 21.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 22.Ho L-J, Hung L-F, Weng C-Y, Wu W-L, Chou P, Lin Y-L, et al. Dengue virus type 2 antagonizes IFN-alpha but not IFN-gamma antiviral effect via down-regulating Tyk2-STAT signaling in the human dendritic cell. J Immunol. 2005;174:8163–72. doi: 10.4049/jimmunol.174.12.8163. [DOI] [PubMed] [Google Scholar]
- 23.Di Domizio J, Blum A, Gallagher-Gambarelli M, Molens J-P, Chaperot L, Plumas J. TLR7 stimulation in human plasmacytoid dendritic cells leads to the induction of early IFN-inducible genes in the absence of type I IFN. Blood. 2009;114:1794–802. doi: 10.1182/blood-2009-04-216770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lasagni L, Francalanci M, Annunziato F, Lazzeri E, Giannini S, Cosmi L, et al. An alternatively spliced variant of CXCR3 mediates the inhibition of endothelial cell growth induced by IP-10, Mig, and I-TAC, and acts as functional receptor for platelet factor 4. J Exp Med. 2003;197:1537–49. doi: 10.1084/jem.20021897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Struyf S, Salogni L, Burdick MD, Vandercappellen J, Gouwy M, Noppen S, et al. Angiostatic and chemotactic activities of the CXC chemokine CXCL4L1 (platelet factor-4 variant) are mediated by CXCR3. Blood. 2011;117:480–8. doi: 10.1182/blood-2009-11-253591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castello G, Scala S, Palmieri G, Curley SA, Izzo F. HCV-related hepatocellular carcinoma: From chronic inflammation to cancer. Clin Immunol. 2010;134:237–50. doi: 10.1016/j.clim.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 27.Bonacchi A. Signal Transduction by the Chemokine Receptor CXCR3. ACTIVATION OF Ras/ERK, Src, AND PHOSPHATIDYLINOSITOL 3-KINASE/Akt CONTROLS CELL MIGRATION AND PROLIFERATION IN HUMAN VASCULAR PERICYTES. Journal of Biological Chemistry. 2001;276:9945–54. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- 28.García-López MA, Sánchez-Madrid F, Rodríguez-Frade JM, Mellado M, Acevedo A, García MI, et al. CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab Invest. 2001;81:409–18. doi: 10.1038/labinvest.3780248. [DOI] [PubMed] [Google Scholar]
- 29.Cheent K, Khakoo SI. Natural killer cells and hepatitis C: action and reaction. Gut. 2011;60:268–78. doi: 10.1136/gut.2010.212555. [DOI] [PubMed] [Google Scholar]
- 30.Cohen-Naftaly M, Friedman SL. Current status of novel antifibrotic therapies in patients with chronic liver disease. Therap Adv Gastroenterol. 2011;4:391–417. doi: 10.1177/1756283X11413002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hosomura N, Kono H, Tsuchiya M, Ishii K, Ogiku M, Matsuda M, et al. HCV-related proteins activate Kupffer cells isolated from human liver tissues. Dig Dis Sci. 2011;56:1057–64. doi: 10.1007/s10620-010-1395-y. [DOI] [PubMed] [Google Scholar]
- 33.Paik Y-H, Lee KS, Lee HJ, Yang KM, Lee SJ, Lee DK, et al. Hepatic stellate cells primed with cytokines upregulate inflammation in response to peptidoglycan or lipoteichoic acid. Lab Invest. 2006;86:676–86. doi: 10.1038/labinvest.3700422. [DOI] [PubMed] [Google Scholar]
- 34.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, et al. Hepatitis C core and nonstructural 3 proteins trigger toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology. 2004;127:1513–24. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 35.Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, et al. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48:914–22. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 36.Cusick MF, Yang M, Gill JC, Eckels DD. Naturally occurring CD4+ T-cell epitope variants act as altered peptide ligands leading to impaired helper T-cell responses in hepatitis C virus infection. Hum Immunol. 2011;72:379–85. doi: 10.1016/j.humimm.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang JH, Layden TJ, Eckels DD. Modulation of the peripheral T-Cell response by CD4 mutants of hepatitis C virus: transition from a Th1 to a Th2 response. Hum Immunol. 2003;64:662–73. doi: 10.1016/s0198-8859(03)00070-3. [DOI] [PubMed] [Google Scholar]
- 38.Penna A, Pilli M, Zerbini A, Orlandini A, Mezzadri S, Sacchelli L, et al. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology (Baltimore, Md ) 2007;45:588–601. doi: 10.1002/hep.21541. [DOI] [PubMed] [Google Scholar]
- 39.Losikoff PT, Self AA, Gregory SH. Dendritic cells, regulatory T cells and the pathogenesis of chronic hepatitis C. Virulence. 2012:3. doi: 10.4161/viru.21823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casrouge A, Decalf J, Ahloulay M, Lababidi C, Mansour H, Vallet-Pichard A, et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest. 2011;121:308–17. doi: 10.1172/JCI40594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hartling HJ, Gaardbo JC, Ronit A, Knudsen LS, Ullum H, Vainer B, et al. CD43 and CD83 regulatory T cells (Tregs) are elevated and display an active phenotype in patients with chronic HCV mono-infection and HIV/HCV co-infection. Scand J Immunol. 2012;76:294–305. doi: 10.1111/j.1365-3083.2012.02725.x. [DOI] [PubMed] [Google Scholar]
- 42.Foy E, Li K, Sumpter R, Loo Y-M, Johnson CL, Wang C, et al. Control of antiviral defenses through hepatitis C virus disruption of retinoic acid-inducible gene-I signaling. Proc Natl Acad Sci US A. 2005;102:2986–91. doi: 10.1073/pnas.0408707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon ACM, Ikeda M, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci US A. 2005;102:2992–7. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review) Oncol Lett. 2011;2:583–9. doi: 10.3892/ol.2011.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monteagudo C, Martin JM, Jorda E, Llombart-Bosch A. CXCR3 chemokine receptor immunoreactivity in primary cutaneous malignant melanoma: correlation with clinicopathological prognostic factors. J Clin Pathol. 2007;60:596–9. doi: 10.1136/jcp.2005.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu Q, Dhir R, Wells A. Altered CXCR3 isoform expression regulates prostate cancer cell migration and invasion. Mol Cancer. 2012;11:3. doi: 10.1186/1476-4598-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datta S, Novotny M, Pavicic PG, Zhao C, Herjan T, Hartupee J, et al. IL-17 regulates CXCL1 mRNA stability via an AUUUA/tristetraprolin-independent sequence. J Immunol. 2010;184:1484–91. doi: 10.4049/jimmunol.0902423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furuya M, Yoneyama T, Miyagi E, Tanaka R, Nagahama K, Miyagi Y, et al. Differential expression patterns of CXCR3 variants and corresponding CXC chemokines in clear cell ovarian cancers and endometriosis. Gynecol Oncol. 2011;122:648–55. doi: 10.1016/j.ygyno.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 49.Smith LS, Nelson M, Naik S, Woten J. Telaprevir: An NS3/4A Protease Inhibitor for the Treatment of Chronic Hepatitis C. Ann Pharmacother. 2011;45:639–48. doi: 10.1345/aph.1P430. [DOI] [PubMed] [Google Scholar]
- 50.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, et al. oceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1207–17. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yee HS, Chang MF, Pocha C, Lim J, Ross D, Morgan TR, et al. Update on the management and treatment of hepatitis C virus infection: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program Office. Am J Gastroenterol. 2012:669–89. doi: 10.1038/ajg.2012.48. quiz 690. [DOI] [PubMed] [Google Scholar]
- 52.Wasmuth J-C, Rockstroh JK, Hardy WD. Drug safety evaluation of maraviroc for the treatment of HIV infection. Expert Opin Drug Saf. 2012;11:161–74. doi: 10.1517/14740338.2012.640670. [DOI] [PubMed] [Google Scholar]
- 53.Lim JK, McDermott DH, Lisco A, Foster GA, Krysztof D, Follmann D, et al. CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J Infect Dis. 2010;201:178–85. doi: 10.1086/649426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J-K, Kim J-H, Shin HK. Therapeutic effects of the oriental herbal medicine Sho-saiko-to on liver cirrhosis and carcinoma. Hepatol Res. 2011;41:825–37. doi: 10.1111/j.1872-034X.2011.00829.x. [DOI] [PubMed] [Google Scholar]
- 56.Polyak SJ, Ferenci P, Pawlotsky J-M. Hepatoprotective and Antiviral Functions of Silymarin Components in HCV Infection. Hepatology (Baltimore, Md) 2012 doi: 10.1002/hep.26179. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colmenero J, Bataller R, Sancho-Bru P, Domínguez M, Moreno M, Forns X, et al. Effects of losartan on hepatic expression of nonphagocytic NADPH oxidase and fibrogenic genes in patients with chronic hepatitis C. Am J Physiol Gastrointest Liver Physiol. 2009;297:G726–34. doi: 10.1152/ajpgi.00162.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ishibashi H, Komori A, Shimoda S, Gershwin ME. Guidelines for therapy of autoimmune liver disease. Semin Liver Dis. 2007:214–26. doi: 10.1055/s-2007-979472. [DOI] [PubMed] [Google Scholar]
- 59.Wang Y, Gao J, Zhang D, Zhang J, Ma J, Jiang H. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132–44. doi: 10.1016/j.jhep.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 60.Davis GL, Albright JE, Cook SF, Rosenberg DM. Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 2003;9:331–8. doi: 10.1053/jlts.2003.50073. [DOI] [PubMed] [Google Scholar]