

Abstract
During cardiogenesis, a subset of epicardial cells undergoes epithelial-mesenchymal-transition (EMT) and the resulting epicardial derived cells (EPDCs) contribute to the formation of coronary vessels. Our previous data showed hypoxia inducible factor-1α (HIF-1α) expression at specific sites within the epicardium and support a link between hypoxia inducible factors (HIFs) and the patterning of coronary vasculogenesis. To better understand the autocrine role of HIFs in the epicardium, we transduced adenovirus mediated expression of constitutively active HIF-1α (AdcaHIF1α) into the embryonic avian epicardium where the vascular precursors reside. We found that introducing caHIF1α into the epicardial mesothelium prevented EPDCs from proper migration into the myocardium. In vitro collagen gel assays and ex vivo organ culture data further confirmed that infection with AdcaHIF1α impaired the ability of EPDCs to invade. However, the proficiency of epicardial cells to undergo EMT was enhanced while the movement of EPDCs within the sub-epicardium and their differentiation into smooth muscle cells were not disrupted by caHIF1α. We also showed that the transcript level of Flt-1 (VEGFR1), which can act as a VEGF signaling inhibitor, increased several fold after introducing caHIF1α into epicardial cells. Blocking the activation of the VEGF pathway in epicardial cells recapitulated the inhibition of EPDC invasion. These results suggest that caHIF1α mediated up-regulation of Flt-1, which blocks the activation of the VEGF pathway, is responsible for the inhibition of EPDC myocardial migration. In conclusion, our studies demonstrate that HIF signaling potentially regulates the degree of epicardial EMT and the extent of EPDC migration into the myocardium, both of which are likely critical in patterning the coronary vasculature during early cardiac vasculogenesis. These signals could explain why the larger coronaries appear and remain on the epicardial surface.
Keywords: Epithelial to mesenchymal transformation, Hypoxia inducible factor, Vascular growth factor, Epicardium, Invasion
Introduction
The epicardium is the last cardiac layer to emerge, arising from the pro-epicardial serosa to form the outermost layer of the heart. This tissue plays crucial roles during embryonic heart development, especially in coronary vasculogenesis (Hiruma and Hirakow, 1989; Mikawa and Fischman, 1992; Poelmann et al., 1993; Viragh and Challice, 1981; Winter and Gittenberger-de Groot, 2007). Concurrent with the formation of the epicardium, a subset of epicardial cells undergoes epithelial-mesenchymal transformation (EMT) and starts migration into the sub-epicardial matrix. Some of these epicardial derived cells (EPDCs) remain within the sub-epicardium whereas a subpopulation migrate farther into the compact zone of the myocardium. EPDCs have the ability to differentiate and give rise to a variety of cell types, including cellular elements of the coronary vasculature (Mikawa and Fischman, 1992; Olivey et al., 2004; Poelmann et al., 1993; Wessels and Perez-Pomares, 2004). In vivo and in vitro assays reveal the functions of some important signals directing coronary vessel formation, involving adhesion molecules, transcription factors and several growth factors (Lee et al., 2006; Morabito et al., 2001; Olivey and Svensson, 2010). These factors, which are important for epicardial EMT and further steps in coronary vessel formation, could inherently originate from the epicardium and EPDCs as well as from the myocardium (Olivey and Svensson, 2010). Although some elements involved in EMT and subsequent steps in epicardial cell differentiation and migration have been identified, the mechanisms that drive the specific spatial and temporal pattern of coronary vasculogenesis are largely unknown.
It has been proposed that hypoxia triggers coronary vascular development. A range of cellular responses to hypoxia is mediated by hypoxia-inducible factors (HIFs), a heterodimer composed of a constitutively expressed HIFβ subunit and an oxygen sensitive HIFα subunit (Wenger, 2002). To date, three HIFα genes and over 100 HIF regulated genes have been identified (Wenger et al., 2005). Components of the HIF complex have been described to be required for normal development and patterning of the cardiovascular system (Dunwoodie, 2009; Ramirez-Bergeron and Simon, 2001). Loss of hypoxia inducible factor-1α (HIF-1α) in the mouse severely disrupts myocardial and vascular endothelial development and embryos die around E10 (Iyer et al., 1998; Ryan et al., 1998). It was also reported that the absence of HIF1β (ARNT) in mouse results in abnormal cardiac morphogenesis and embryonic lethality by E10 (Adelman et al., 2000; Maltepe et al., 1997). Using the avian model, it was shown that environmental oxygen influences embryonic angiogenesis and hypoxia treatment causes myocardial and coronary artery anomalies and an increase of capillary density in hypoxic regions (Nanka et al., 2008). Our previous studies utilizing the hypoxia marker EF5 [2-(2-nitro-1H-imidazol-1-yl)-N-(2,2,3,3,3 pentafluoropropyl)acetamide] revealed the atrioventricular junction (AVJ), ventricular apex, and interventricular septum (IVS) are relatively hypoxic in embryonic hearts. Many of these regions corresponded to the sites where major coronary vessels develop. Furthermore, hypoxia indicators and nuclear labeling of HIF-1α were co-localized in these hypoxic regions (Wikenheiser et al., 2006). Altered HIF-1α expression levels in the myocardium disrupted normal patterning of coronary vessels, suggesting that differential levels of hypoxia within the embryonic myocardium modulate coronary vessel development through transcriptional regulation by HIF-1 (Wikenheiser et al., 2012; Wikenheiser et al., 2009).
Though the importance of myocardial HIF in regulating coronary vasculogenesis has long been accepted, the functions of epicardial-HIF during coronary vessel development have not been studied (Tomanek et al., 2003). To unravel the in vivo roles of epicardial-HIF signaling, we disturbed HIF-1α gene expression and analyzed the consequences in ovo, in ex vivo explant culture, and also in various in vitro systems. Here, we present evidence that HIF1 has a complex regulatory role during specific steps of epicardial development. While EMT was stimulated by the forced expression of constitutively active HIF-1α (caHIF1α), EPDCs transduced with caHIF1α displayed profoundly impaired invasion into and migration within the myocardium. Our findings support the potential for micro-environmental hypoxia via HIFs and the VEGF pathway to regulate both EMT and their ability to migrate within the myocardium.
Materials and Methods
Chicken and quail fertilized eggs
Fertile White Leghorn chicken (Gallus gallus domesticus) eggs were obtained from Case Western Reserve University’s Squire Valleevue Farm (Cleveland, OH, USA) or from Charles River (MA, USA). Fertile quail (Coturnix coturnix communis) eggs were purchased from (Boyd’s Bird Company, Inc. Pullman, WA, USA). Eggs were incubated in a humidified forced draft hatching incubator (G.Q.F. Manufacturing Co., Savannah, GA, USA) at 38°C to the appropriate stages. The embryos were staged according to the method of Hamburger and Hamilton (HH, Hamburger and Hamilton, 1951).
In ovo injection of engineered adenoviruses into the pericardial space and assessment of EPDC migration
Using a modification of a previously described method (Fisher et al., 1997; Fisher and Watanabe, 1996), 0.5 μl adenovirus containing AdcaHIF1α or AdGFP (1×109 pfu/ml) was injected into the pericardial space of the avian heart at stage HH 24 (ED 4), when the surface of the heart is largely covered by the epicardium. The AdcaHIF1α virus bears a constitutively active form of human HIF-1α (caHIF1α) and green fluorescent protein (GFP) sequence which were driven by a dual CMV promoter (Kelly et al., 2003). The caHIF1α contains a deletion of the oxygen-dependent degradation domain (residues 392–520) and two missense mutations (Pro567Thr and Pro658Gln). AdGFP containing the CMV promoter that drives expression of GFP was used as a control. After injection, eggs were returned to the incubator and embryos were harvested at stage HH26 (ED 5) and HH 28 (ED 5.5–6). GFP positive cells located in different regions and positions were scored as follows. Comparable areas were randomly chosen from non-serial sections for each group. The number of positive cells containing distinct DAPI stained nuclei within these areas were counted for each sample. At least 10 areas were scored for each different region and the average number was determined by the total number of positive cells divided by the total number of areas counted in each of six independent experiments for each stage (HH26 and HH28). DAPI positive cells at AVJ were also counted using the same method as described before.
Collagen gel assay and quantification of migration
The collagen gel assay was performed as previously described with some modifications (Dokic and Dettman, 2006). Briefly, neutralized collagen I gel was added to 24 well plates (700μl/well) and allowed to solidify at 37°C in the incubator for 60 min. Stage HH25 (ED 4.5) avian hearts were harvested in sterile PBS and placed onto the surface of the gels which were covered by serum free M199 medium. After 24 h culturing at 37°C, 5% CO2, hearts were removed and epicardial monolayers were allowed to grow out on the surface of the gel in serum free medium for 24 h. For virus infection experiments, monolayers were incubated overnight with serum free M199 containing AdGFP or AdcaHIF1α (1×109pfu/ml) and the medium was then replaced with M199 supplemented with 10% fetal bovine serum (FBS) or 10% FBS+50 ng/ml VEGFA to trigger migration. After 36 h incubation, gels were fixed in 4% paraformaldehyde (PFA) in 1xPBS, washed and embedded in 1.5% agarose. Embedded samples were equilibrated in 30% sucrose (w/v) overnight at 4°C and maintained at −80°C. Cryosections (20μm) were further stained with DAPI and imaged on a Leica DM2500 microscope. For inhibition experiments, epicardial monolayers were cultured in 10% FBS M199 medium containing DMSO (Fisher Scientific, MA, USA), DFO (150 μM, Sigma-Aldrich, MO, USA), SU5416 (15 μM, Calbiochem, CA, USA), MAZ51 (5 μM, EMD4 Biosciences, CA, USA), BSA(30 μg/ml, Fisher), Fc-sFlt1 (30 μg/ml, R&D systems, MN, USA) for 2 days before analysis.
To evaluate cell migration, the Image J software (NIH) was used to measure the distance for each GFP positive (for adenovirus experiments) or DAPI stained (for inhibition experiments) cell that migrated from the surface into the gel. At least 15 comparable non-serial sections were chosen and analyzed for each group per independent experiment. For each treatment group, the average distance of migration was calculated by the total distance of cell migration divided by the total number of cells counted. Three independent experiments were performed for each treatment.
Organ culture assay
For ex ovo migration assays, stage HH25 (ED 4.5) avian hearts were excised from embryos in sterile PBS and cultured overnight at 37°C in serum free M199 medium added adenovirus (AdGFP or AdcaHIF1α, 1×109pfu/ml). Then hearts were placed in fresh M199 medium supplemented with 10%FBS and cultured for 1.5 days before harvest. For in vitro inhibition experiments, dissected stage HH25 hearts were placed in serum-free M199 medium with adenovirus (AdGFP, 1×109pfu/ml) and also control [DMSO, BSA (30 μg/ml)], DFO (150 μM), or inhibitors [SU5416 (15 μM), MAZ51(5 μM), Fc-sFlt1 (30 μg/ml)] and incubated at 37°C overnight, then medium was replaced with 10%FBS M199 added the same control or inhibitors and hearts were continually cultured for 1.5 days before fixation and cutting.
Cell culture and adenovirus infection
For primary avian epicardial cell culture, stage HH25 (ED 4.5) chicken or quail hearts were dissected and explanted into tissue culture-treated dishes with M199 medium containing 10% FBS. Hearts were removed after 24 h and the attached epicardial monolayers were allowed to grow in fresh medium. Immortalized cells derived from E13.5 mouse hearts (Obtained from Dr. Barnett, Vanderbilt University, USA) were propagated at 33°C on collagen I coated dishes in DMEM supplemented with 10% FBS, insulin-transferrin-selenium (Invitrogen) and 10 units/ml mouse IFN-gamma (Peprotech, NJ, USA) (Austin et al., 2008). Before starting the next experiment, cells were transferred to DMEM medium without ITS and IFN-γ and with 5% FBS. To test the HIF-1α protein levels, mouse epicardial cells were treated with DFO (150 μM) or incubated under 1% O2 (Heracell 150 inbubators, Thermo scientific, IL, USA) for 2 h before harvest. Immortalized rat epicardial cells (Obtained from Dr. Bader, Vanderbilt University, USA) were cultured in DMEM medium supplemented with 10% FBS (Dokic and Dettman, 2006; Eid et al., 1992). All cells were cultured at 37°C in a 5% CO2 culture incubator. Prior to infection, cells were washed with sterile PBS and then incubated with serum free medium containing adenovirus (AdGFP or AdcaHIF1α, 1×109pfu/ml) overnight at 37°C. GFP expression in cells was monitored to determine the transfection efficiency. Virus infected primary quail epicardial cells were incubated with recombinant human TGFβ1 (final concentration 250 pM, Peprotech) for 48 h to stimulate smooth muscle cell differentiation.
Immunohistochemistry
Dissected hearts or heart explants were fixed in fresh 4% PFA for 1 h at room temperature, then washed in 1XPBS and equilibrated in 30% sucrose overnight at 4°C before embedding in OCT. 10μm thick sections were collected on treated slides (Plus, Fisher) and immunostained with antibodies. For HIF-1α and VEGFA staining, primary antibody was detected with goat anti-rabbit IgG secondary antibody conjugated with biotin (Vector, Burlingame, CA) at 1:400 (HIF-1α) or 1: 100 (VEGFA) dilution, and the signal was amplified with the TSA systems with fluorescein tyramide signal (Perkin Elmer, Boston, MA) (HIF-1α) or DAB system (Vector) (VEGFA). VEGFA staining was quantified using Image J software. Cells were washed in PBS and fixed with 4% PFA for 10min at room temperature, then washed in PBS and permeabilized with 0.2% Triton X-100 for 5 min. After blocking with 2% bovine serum albumin in PBS for 1 h, cells were incubated with diluted primary antibodies overnight at 4°C and detected with appropriate secondary antibodies.
The following antibodies were used: anti-ZO1 antibody (1:100, Invitrogen), anti-β catenin antibody (1:50, Cell Signaling, MA, USA), anti-HIF-1α (1:600, gift of Dr. Faton Agani), anti-PH3 antibody (1:100, Cell Signaling), anti-VEGFA antibody (1:50, Neomarkers, MI, USA), anti-SM22α antibody (1:200, Abcam, MA, USA), anti-α smooth muscle actin conjugated with Cy3 (1:200, Sigma-Aldrich, MO, USA). MF-20 antibody (1:50) were obtained from the Developmental Studies Hybridoma Bank created under the auspices of the NICHD and maintained by the University of Iowa Department of Biology, Iowa City, IA 522422. Texas Red-phalloidin (1:40, Invitrogen) was used to label F-actin to delineate the cellular cytoskeleton. Terminal deoxynucleotidyltransferase (TdT) analysis (TUNEL) was carried out on frozen sections or fixed cells and developed with DAB according to the manufacturer’s protocol (In situ cell death detection kit, Roche, IN, USA). Photomicrographs were captured with an inverted fluorescence microscope (Leica DM2500) and QCapture Pro software.
For quantification of cell numbers, comparable areas were randomly chosen from each group and numbers of positive cells were counted. At least 6 areas were scored for each sample.
Western blotting
Proteins were quantified using a Bradford assay (Pierce Biotech, IL, USA) and equal amounts were loaded onto 10% SDS-PAGE gels (Bio-Rad, CA, USA) and transferred to a PVDF membrane (Millipore, MA, USA). Membranes were blocked for 1 h at room temperature followed by incubation with primary antibodies overnight at 4°C. The following antibodies were used: anti-β actin (1:10,0000, Sigma), anti-E-cadherin (1:1000, BD Biosciences, CA, USA), anti-HIF-1α (1:500, Novus, CO, USA). Then the blots were incubated with appropriate secondary anbibodies for 1 h at room temperature. Signals were then visualized with a HRP detection kit (Pierce).
Real time RT-PCR
Total RNA was isolated from cultured avian epicardial cells using the RNeasy mini kit (Qiagen, CA, USA) and 1 μg of total RNA was used for cDNA synthesis with the iscriptcDNA synthesis kit (Bio-Rad) according to the manufacturer’s instructions. Real-time PCR was performed using SYBR (SYBR supermix, Bio-Rad). 18S was used as the internal control. The sequences of the primers were as follows:
| Gene | Sense primer (5′-3′) | Anti-sense primer (5′-3′) |
|---|---|---|
|
| ||
| 18S | ATGGCCGTTCTTAGTTGGTG | GAACGCCACTTGTCCCTCTA |
| Integrin α4 | TCCCAGAAACTGTAATGCACCGGA | CCAGGTGCAACTGAATGGTTGCTT |
| Integrin β1 | AAGTCGAGACAAATTGCCACAGCC | AGAACCAGCAGTCATCAACGTCCT |
| Twist1 | ATTCCCAAGAGGTCGTGCCTATGA | GCTTTGGTTGGGTGCTTTGCTTTC |
| Twist2 | TGAACCTTCGCACCATCTCTCCAA | ACCAAACACCCAGACAAACCACAC |
|
| ||
| VEGFA | TCTGCAGGACAATTGAGACCC | AACCCGCACATCTCATCAGAG |
| Flt-1 | TAACGCACCGAGAGACCAACACAA | GCTGCCTTGACAGTGCAGTTGATT |
| Flk-1 | TGGGATGGTTCTTGCATCTGA | GACTCATTGCTTTTGCTGGGC |
Transwell experiments
Adenovirus infected primary quail epicardial cells were starved in serum free medium for 18 h and seeded at a density of 1 × 104 cells/well onto the upper insert (non-coated) of the transwell chamber (Corning, MA, USA). The bottom chamber was filled with M199 medium (700μl/well) supplemented with 10%FBS or growth factors: VEGFA (15 ng/ml, Invitrogen) or TGFβ1 (5 ng/ml) as the chemical attractant. Cells were permitted to migrate for 12 h. Then the non-migrated cells on the upper side were removed carefully with a cotton swab and the filters were fixed with 4% PFA before staining with DAPI solution. Migrated cells were counted in five randomly chosen fields under fluorescent microscope at 100X magnification. Three independent experiments were performed for each treatment.
Statistical analysis
Data are presented as means ± SEM. All Statistical analysis was performed using student’s paired t test. P value < 0.05 was considered significant.
Results
Expression of constitutively active HIF-13 in the epicardium inhibits EPDC migration into the myocardium
During avian embryo development, the heart is largely covered by epicardial cells by stage HH24 (Hiruma and Hirakow, 1989). Endogenous HIF-1α expression in the epicardium and subepicardium from stage HH24 to stage HH28 was examined by immunofluorescence staining (Figs. 1, S7). Few ventricular epicardial or sub-epicardial cells were HIF-1α positive at stage HH24 to stage HH28 (Figs 1A–D, 1M-P). Most of the HIF-1α positive staining in the epicardium or subepicardium was concentrated at the atrioventricular junction (AVJ) at these stages (Figs. 1E–L). Besides the staining in the epicardium and subepicardium, an increase of the number of HIF-1α positive nuclei in the myocardium was also observed during stages 26–28 (Figs. 1E–P). In order to investigate the intrinsic role of epicardial HIF-1 during EMT and subsequent EPDC migration in vivo, we utilized a replication defective adenovirus carrying a mutated form of HIF-1α that eliminates key regulatory oxygen-sensitive sites inhibiting protein turnover and rendering it constitutively active (caHIF1α) (Kelly et al., 2003). We injected the caHIF1α co-expressing a GFP reporter gene or the gene for GFP alone (negative control) into the pericardial space of avian embryos at stage HH24. This injection strategy ensured that the epicardium, but not the underlying myocardium was infected. We were able to determine whether the gain of function of HIF-1α (caHIF1α) had an effect on EPDC migration by comparing the location in histological sections of AdcaHIF1α- and the AdGFP-infected cells; both sets of infected cells expressed the GFP tag. We observed that avian hearts transduced with AdcaHIF1α had reduced numbers of cells expressing the GFP marker within the myocardium (Fig. 2). In quails this defect was first identified at stage HH26 (Figs. 2A, B), and became more obvious at later stages (Figs. 2C, D). A similar deficiency of GFP+ cell numbers in the myocardium was also detected in another avian species, chicken embryos, injected with caHIF1α (Figs. 2E, F). GFP+ cells were quantified in comparable areas within the myocardium of AdcaHIF1α- and AdGFP- infected quail embryos at various developmental stages. We discovered that the most significant decrease in the number of EPDCs infected with AdcaHIF1α occurred in the ventricular myocardium (P<0.01, Fig. 2G). This reduction was less prominent in the atrial myocardium than in the myocardium of the ventricle and at the AVJ (Fig. 2G).
Fig 1. HIF-1α expression in the epicardium and subepicardium.

Costaining for HIF-1α and the myocardium marker MF20 reveals the HIF-1α staining in the epicardium and subepicardium during stage HH24-28. HIF-1α expression was observed in a few ventricular epicardial cells at stage HH24 (A–D) and stage HH28 (M–P). (E–L) HIF-1α is especially intense in the epicardium and subepicardium at the AVJ at stage HH26 (E–H) and continued to be nuclear-localized in this area to stage HH28 (I–L). HIF-1α was also detected in myocardial cells (E–P). Dashed lines delineate the border between the myocardium and the epicardium/subepicardium. Arrows point to HIF-1α positive epicardial or subepicardial cells and arrowheads point to HIF-1α positive myocardial cells. Bar = 20 μm for A–H and M–P, Bar = 40 μm for I–L.
Fig 2. Few EPDCs expressing constitutively active HIF-1α were found in the myocardium after in ovo transfection.
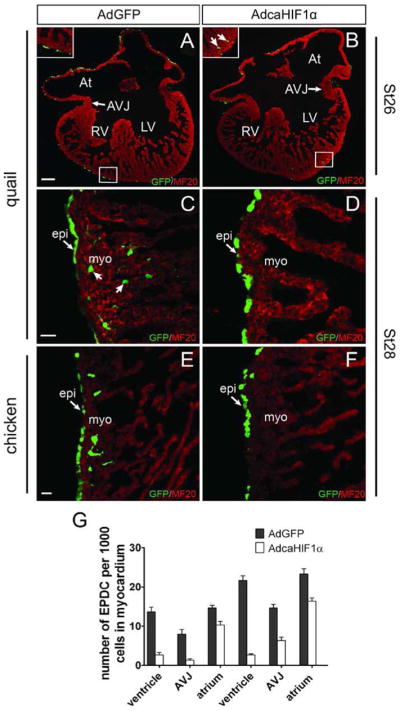
Sections of virally transfected embryonic quail (A–D) or chicken (E, F) hearts were stained with anti-MF20 (red) antibody to label myocardium. Many labeled EPDCs were found in the myocardium of control AdGFP transfected hearts (A, C, E), while only a few AdcaHIF1α-infected cells (green) were detected in the myocardium (B, D, F). Arrows point to GFP+EPDCs. At: atrium; AVJ: atrioventricular junction; LV: left ventricle; RV: right ventricle; epi: epicardium; myo: myocardium. Bar = 100 μm for A and B and 25 μm for C–F. (G) GFP+ cell numbers detected in the myocardium of control AdGFP- and AdcaHIF1α-infected hearts, n=6. * P < 0.05; ** P < 0.01.
We further performed an ex vivo assay to examine whether we can observe similar defects in this more accessible system where the viral infection was more consistent. Quail hearts were dissected and infected with adenovirus in vitro, then processed for immunohistochemistry. The results confirmed that AdcaHIF1α greatly inhibited the migration of EPDCs into the myocardium in this ex vivo environment (Figs. 3A, B, D). Furthermore, we observed similar inhibition of EPDC migration in cultured hearts treated with desferrioxamine (DFO), a hypoxia-mimetic agent that induces HIF-1α protein (Fig. 3C, D). This defect was also reproduced in AdcaHIF1α infected chicken embryo heart explants cultures (data not shown).
Fig 3. In vitro experiments demonstrated that expression of HIF-1α inhibited the invasion of epicardial cell derivatives.

(A–D) In the heart explant culture using quail hearts, penetration into the myocardium was significantly deeper for EPDCs with the control AdGFP (A) than those infected with the AdcaHIF1α (B) or treated with DFO (C); (D) Quantitative data show the number of GFP+ EPDCs in ventricular myocardium significantly decreased in AdcaHIF1α infected or DFO treated group compared to the AdGFP control. EPDCs were marked by GFP (green) and the depth reached by EPDCs is indicated by dashed lines. Scale bar represents 25 μm for A –C. (E–J) Sections of collagen gels containing adenovirus infected (E, F) or DFO treated (H, I) epicardial cell monolayers (from stage HH25 quail hearts) were stained with DAPI. Dashed lines delineate the surface of the gel, arrow indicates the direction of invasion by the epicardial cells into the collagen gel and arrowheads point to the migrated cells without GFP. Bar = 20 μm for E–F and H–I; (G) Quantification of the average distance of virus infected quail EPDC migration into gels (48 h, n=3); (J) Quantification of the average migration distance of quail EPDC treated with DMSO control or HIF-1α inducer DFO (48 h, n=3). (K) Western blotting result showing HIF-1α protein levels in mouse epicardial cells treated with hypoxia, DFO or infected with AdcaHIF1α. Actin was used as the loading control.
To test the intrinsic biological effects of caHIF1α epicardial cells devoid of influences from other types of cells which exists in ovo, in vitro 3-dimensional collagen gel assays were performed. Epicardial monolayers derived from stage HH25 quail hearts were cultured on collagen gels and infected with adenovirus. GFP was used to track epicardial cell invasion into the matrix. GFP+ cells from AdGFP infected control cultures were found to penetrate deep into the gel (Fig. 3E). In contrast, GFP+ cells from AdcaHIF1α infected cultures failed to infiltrate the collagen gel (Fig. 3F). Importantly, we observed that uninfected GFP- cells detected by DAPI staining were able to migrate extensively even in the presence of AdcaHIF1α infected cells (arrowheads, Fig. 3F), indicating that the inhibition was cell autonomous. Measurements of the distances traveled by GFP+ EPDCs in the collagen gels demonstrated a statistically significant decrease (about 60%, 35.4±5.6 μm for AdGFP infected cells and 16.4±5.5 μm for AdcaHIF1α control) in the invasion of AdcaHIF1α infected cells (P < 0.01, Fig. 2G). Furthermore, monolayers treated with DFO also exhibited impaired invasion of epicardial cells with cell migration distances significantly decreased by 50% (P < 0.05, Figs. 3H–J). Western blot of mouse epicardial cell lysates indicated that AdcaHIF1α infection and the DFO are effective methods mimicking the hypoxic condition by inducing the accumulation of HIF-1α protein (Fig. 3K). These ex ovo and in vitro data closely mirrored our in ovo experiments and strongly indicate that over-expression of stable HIF-1α in the epicardium impairs the ability of epicardial-derived cells to travel a distance into matrices in a complex active or inactive myocardium or in a simple collagen gel.
Forced expression of HIF-1α does not affect cell proliferation and survival but stimulates epithelial-mesenchymal transformation
There are many potential explanations for fewer AdcaHIF1α infected cells in the myocardium compared to AdGFP cells. AdcaHIF1α infected cells (1) cannot proliferate or survive before they migrate, (2) are unable to migrate at all, (3) are not capable of undergoing EMT, (4) fail to respond to extrinsic signals, and/or (5) may undergo EMT and travel within the epicardial matrix but are incapable of migrating to and/or within the myocardium.
The total number of cells in the epicardium and subepicardium at AVJ was evaluated by DAPI staining and no obvious difference was detected between the control and AdcaHIF1α group, suggesting that cell proliferation and survival are not influenced by AdcaHIF1α infection (Fig. 4A). To further determine whether the reduction in number of AdcaHIF1αGFP+ cells within the myocardium resulted from reduced cell proliferation of epicardial cells or EPDCs, quail heart sections were stained with an anti-phosphohistone H3 (PH-3), a sensitive marker for mitosis. There was no significant difference in the number of PH-3 positive cells in the epicardium infected with either AdGFP or AdcaHIF1α (Figs. S1A, E, I). Likewise, the data showed no significant difference in TUNEL+ immunostained apoptotic cell numbers between the AdGFP and AdcaHIF1α infected hearts (Figs. S1B, F, I). Furthermore, in vitro cultured quail epicardial cells displayed no alteration of proliferation or survival in AdcaHIF1α transfected cells (Figs. S1C, D, G, H, J). According to these results, changes in cell proliferation and survival could not explain the reduced numbers of HIF-1α transduced EPDCs within the myocardium.
Fig 4. EPDCs infected with AdcaHIF1α invaded the subepicardium.

Cell migration into and within the sub-epicardium was not affected even within the thick subepicardium at the AVJ (A–C). (A) Total cell numbers in the epicardium and subepicardium at AVJ counted in DAPI stained sections were not significantly different between control and AdcaHIF1α infected hearts. (B, C) GFP positive cells were found in the thick sub-epicardium of the AVJ both in AdGFP and AdcaHIF1α infected quail hearts with no obvious differences in numbers or location. epi: epicardium; se: sub-epicardium. Bar = 25 μm for B and C. (D) Quantitative analysis indicated that the number of AdcaHIF1α infected cells located in the sub-epicardium of the AVJ was not significantly different from the number of AdGFP infected cells assayed at two stages, n=6. (E) Transwell experiments demonstrated that the initial response to growth factor stimuli, the release from neighboring epicardial cells and the short migration through the 8 μm pore through the 10 μm thick Transwell membrane, was not altered in epicardial cells with caHIF1α. SFM: serum free medium.
Based on our immunostaining data, EPDC movement into the connective tissue of the subepicardium was grossly normal despite the fact that their migration into and within the myocardium was inhibited. In both AdcaHIF1α and control AdGFP infected quail hearts, GFP+ cells were detected populating the width of the subepicardial space of the AVJ region even where the epicardium is especially thick (maximal thickness is around 75μm, Figs. 4B, C). There were no significant differences in the number of EPDCs in the subepicardium of the AVJ between the control AdGFP and the AdcaHIF1α transduced hearts (Fig. 4D). While evaluation of GFP+ cell migration within the subepicardium covering the ventricle and atrium was difficult due to the thinner epicardium than at the AVJ, no discernable gross differences were observed. To examine the intrinsic motility of AdcaHIF1α cells, we utilized a Transwell assay thus eliminating matrix invasion requirements and exclusively assayed their response to growth factor stimuli. Infected primary cultures of quail epicardial cells were seeded onto the un-coated top chambers and exposed for 12 h to the medium in the lower wells containing FBS, vascular endothelial growth factor A (VEGFA, 15 ng/ml) or transforming growth factor beta-1 (TGF-β1, 5 ng/ml) (Han et al., 2012; Kim et al., 2006). Cells that migrated through the pores of the filter to the underside were microscopically quantified. There was no significant difference in the number of migrated cells in the AdGFP and AdcaHIF1α cultures, suggesting that the expression of caHIF1α had no influence on the initial chemotactic responses of the epicardial cell, that is, their ability to release and migrate a limited distance in the direction of the source of the growth factors (Fig. 4E).
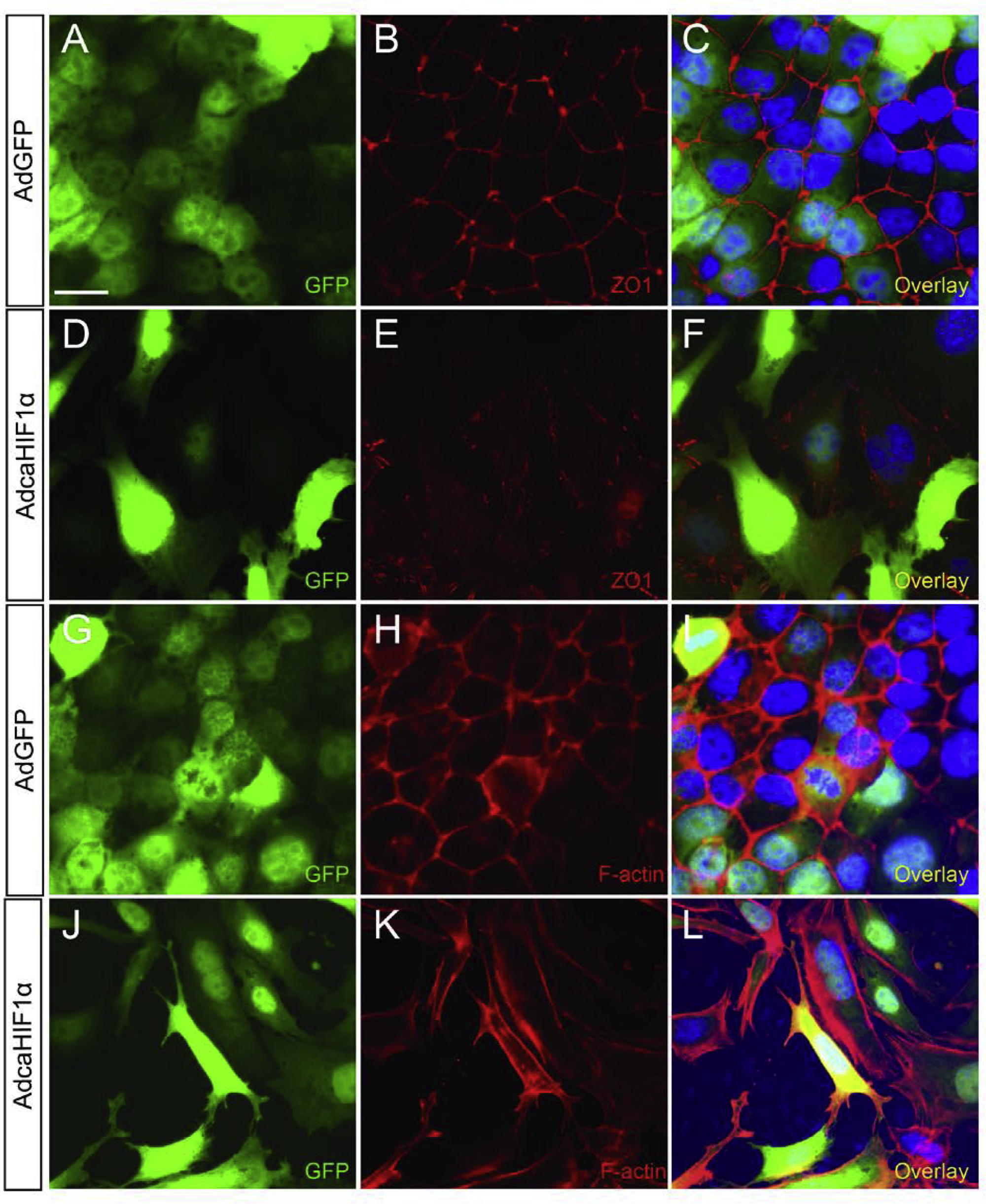
As epicardial precursors contribute to the coronary vasculature, EPDCs need to undergo EMT to acquire the ability to migrate. Unchanged EPDC invasion into and within the subepicardium and responses to growth factor stimuli directed us to postulate that epicardial EMT was not disrupted due to the expression of caHIF1α. To test this hypothesis, quail epicardial monolayers from stage HH25 hearts were cultured and transfected with AdGFP and AdcaHIF1α. Since loss of cell-cell adhesion is an important early step in epicardial EMT, we stained the cells with an antibody to zonula occludens-1 (ZO-1), a marker for tight junctions that directly interact with the actin cytoskeleton. Primary epicardial cells infected with AdcaHIF1α lost strong immunostaining for ZO-1 at cell-cell borders when compared to the AdGFP-infected cells (Figs. 5A–F). This feature was replicated in chicken epicardial cell cultures (data not shown). Moreover, we detected a decreased expression of ZO-1 and elongation of cell shape in immortalized mouse embryonic epicardial cells infected with AdcaHIF1α (Figs. S2A–F). Phalloidin staining further indicated that stress fibers were increased in cells with forced expression of caHIF1α when compared to controls (Figs. S2G–L). In the rat adult epicardial cell culture system, we further observed an even distribution of β-catenin at cell membranes at cell-cell junctions in AdGFP infected control cells (Figs. S3A–C), whereas robust staining was detected in the cytoplasm in cells expressing caHIF1α (Figs. S3D–F). These findings suggest that forced expression of caHIF1α results in the altered expression and distribution of proteins important in cell-cell connections, a characteristic of cells undergoing EMT.
Fig 5. caHIF1α expression promotes characteristics of EMT in epicardial cells.

(A–F) Loss of tight junctions in AdcaHIF1α treated primary quail epicardial cells. Quail monolayers derived from stage HH25 hearts were incubated with AdGFP (A–C) or AdcaHIF1α (D–F) and fixed for examination of ZO-1 (red) immunostaining. AdcaHIF1α infected cells displayed loss of cell-cell contacts and ZO-1 expression at cell-cell borders (E, F). Scale bar represents 30 μm for A–F. (G) Quantitative RT-PCR analysis of expression of some EMT associated markers in chicken epicardial cells transfected with AdGFP and AdcaHIF1α. * P < 0.05; ** P < 0.01.
Previous studies supported that EMT is accompanied by enhanced expression of twist and a decrease of E-cadherin, while integrin α4 is reported to be an inhibitor of EMT (Dettman et al., 2003; Lee et al., 2006). We further examined the transcript levels of some important EMT markers in primary chicken epicardial monolayer cultures by quantitative real-time PCR. In AdcaHIF1α infected cells, there were statistically significant changes in the expression levels of a subset of EMT markers including the increased expression of twist2 and reduced levels of integrin α4 and β1 transcripts, as compared to the AdGFP controls (Fig. 5G). No significant changes were observed in the expression of twist1 and vimentin. Furthermore, E-cadherin protein levels were decreased in rat adult epicardial cells transfected with AdcaHIF1α (Fig. S3G). The alteration of the tested EMT markers is consistent with the loss of epithelial characteristics detected from immunostaining experiments, indicating that expression of caHIF1α in epicardial cells promotes EMT. Therefore because EMT itself is not inhibited but in fact enhanced, the defective invasion/migration of HIF-1α transduced epicardial cells may be due to changes that inhibit cells from migrating substantial distances into and interacting with substrates after they undergo EMT.
Differentiation of EPDCs into smooth muscle cells was not affected in cells infected with AdcaHIF1α
Another point to consider while investigating the role of HIF1 in regulating the commitment of EPDC to coronary vasculogenesis is the differentiation of the EPDC into diverse cell types. We utilized an in vitro culture system to address whether EPDCs with caHIF1α can give rise to vascular smooth muscle cells (VSMCs). It has been shown that TGFβ1 triggers epicardial EMT and the differentiation of epicardial cells into VSMCs (Austin et al., 2008; Olivey et al., 2006). Here, quail monolayers were treated with TGFβ1 for 48 h and then analyzed for differentiation into VSMCs by staining with two different markers: anti-smooth muscle actin (SMA) and SM22α. Expression of SMA or SM22α was observed in both AdGFP and AdcaHIF1a infected cells in response to TGFβ1 (Figs. 6A–F, S4A–F), suggesting that forced expression of caHIF1α did not disrupt the differentiation of EPDCs into VSMCs. Quantification further confirmed no significant difference between AdGFP and AdcaHIF1α groups in terms of the percent of SMA+GFP+ cells (Fig. 6G, S4G). In summary, caHIF1α expression in epicardial cells had no effect on EPDC differentiation into vascular smooth muscle cells.
Fig 6. EPDC differentiation into smooth muscle cells was not disrupted by expression of caHIF1α.

(A–F) TGFβ1 (250 pM) induced SMA expression (red) in stress fibers in both AdGFP (A–C) and AdcaHIF1α treated quail epicardial cells (D–F). Scale bar represents 30 μm for A–F. (G) Quantification of the percent of SMA GFP positive cells in total GFP+ cells.
The up-regulation of Flt-1 in epicardial cells expressing constitutively active HIF1α contributes to their altered migration
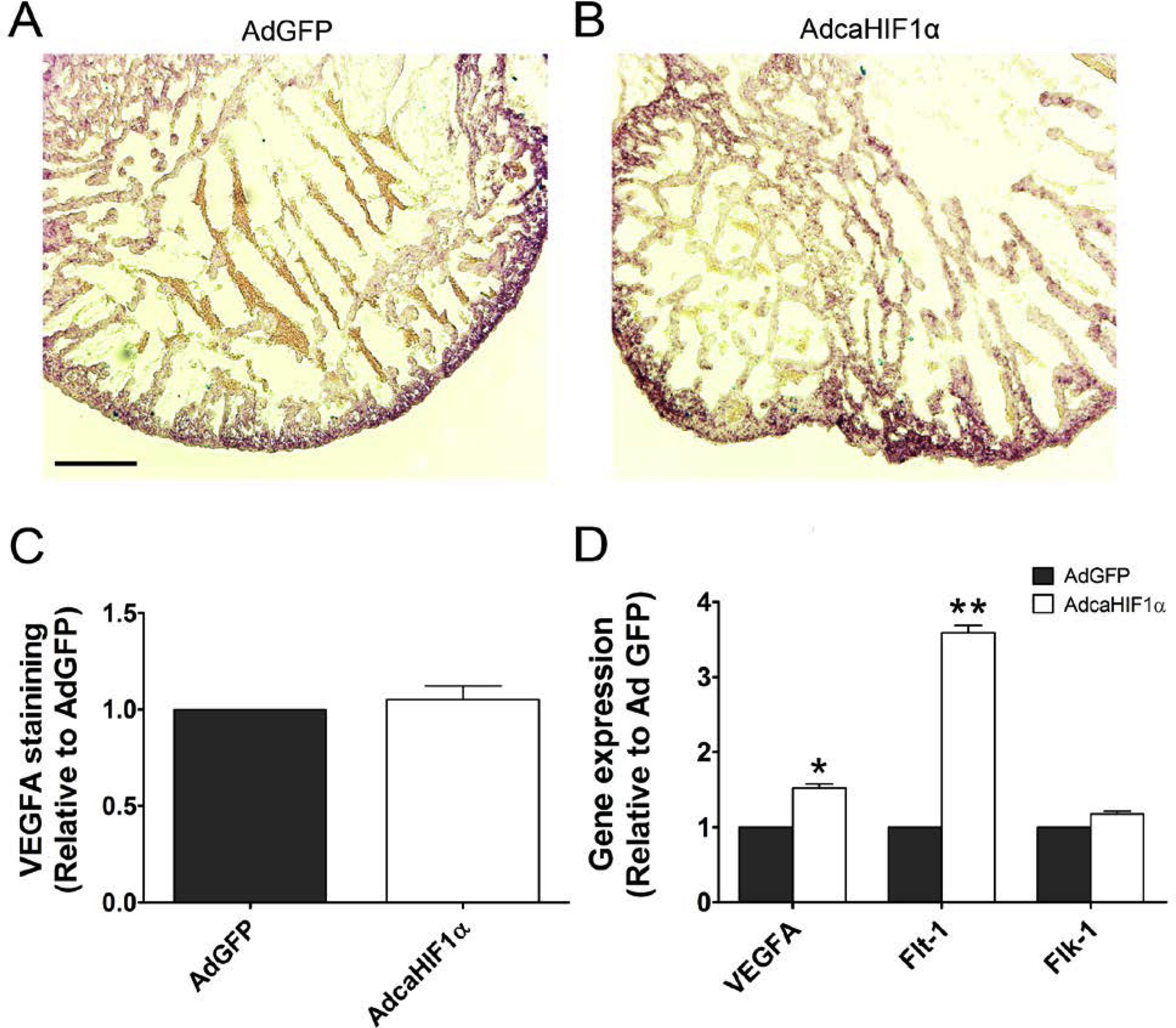
Many of the pathways that are involved in epicardial EMT and in directing EPDC migration and differentiation are activated by growth factors, including VEGF (Olivey and Svensson, 2010). VEGFA and vascular endothelial growth factor receptor 1 (VEGFR1, Flt-1) are known HIF1 target genes (Forsythe et al., 1996; Gerber et al., 1997; Wenger et al., 2005). We used quantitative real time RT-PCR to examine the expression levels of VEGF and VEGF receptors in primary cultures of chicken epicardial cells with caHIF1α expression. We found that cells infected with AdcaHIF1α expressed 3.7 fold more Flt-1 transcript levels than those infected with the control AdGFP (Fig. 7A). By contrast, transcript levels of Flk-1 did not increase significantly (Fig. 7A). VegfA expression was also up-regulated, although modestly (1.5 fold, P<0.05) (Fig. 7A). Immunohistochemical analysis further confirmed that VEGFA protein levels were not markedly increased nor were patterns of expression changed by AdcaHIF1α (Figs. S5A–C). Primary cultures of quail epicardial cells demonstrated the same expression pattern for these transcripts as the chicken epicardial cells (Fig. S5D). Given that Flk-1 mediates most of the responses to VEGF and the soluble form of Flt-1 acts as a decoy receptor that sequesters VEGF from Flk-1 (Shibuya, 2006), the above data imply that the up-regulation of Flt-1 is a possible underlying mechanism associated with the aberrant EPDC migration within the myocardium. On this basis, we proposed that the increased expression of Flt-1 induced by AdcaHIF1α negatively regulated the VEGF intracellular signaling mediated by Flk-1, which resulted in the impaired invasion of EPDCs into and within the myocardium.
Fig 7. Increased levels of Flt-1 that inhibits Flk-1 signaling could be the mechanism for the inhibition of EPDC migration into the myocardium.

(A) Expression of important factors involved in the VEGF signaling pathway was evaluated using qRT-PCR analysis in chicken epicardial cells, n=3. The level of Flt-1 mRNA expression was much higher in AdcaHIF1α transfected cells compared with the AdGFP transfected cells. (B–K) Data from in vitro collagen gel assays (B–F) or heart explant cultures (G–K) showed that chicken epicardial cells invaded efficiently in the vehicle control DMSO (B, G), with the BSA alone control (E, J), and even with the addition of the VEGFR3 specific inhibitor MAZ51 (D, I). In contrast, the invasion of epicardial cells into gels or the myocardium was inhibited by the Flk-1 specific inhibitor SU5416 (C, H), or Fc-sFlt1 which was utilized to trap VEGF ligands (F, K). Treatments are indicated at the top of the panel. Scale bars represent 20 μm for B–F and 25 μm for G–K. (L) Quantification of average migration distances of epicardial cells in collagen gel assay (Normalized to comparable control group: DMSO or BSA group). (M) Collagen gel assay indicated that inhibition of EPDC migration was partially rescued by VEGFA (50 ng/ml) treatment.
To test our hypothesis, we used the collagen gel assay to examine the in vitro migration of EPDCs in the presence of inhibitors for the VEGF signaling pathway. Chicken epicardial cells were cultured in 10% FBS medium with or without VEGF signaling pathway inhibitors. FBS significantly stimulated epicardial cell invasion into collagen gels in the DMSO vehicle control (Fig. 7B). In contrast, when monolayers were treated with a specific intrinsic inhibitor (SU5416) of Flk-1 tyrosine kinase activity, the migration of cells was strongly reduced (Fig. 7C). The average distance traveled from the surface by cells treated with SU5416 was 35% that of the DMSO control group (Fig. 7L). The recombinant soluble Fc-sFlt1 chimera was utilized to block extrinsic VEGF signaling by trapping VEGF and further preventing its binding and activation of cellular receptors (Ambati et al., 2006; Roberts et al., 2004). We observed that addition of Fc-sFlt1 inhibited the EPDC invasion stimulated by FBS (Figs. 7E, F). Although the effect was not as robust as with SU5416, the migration distance was significantly diminished (by 40%) (Fig. 7L). MAZ51, an inhibitor specific for VEGFR3 when used at low concentration had no impact on the cell invasion triggered by FBS (Fig. 7B, D), suggesting that the inhibition was specifically taking effect through signaling pathways mediated by VEGF:Flk-1/Flt-1.
To replicate our experiments in an organ model, the ex vivo whole heart explant culture system was used to test the effect of VEGF inhibitors on EPDC migration in the myocardium. The results demonstrated that SU5416 (Fig. 7H) and Fc-sFlt1 (Fig. 7K) treatment blocked the EPDC invasion into the myocardium when compared to DMSO (Fig. 7G) and BSA (Fig. 7J) control groups. Moreover, we again did not identify any inhibition of migration in the MAZ51-treated hearts (Fig. 7I). The collagen gel system was further used to examine whether VEGFA can rescue the inhibition of cell invasion. Evaluation of the migration distance clearly indicated that the average migration distance significantly increased in VEGFA treated AdcaHIF1αGFP+ cells compared to the AdcaHIF1α control cells (Figs. 7M, S6). Together these data support the hypothesis that enhanced expression of Flt-1 in EPDCs as a result of expressing caHIF1α inhibited cell invasion into and within the myocardium via the inhibition of the VEGF pathway.
Discussion
HIF regulates EPDC invasion
Although previous studies provide some clues about the regulatory system that drives EPDC invasion, the mechanisms which spatio-temporally regulate EPDC migration to their final destination have not been defined. In this study, we present evidence that HIF-1 regulates EPDC invasion and hypoxia is a potential guiding signal required for proper coronary vessel formation (Fig. 8). Our immunostaining data and previous studies support that during heart development, differential levels of hypoxia exist in specific regions thereby establishing a dynamic process for moderating the levels of HIF-1α expression in the epicardium and myocardium that coordinate the transcriptional regulation of multiple target genes essential for proper cardiac development, including EPDCs’ proficiency to form coronary vessels. At the stages we investigated, HIF-1α expression in the epicardium and subepicardium is always high at the AVJ but lower on the ventricle. However, its expression in the myocardium increases during heart development. Indeed, a current hypothesis is that the local hypoxic microenvironment influences the maintenance and differentiation of both embryonic and adult stem cells (Mohyeldin et al., 2010; Simon and Keith, 2008). We postulate that by affecting the normal dynamic expression of HIF-1 protein in AdcaHIF1α infected embryonic epicardium, the inability to moderate the temporal-spatial expression of HIF-1 exacerbates intrinsic signals that promote the proper migration of EPDCs (Fig. 8).
Fig 8. Model of how hypoxia regulates EPDCs invasion into the ventricular myocardium.

In AdGFP-injected controls, HIF-1α is expressed in some epicardial cells at an early stage (stage HH24) and its expression in the myocardium increases at later stages (i.e., stage HH28). This pattern guides EPDCs invasion through the subepicardium and myocardium. However, disruption of the normal pattern of HIF expression with a sustained expression in the epicardium mediated by adenovirus caHIF1α results in the up-regulation of expression of Flt-1 (possibly for both membrane and soluble forms), which leads to the inhibition of EPDC migration into the myocardium.
The major coronary vessels form with predictable patterns in species with four-chambered hearts. However, coronary anomalies associated with negative consequences for morbidity and mortality do occur (Frommelt and Frommelt, 2004; Jureidini et al., 1998; von Ludinghausen, 2003). The underlying mechanisms for coronary anomalies arising during cardiogenesis have not been understood. Here, we indicate that HIF-1 is crucial for regulating epicardial EMT and also regulates the invasion of the progenitor cells of vessels into the myocardium. Very hypoxic sulcus regions, as detected by hypoxia indicators and nuclear-localized HIF1a, are also the regions where major coronary vessels develop (Mancini et al., 1991; Nanka et al., 2006; Wikenheiser et al., 2009). Such major coronary vessels are generally found within the epicardium at sulcus regions but in rare conditions, such as myocardial bridging, a segment of a major coronary vessel can dive into the myocardium (Loukas et al., 2011). Our findings provide a potential explanation for why the major coronary vessels usually stay within the epicardium. The sulcus regions in the embryonic heart would have more HIF-1 positive epicardial cells than other regions. Thus these cells would undergo EMT more readily and be inhibited from migrating into the myocardium. This would result in a dense population of precursor cells within the epicardium at sulcus regions that would be available for differentiation and incorporation into large vessels. In other epicardial regions of the heart, EMT would occur later but without the high levels of HIF1 in epicardial cells, the precursors would be able to travel substantial distances into the myocardium. In this scenario, the level of epicardial HIF would control the location of large, medium and small vessels within the myocardial wall by controlling the depth reached by progenitor cells.
The VEGF signaling pathway and EPDC migration in the myocardium
Our findings support that caHIF1α inhibits EPDC invasion through the disruption of VEGF signaling pathway. VEGF binds to their tyrosine kinase receptors and plays a major role in vasculogenesis and angiogenesis (Shibuya, 2006). It has been recognized that expression of some crucial components of the VEGF pathway are regulated by hypoxia (Elvert et al., 2003; Forsythe et al., 1996; Gerber et al., 1997; Han et al., 2010). Investigating the relationship between hypoxia, VEGF, and the development of coronary vasculature reveals that vascular growth is accelerated in heart explants incubated under hypoxic conditions, accompanied by the up-regulation of VegfA mRNA (Yue and Tomanek, 1999). In ovo injection of VEGF-Trap or soluble VEGFRs precludes or reduces the formation of coronary arteries (Tomanek et al., 2006). In addition, in vitro treatment with VEGF164 increases epicardial EMT whereas blocking the VEGF signaling pathway via a Flk-1 specific inhibitor decreases endothelial tube formation (Nesbitt et al., 2009). These published studies further support a critical correlation among HIF, VEGF and coronary vasculogenesis.
Our data document that forced expression of caHIF1α in epicardial cells increased the expression of Flt-1 transcripts. Although some controversy exists, Flk-1 and Flt-1 are considered positive and negative regulators of VEGF signaling, respectively (Shibuya, 2006). While membrane bound Flt-1 possesses higher affinity for VEGFA than Flk-1, it has relatively weak tyrosine kinase activity (de Vries et al., 1992). In its soluble form, sFlt1, competes with Flk-1 by binding to VEGFA and can be considered to be of great importance in generating a VEGF gradient. Indeed, levels of VEGF signaling are well documented in controlling sprouting of endothelial cells (Chappell et al., 2009; Han et al., 2010; Kappas et al., 2008). In our study, blocking VEGF signaling using either cell intrinsic or extrinsic inhibitors resulted in inhibition of EPDC migration similar to that observed when epicardial cells were transduced with caHIF1α. These results indicate that VEGF signaling is required for EPDC movement within the myocardium and caHIF1α inhibits this process by elevating the levels of Flt-1.
Distinct roles of VEGF signaling during valve development have been described in a transgenic mouse study indicating that Flt-1 is required for endocardial cell EMT in the outflow tract (OFT) while Flk-1 plays a role in morphogenesis of atrioventricular canal (AVC) cushions (Stankunas et al., 2010). Another significant finding in human fetal hearts shows that endothelial cells lining epicardial coronary arteries exclusively express Flt-1 but those lining myocardial capillaries express both Flt-1 and Flk-1 (Partanen et al., 1999). While Flt-1 have been shown to be HIF-1α and HIF-2α transcriptional targets, Flk-1 is considered to be only a HIF-2 target (Elvert et al., 2003; Kappel et al., 1999). Considering these data, we propose HIF-1 and HIF-2 may have distinct roles in regulating the spatiotemporal levels of Flt-1 and Flk-1 and the intrinsic Flt-1:Flk-1 ratio strongly influences the final location of EPDCs and their derivatives within the depth of the heart wall.
Although myocardially expressed VEGF has been recognized to potently stimulate epicardial EMT and further induce coronary vessel formation by a paracrine process (Tomanek et al., 2006), we believe this to be the first report supporting that cell autonomous VEGF signaling is involved in regulating EPDC invasion of the myocardium. It is valuable to further unravel the downstream factors associated with the VEGF pathway whose function is indispensable for proper EPDC migration into the myocardium.
Supplementary Material
Immunofluorescent staining for phospho-histone H3 (PH3, red) or TUNEL staining were carried out in quail heart sections (A, B, E, F) or cultured quail epicardial cells (C, D, G, H). No obvious differences were observed between the AdGFP-control (A–D) and AdcaHIF1α-(E–F) infected groups. Nuclei were counterstained using DAPI (blue). Arrows pointed to PH3 (A, E, C, G) or TUNEL positive cells (B, F, D, H). Scale bars are 30 μm. (I, J) Numbers of PH3 or TUNEL positive cells were quantified in heart sections (I) or cultured cells (J) respectively.
{kind=link}
Immortalized mouse epicardial cells were cultured on collagen I coated coverslips and transfected with adenovirus. Fixed cells were immunostained for ZO-1 and F-actin. Nuclei were counterstained with DAPI (blue). Compared with cells incubated with AdGFP which maintained the cobblestone epithelial phenotype (A–C, G–I), cells incubated with AdcaHIF1α lost tight junctions at cell-cell borders (D–F) and some of them displayed elongated morphology and increased stress fibers (J–L) as found in migratory mesenchymal cells. Bar = 30 μm for A–L.
{kind=link}
Adult rat epicardial cells were grown on glass slides and infected with AdGFP or AdcaHIF1α. (A) Control cells infected by AdGFP grew with an epithelial morphology, tending to form sheets of cells with a regular, cobblestone pattern. (B, C) Immunostaining of β-catenin was evenly distributed on cell membranes at cell-cell interfaces in control cells. (D) Cells infected with AdcaHIF1α tended to grow as individual cells, rather than in sheets, and had a more spindle-like morphology. (E, F) The most intense staining for β-catenin was located in the cytoplasm in cells infected with AdcaHIF1α (shown with arrow). Bar = 30 μm for A–F. (G) Western blotting showed that the E-cadherin protein that functions in cell-cell adhesions was reduced in rat cells after infection with AdcaHIF1α. Actin was used as the loading control.
{kind=link}
(A–F) TGFβ1 (250 pM) induced SM22α expression (red) in both AdGFP- (A–C) and AdcaHIF1α-infected quail epicardial cells (D–F). Bar = 30 μm for A–F. (G) Quantification of the percent of SM22α GFP positive cells in total GFP+ cells.
{kind=link}
(A, B) Immunohistochemical staining for VEGFA levels in quail hearts infected with AdGFP (A) or AdcaHIF1α (B). Scale bar = 100 μm for A and B. (C) Quantitative analysis indicated no significant difference in VEGFA staining between AdGFP- and AdcaHIF1α-infected hearts. (D) RT-PCR quantification of fold changes in VEGFA, Flt-1 and Flk-1 expression for quail epicardial cells infected with AdGFP control and AdcaHIF1α virus. * P < 0.05; **P < 0.01.
{kind=link}
GFP+ cells invaded the gel matrix normally in AdGFP control (A) whereas the invasion ability of AdcaHIF1αGFP+ cells was impaired (B). (C) Adding VEGFA (50ng/ml) compromised the invasion inhibition effect of AdcaHIF1α. Dashed lines delineate the surface of the gel and arrow indicates the direction of invasion by the epicardial cells into the collagen gel. Bar = 20 μm for A–C.
{kind=link}
(A, C, E) HIF1α signals were located in the nuclei of epicardial cells in AVJ (pointed by arrows), (B, D, F) but no real positive staining was identified in the negative control (serum only). Bar = 30 μm for A–F.
{kind=link}
Epicardial HIF signaling regulates EPDC invasion;
The VEGF signaling pathway is involved in the HIF regulated EPDC migration;
HIF stimulates epicardial EMT in vitro.
Acknowledgments
We thank Dr. David M. Bader and his laboratory for providing rat epicardial cells, Dr. Joey V. Barnett and his laboratory for providing mouse epicardial cells, Dr. Gregg L. Semenza for providing caHIF1α adenovirus. We thank Dr. Robert J. Tomanek and Dr. Aaron Proweller for valuable discussions. We also thank Drs. Ganga Karunamuni and Yu Han for help with the experiments. This work was supported by NIH grant HL091171 ARRA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelman DM, Gertsenstein M, Nagy A, Simon MC, Maltepe E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–3203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambati BK, Nozaki M, Singh N, Takeda A, Jani PD, Suthar T, Albuquerque RJ, Richter E, Sakurai E, Newcomb MT, Kleinman ME, Caldwell RB, Lin Q, Ogura Y, Orecchia A, Samuelson DA, Agnew DW, St Leger J, Green WR, Mahasreshti PJ, Curiel DT, Kwan D, Marsh H, Ikeda S, Leiper LJ, Collinson JM, Bogdanovich S, Khurana TS, Shibuya M, Baldwin ME, Ferrara N, Gerber HP, De Falco S, Witta J, Baffi JZ, Raisler BJ, Ambati J. Corneal avascularity is due to soluble VEGF receptor-1. Nature. 2006;443:993–997. doi: 10.1038/nature05249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin AF, Compton LA, Love JD, Brown CB, Barnett JV. Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFbeta. Dev Dyn. 2008;237:366–376. doi: 10.1002/dvdy.21421. [DOI] [PubMed] [Google Scholar]
- Chappell JC, Taylor SM, Ferrara N, Bautch VL. Local guidance of emerging vessel sprouts requires soluble Flt-1. Dev Cell. 2009;17:377–386. doi: 10.1016/j.devcel.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science. 1992;255:989–991. doi: 10.1126/science.1312256. [DOI] [PubMed] [Google Scholar]
- Dettman RW, Pae SH, Morabito C, Bristow J. Inhibition of alpha4-integrin stimulates epicardial-mesenchymal transformation and alters migration and cell fate of epicardially derived mesenchyme. Dev Biol. 2003;257:315–328. doi: 10.1016/s0012-1606(03)00064-2. [DOI] [PubMed] [Google Scholar]
- Dokic D, Dettman RW. VCAM-1 inhibits TGFbeta stimulated epithelial-mesenchymal transformation by modulating Rho activity and stabilizing intercellular adhesion in epicardial mesothelial cells. Dev Biol. 2006;299:489–504. doi: 10.1016/j.ydbio.2006.08.054. [DOI] [PubMed] [Google Scholar]
- Dunwoodie SL. The role of hypoxia in development of the Mammalian embryo. Dev Cell. 2009;17:755–773. doi: 10.1016/j.devcel.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Eid H, Larson DM, Springhorn JP, Attawia MA, Nayak RC, Smith TW, Kelly RA. Role of epicardial mesothelial cells in the modification of phenotype and function of adult rat ventricular myocytes in primary coculture. Circ Res. 1992;71:40–50. doi: 10.1161/01.res.71.1.40. [DOI] [PubMed] [Google Scholar]
- Elvert G, Kappel A, Heidenreich R, Englmeier U, Lanz S, Acker T, Rauter M, Plate K, Sieweke M, Breier G, Flamme I. Cooperative interaction of hypoxia-inducible factor-2alpha (HIF-2alpha) and Ets-1 in the transcriptional activation of vascular endothelial growth factor receptor-2 (Flk-1) J Biol Chem. 2003;278:7520–7530. doi: 10.1074/jbc.M211298200. [DOI] [PubMed] [Google Scholar]
- Fisher SA, Siwik E, Branellec D, Walsh K, Watanabe M. Forced expression of the homeodomain protein Gax inhibits cardiomyocyte proliferation and perturbs heart morphogenesis. Development. 1997;124:4405–4413. doi: 10.1242/dev.124.21.4405. [DOI] [PubMed] [Google Scholar]
- Fisher SA, Watanabe M. Expression of exogenous protein and analysis of morphogenesis in the developing chicken heart using an adenoviral vector. Cardiovasc Res. 1996;31(Spec No):E86–95. [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommelt PC, Frommelt MA. Congenital coronary artery anomalies. Pediatr Clin North Am. 2004;51:1273–1288. doi: 10.1016/j.pcl.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem. 1997;272:23659–23667. doi: 10.1074/jbc.272.38.23659. [DOI] [PubMed] [Google Scholar]
- Han Y, Kuang SZ, Gomer A, Ramirez-Bergeron DL. Hypoxia influences the vascular expansion and differentiation of embryonic stem cell cultures through the temporal expression of vascular endothelial growth factor receptors in an ARNT-dependent manner. Stem Cells. 2010;28:799–809. doi: 10.1002/stem.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Yang K, Proweller A, Zhou G, Jain MK, Ramirez-Bergeron DL. Inhibition of ARNT severely compromises endothelial cell viability and function in response to moderate hypoxia. Angiogenesis. 2012;15:409–420. doi: 10.1007/s10456-012-9269-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma T, Hirakow R. Epicardial formation in embryonic chick heart: computer-aided reconstruction, scanning, and transmission electron microscopic studies. Am J Anat. 1989;184:129–138. doi: 10.1002/aja.1001840204. [DOI] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jureidini SB, Marino CJ, Rao PS. Congenital coronary artery abnormalities. Ind J Pediatr. 1998;65:217–229. doi: 10.1007/BF02752298. [DOI] [PubMed] [Google Scholar]
- Kappas NC, Zeng G, Chappell JC, Kearney JB, Hazarika S, Kallianos KG, Patterson C, Annex BH, Bautch VL. The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J Cell Biol. 2008;181:847–858. doi: 10.1083/jcb.200709114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel A, Ronicke V, Damert A, Flamme I, Risau W, Breier G. Identification of vascular endothelial growth factor (VEGF) receptor-2 (Flk-1) promoter/enhancer sequences sufficient for angioblast and endothelial cell-specific transcription in transgenic mice. Blood. 1999;93:4284–4292. [PubMed] [Google Scholar]
- Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kim JG, Moon MY, Jeon CY, Won HY, Kim HJ, Jeon YJ, Seo JY, Kim JI, Kim J, Lee JY, Kim PH, Park JB. Transforming growth factor-beta1 regulates macrophage migration via RhoA. Blood. 2006;108:1821–1829. doi: 10.1182/blood-2005-10-009191. [DOI] [PubMed] [Google Scholar]
- Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loukas M, Von Kriegenbergh K, Gilkes M, Tubbs RS, Walker C, Malaiyandi D, Anderson RH. Myocardial bridges: A review. Clin Anat. 2011;24:675–683. doi: 10.1002/ca.21150. [DOI] [PubMed] [Google Scholar]
- Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- Mancini L, Bertossi M, Ribatti D, Bartoli F, Nico B, Lozupone E, Roncali L. The effects of long-term hypoxia on epicardium and myocardium in developing chick embryo hearts. Int J Microcir Clin Exp. 1991;10:359–371. [PubMed] [Google Scholar]
- Mikawa T, Fischman DA. Retroviral analysis of cardiac morphogenesis: discontinuous formation of coronary vessels. Proc Natl Acad Sci. 1992;89:9504–9508. doi: 10.1073/pnas.89.20.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell stem cell. 2010;7:150–161. doi: 10.1016/j.stem.2010.07.007. [DOI] [PubMed] [Google Scholar]
- Morabito CJ, Dettman RW, Kattan J, Collier JM, Bristow J. Positive and negative regulation of epicardial-mesenchymal transformation during avian heart development. Dev Biol. 2001;234:204–215. doi: 10.1006/dbio.2001.0254. [DOI] [PubMed] [Google Scholar]
- Nanka O, Krizova P, Fikrle M, Tuma M, Blaha M, Grim M, Sedmera D. Abnormal myocardial and coronary vasculature development in experimental hypoxia. Anat Rec (Hoboken) 2008;291:1187–1199. doi: 10.1002/ar.20738. [DOI] [PubMed] [Google Scholar]
- Nanka O, Valasek P, Dvorakova M, Grim M. Experimental hypoxia and embryonic angiogenesis. Dev Dyn. 2006;235:723–733. doi: 10.1002/dvdy.20689. [DOI] [PubMed] [Google Scholar]
- Nesbitt TL, Roberts A, Tan H, Junor L, Yost MJ, Potts JD, Dettman RW, Goodwin RL. Coronary endothelial proliferation and morphogenesis are regulated by a VEGF-mediated pathway. Dev Dyn. 2009;238:423–430. doi: 10.1002/dvdy.21847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey HE, Compton LA, Barnett JV. Coronary vessel development: the epicardium delivers. Trends Cardiovasc Med. 2004;14:247–251. doi: 10.1016/j.tcm.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Olivey HE, Mundell NA, Austin AF, Barnett JV. Transforming growth factor-beta stimulates epithelial-mesenchymal transformation in the proepicardium. Dev Dyn. 2006;235:50–59. doi: 10.1002/dvdy.20593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivey HE, Svensson EC. Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res. 2010;106:818–832. doi: 10.1161/CIRCRESAHA.109.209197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partanen TA, Makinen T, Arola J, Suda T, Weich HA, Alitalo K. Endothelial growth factor receptors in human fetal heart. Circulation. 1999;100:583–586. doi: 10.1161/01.cir.100.6.583. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Gittenberger-de Groot AC, Mentink MM, Bokenkamp R, Hogers B. Development of the cardiac coronary vascular endothelium, studied with antiendothelial antibodies, in chicken-quail chimeras. Circ Res. 1993;73:559–568. doi: 10.1161/01.res.73.3.559. [DOI] [PubMed] [Google Scholar]
- Ramirez-Bergeron DL, Simon MC. Hypoxia-inducible factor and the development of stem cells of the cardiovascular system. Stem Cells. 2001;19:279–286. doi: 10.1634/stemcells.19-4-279. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am J Pathol. 2004;164:1531–1535. doi: 10.1016/S0002-9440(10)63711-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–478. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nature reviews Mol Cell Biol. 2008;9:285–296. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Ma GK, Kuhnert FJ, Kuo CJ, Chang CP. VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev Biol. 2010;347:325–336. doi: 10.1016/j.ydbio.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomanek RJ, Ishii Y, Holifield JS, Sjogren CL, Hansen HK, Mikawa T. VEGF family members regulate myocardial tubulogenesis and coronary artery formation in the embryo. Circ Res. 2006;98:947–953. doi: 10.1161/01.RES.0000216974.75994.da. [DOI] [PubMed] [Google Scholar]
- Tomanek RJ, Lund DD, Yue X. Hypoxic induction of myocardial vascularization during development. Adv Exp Med Biol. 2003;543:139–149. doi: 10.1007/978-1-4419-8997-0_10. [DOI] [PubMed] [Google Scholar]
- Viragh S, Challice CE. The origin of the epicardium and the embryonic myocardial circulation in the mouse. Anat Rec. 1981;201:157–168. doi: 10.1002/ar.1092010117. [DOI] [PubMed] [Google Scholar]
- von Ludinghausen M. The clinical anatomy of coronary arteries. Adv Anat Embryol Cell Biol. 2003;167:III–VIII. 1–111. doi: 10.1007/978-3-642-55807-8. [DOI] [PubMed] [Google Scholar]
- Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002;16:1151–1162. doi: 10.1096/fj.01-0944rev. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Science’s STKE. 2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- Wessels A, Perez-Pomares JM. The epicardium and epicardially derived cells (EPDCs) as cardiac stem cells. The anatomical record Part A. Anat Rec A Discov Mol Cell Evol Biol. 2004;276:43–57. doi: 10.1002/ar.a.10129. [DOI] [PubMed] [Google Scholar]
- Wikenheiser J, Doughman YQ, Fisher SA, Watanabe M. Differential levels of tissue hypoxia in the developing chicken heart. Dev Dyn. 2006;235:115–123. doi: 10.1002/dvdy.20499. [DOI] [PubMed] [Google Scholar]
- Wikenheiser J, Karunamuni G, Sloter E, Walker MK, Roy D, Wilson DL, Watanabe M. Altering HIF-1alpha Through 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) Exposure Affects Coronary Vessel Development. Cardiovasc Toxicol. 2012 doi: 10.1007/s12012-012-9194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikenheiser J, Wolfram JA, Gargesha M, Yang K, Karunamuni G, Wilson DL, Semenza GL, Agani F, Fisher SA, Ward N, Watanabe M. Altered hypoxia-inducible factor-1 alpha expression levels correlate with coronary vessel anomalies. Dev Dyn. 2009;238:2688–2700. doi: 10.1002/dvdy.22089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter EM, Gittenberger-de Groot AC. Epicardium-derived cells in cardiogenesis and cardiac regeneration. Cell Mol Life Sci. 2007;64:692–703. doi: 10.1007/s00018-007-6522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Tomanek RJ. Stimulation of coronary vasculogenesis/angiogenesis by hypoxia in cultured embryonic hearts. Dev Dyn. 1999;216:28–36. doi: 10.1002/(SICI)1097-0177(199909)216:1<28::AID-DVDY5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunofluorescent staining for phospho-histone H3 (PH3, red) or TUNEL staining were carried out in quail heart sections (A, B, E, F) or cultured quail epicardial cells (C, D, G, H). No obvious differences were observed between the AdGFP-control (A–D) and AdcaHIF1α-(E–F) infected groups. Nuclei were counterstained using DAPI (blue). Arrows pointed to PH3 (A, E, C, G) or TUNEL positive cells (B, F, D, H). Scale bars are 30 μm. (I, J) Numbers of PH3 or TUNEL positive cells were quantified in heart sections (I) or cultured cells (J) respectively.
Immortalized mouse epicardial cells were cultured on collagen I coated coverslips and transfected with adenovirus. Fixed cells were immunostained for ZO-1 and F-actin. Nuclei were counterstained with DAPI (blue). Compared with cells incubated with AdGFP which maintained the cobblestone epithelial phenotype (A–C, G–I), cells incubated with AdcaHIF1α lost tight junctions at cell-cell borders (D–F) and some of them displayed elongated morphology and increased stress fibers (J–L) as found in migratory mesenchymal cells. Bar = 30 μm for A–L.
Adult rat epicardial cells were grown on glass slides and infected with AdGFP or AdcaHIF1α. (A) Control cells infected by AdGFP grew with an epithelial morphology, tending to form sheets of cells with a regular, cobblestone pattern. (B, C) Immunostaining of β-catenin was evenly distributed on cell membranes at cell-cell interfaces in control cells. (D) Cells infected with AdcaHIF1α tended to grow as individual cells, rather than in sheets, and had a more spindle-like morphology. (E, F) The most intense staining for β-catenin was located in the cytoplasm in cells infected with AdcaHIF1α (shown with arrow). Bar = 30 μm for A–F. (G) Western blotting showed that the E-cadherin protein that functions in cell-cell adhesions was reduced in rat cells after infection with AdcaHIF1α. Actin was used as the loading control.
(A–F) TGFβ1 (250 pM) induced SM22α expression (red) in both AdGFP- (A–C) and AdcaHIF1α-infected quail epicardial cells (D–F). Bar = 30 μm for A–F. (G) Quantification of the percent of SM22α GFP positive cells in total GFP+ cells.
(A, B) Immunohistochemical staining for VEGFA levels in quail hearts infected with AdGFP (A) or AdcaHIF1α (B). Scale bar = 100 μm for A and B. (C) Quantitative analysis indicated no significant difference in VEGFA staining between AdGFP- and AdcaHIF1α-infected hearts. (D) RT-PCR quantification of fold changes in VEGFA, Flt-1 and Flk-1 expression for quail epicardial cells infected with AdGFP control and AdcaHIF1α virus. * P < 0.05; **P < 0.01.
GFP+ cells invaded the gel matrix normally in AdGFP control (A) whereas the invasion ability of AdcaHIF1αGFP+ cells was impaired (B). (C) Adding VEGFA (50ng/ml) compromised the invasion inhibition effect of AdcaHIF1α. Dashed lines delineate the surface of the gel and arrow indicates the direction of invasion by the epicardial cells into the collagen gel. Bar = 20 μm for A–C.
(A, C, E) HIF1α signals were located in the nuclei of epicardial cells in AVJ (pointed by arrows), (B, D, F) but no real positive staining was identified in the negative control (serum only). Bar = 30 μm for A–F.