

Abstract
Amongst the numerous conserved residues in the aldehyde dehydrogenase superfamily, the precise role of Thr-244 remains enigmatic. Crystal structures show that this residue lies at the interface between the coenzyme-binding and substrate-binding sites with the side chain methyl substituent oriented toward the B-face of the nicotinamide ring of the NAD(P)+ coenzyme, when in position for hydride transfer. Site-directed mutagenesis in ALDH1A1 and GAPN has suggested a role for Thr-244 in stabilizing the nicotinamide ring for efficient hydride transfer. Additionally, these studies also revealed a negative effect on cofactor binding which is not fully explained by the interaction with the nicotinamide ring. However, it is suggestive that Thr-244 immediately precedes helix αG, which forms one-half of the primary binding interface for the coenzyme. Hence, in order to more fully investigate the role of this highly conserved residue, we generated valine, alanine, glycine and serine substitutions for Thr-244 in human ALDH2. All four substituted enzymes exhibited reduced catalytic efficiency toward substrate and coenzyme. We also determined the crystal structure of the T244A enzyme in the absence and presence of coenzyme. In the apo-enzyme, the alpha G helix, which is key to NAD binding, exhibits increased temperature factors accompanied by a small displacement toward the active site cysteine. This structural perturbation was reversed in the coenzyme-bound complex. Our studies confirm a role for the Thr-244 beta methyl in the accurate positioning of the nicotinamide ring for efficient catalysis. We also identify a new role for Thr-244 in the stabilization of the N-terminal end of helix αG. This suggests that Thr-244, although less critical than Glu-487, is also an important contributor toward coenzyme binding.
Keywords: Aldehyde dehydrogenase, NAD, disorder, isomerization, coenzyme binding
Introduction
ALDH2 is a member of a broad superfamily of enzymes which are involved in the removal of exogenous and biogenic toxins, primarily via the oxidation of aldehyde intermediates [1]. ALDH2 has long been known for its role in the oxidation of ethanol-derived acetaldehyde [2]. This is in no small part due to the prevalence of the inactive variant in East Asian populations (ALDH2*2) which results in ethanol-induced flushing as well as a higher incidence of head and neck cancers [3,4]. In the last decade, ALDH2 has also been recognized for its role in the bio-activation of nitroglycerin used to treat angina pectoris [5,6]. This catalytic activity is closely related to the esterase activity of ALDH2 [7–9]. Most recently, it has been shown that ALDH2 plays a central role in ethanol-induced cardio protection [10], most likely via detoxication of 4-HNE [11,12], making it an interesting pharmacological target [13–15].
All known ALDH family members share a common tertiary structure and reaction mechanism. Structurally speaking, ALDHs are homotetrameric or homodimeric with each monomer consisting of two Rossmann fold domains and an oligomerization tail (Fig. 1A). The coenzyme NAD(P)+ binds to the first Rossmann fold in an unusual manner, where the adenosine occupies a cleft between the αF and αG helices, the pyrophosphates are solvent exposed, and the nicotinamide mononucleotide has been observed to occupy various positions [16–19] (Fig. 1B). The ALDH tetramer has been conceptualized as a dimer of dimers [18]. Within a dimer, the αG helices from adjacent monomers are in close contact across the dimer interface (Fig. 1A). Previous work from our laboratory has highlighted the importance of the αG helices: in the Asian Variant ALDH2*2, the substitution of lysine for glutamate at position 487 disrupts a salt bridge at the C-terminal end of αG, resulting in disorder at the αG helix [20]. This results in a large energetic barrier to NAD binding that is reflected in the 200-fold increased KM(NAD+) for this isozyme [21].
Fig. 1.
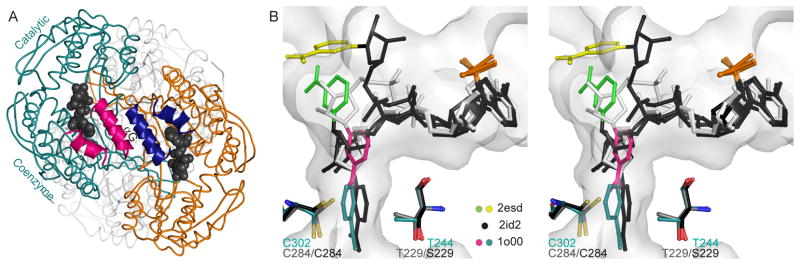
NAD binding in the ALDH superfamily. (A) ALDH2 tetramer with helices αF and αG shown as ribbons and NAD as vdw spheres (PDB code 1o02). (B) Stereo view of the cofactor conformations observed in the wild-type ALDH2-NAD complex (PDB code 1o00), in the E268A GAPN-NADP complex (PDB code 2esd, ref. 16), and the T244S GAPN-NADP complex (PDB code 2id2, ref. 19). Legend refers to color of nicotinamide ring, surface representation shown for 1o00 only. All molecular figures were generated using PyMol [38].
Although amino-acid sequence diversity is high, ALDH family members share a core set of residues, mainly in the active and cofactor binding sites, many of which have been studied extensively (Fig. 2). Cys-302 is the active site nucleophile which forms a covalent adduct with the substrate aldehyde; the resulting thiohemiacetal intermediate is most likely stabilized by Asn-169 [18,22,23]. The substrate hydride is then transferred to the C4N carbon of the nicotinamide ring [24]. It is generally accepted that, in order for the general base Glu-268 to activate a water molecule for attack on the acyl-sulfur bond, the nicotinamide ring must occupy a position different than that ideal for hydride transfer (Fig. 2) [17,25,26]. Product is then released before NADH [23,25] (Scheme 1). In addition to the obvious substrate specificities of ALDH isozymes, different family members also exhibit different rate-limiting steps in this general reaction scheme, in particular, the CoA-dependent ALDHs follow a Ping-Pong mechanism [27,28].
Fig. 2.

Stereo views of the active site of ALDH2 with butyraldehyde modeled in (A) PDB code 1o04 with a transparent vdw surface shown for Thr-244 Cγ2. (B) PDB code 1o01 (absent non-covalent crotonaldehyde). Alternate structures with Glu in “down” position: Apo wt 1o05 (subunits BCEFH) and SM-NAD wt 1cw3 (subunits ADEH).
Scheme 1.
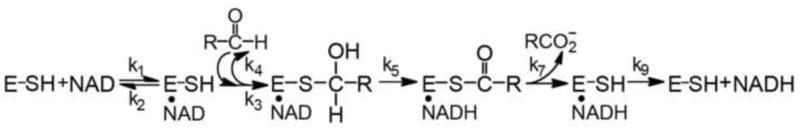
General mechanism for the dehydrogenase reaction catalyzed by ALDH.
Threonine 244 is one highly conserved residue whose function has only recently been examined in detail [19,29,30]. X-ray crystal structures show that Thr-244 lies between the coenzyme- and substrate-binding sites with the beta-methyl positioned to interact with the B-face of the nicotinamide ring, when coenzyme is positioned for hydride transfer (Fig. 1B, 2id2 and 1o00, and Fig. 2) [17,18]. Recently, Pailot, et al. showed that substitution of Thr-244 in non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase (GAPN) resulted in decreased catalytic efficiency, increased KM for cofactor and a small shift in the position of the nicotinamide ring (Fig 1B, 2id2) [19]. Similarly, we have shown that the T244S in human ALDH1A1 resulted in decreased catalytic efficiency and increased KM for NAD+ [29]. In both cases, the binding of NADH was only slightly impaired [19,29]. Finally, we have also reported that substitution of Thr-244 reduces catalytic efficiency in human ALDH3A2 as well [30].
The suggestion that substitution of Thr-244 may alter the interaction with coenzyme is interesting because Thr-244 immediately precedes the αG helix which forms one-half of the primary binding interface for NAD. We have previously shown that disruption of the C-terminal salt-bridge by the E487K or R475Q substitutions not only results in disorder of helix αG, but that the disorder is propagated all through the subunit interface and into the catalytic site, reducing catalytic efficiency and NAD+ binding affinity [20,31]. Furthermore, similar to what was observed for the Thr-244 substituted enzymes described above, with both the E487K and E475Q substitutions, the negative impact on binding affinity was larger for NAD+ than it was for NADH [21]. Hence, we set out to examine the structural and kinetic effects of Thr-244 substitution in ALDH2. Our work, taken together with the aforementioned studies in GAPN and ALDH1A1, suggests that Thr-244 may have larger role in catalysis than previously assumed.
2. Materials and Methods
2.1. Chemicals
Tryptone and yeast extract were obtained from Difco Laboratories (Detroit, MI). The PEG 6000 used for crystallization was purchased from Hampton Research (Aliso Viejo, CA). HPLC purified oligonucleotide primers for site-directed mutagenesis and sequencing were purchased from IDT (Coralville, IA). The QuickChange II Mutagenesis kit was supplied by Stratagene (La Jolla, CA). Reagent kits for plasmid isolation and purification were obtained from Qiagen (Valencia, CA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO). Unless otherwise stated, all chemicals were used without further purification.
2.2. Sequence analysis and structural comparisons
To find sequences similar to human ALDH2, we conducted two BLAST [32] iterations using the PSI-BLAST algorithm [33] against the database of non-redundant Swissprot sequences [34]. Mature human ALDH2 was used as the query sequence and default parameters were used with the exception that the maximum number of target hits was raised to 5,000. The Constraint-based Multiple Alignment Tool (COBALT) was then used to obtain the multiple sequence alignment using default parameters [35]. Four redundant sequences and five short fragments were manually removed from the alignment. The initial 995 non-redundant hits encompassed distantly related proteins, down to 13.5% identity and an Evalue of 1×10−8. This initial set included human ALDH18A1 (P5CS, 16.5% identity to ALDH2) and over 400 other members of the gamma-glutamyl phosphate reductase (GPR, EC 1.2.1.41) family. Hence, the alignment was truncated to the 539 sequences which share more than 20% pairwise sequence identity to human ALDH2. This excluded all of the phosphorylating ALDHs, including 450 with Arg-244 and 6 with Thr-244. Aligned sequences were imported into Excel to obtain residue counts, GeneDoc [36] and JalView [37] were used to obtain consensus sequences and degap the truncated alignment. For structural comparison of Lys-178 and Thr-244, 97 ALDH structures representing 35 unique enzymes were downloaded from the Protein Data Bank on Aug, 16 2010. PyMol was used to align each structure to the wild-type ALDH2 tetramer and to measure interatomic distances [38].
2.3. Plasmids, mutagenesis, protein expression
ALDH2 cDNA cloned into the pT7-7 vector [39] was used as template for mutagenesis using the QuickChange II Mutagenesis kit. Thr-244 was replaced through site-directed mutagenesis with one of four residues; Gly, Ala, Val or Ser. The primer pair 5′-GACAAAGTGGCATTCNNNGGCTCCACTGAGATTGG-3′ and 5′-CCAATCTCAGTGGAGCCN1N1N1GAATGCCACTTTGTC-3′ was used with base sequences NNN/N1N1N1 as GGA/TCC for Gly, GCA/TGC for Ala, TCA/TGA for Ser, and GTA/TAC for Val. Plasmids isolated after transformation into DH5α cells were screened for the desired substitution by DNA sequencing. Positive clones were transformed into the Escherichia coli strain BL21(DE3)pLysS for protein expression as described previously [39].
2.4. Purification of native and Thr-244 substituted enzymes
All enzymes were purified as described previously [40]. Briefly, recombinantly expressed enzymes were first subjected to protamine sulfate treatment (1.25 mg/ml) followed by DEAE- cellulose column. Then the ALDH2 native or Thr-244 substituted enzymes were purified by 5′ AMP Sepharose 4B and p-hydroxyacetophenone (HAP) affinity chromatography, respectively. The homogeneity of individual enzymes was indicated by SDS-PAGE. All the purified enzymes were found to be homogeneous, yielding a single band of molecular weight ~55 kD on SDS-PAGE. Fractions of individual purified enzymes were pooled, concentrated by Centricon centrifugal filters (Amicon) and stored in 50% glycerol at −20 °C, prior to characterization. The T244G enzyme bound poorly to the 4-hydroxyacetophenone affinity column which, In this case, the T244G enzyme was further purified by substituting a second 5′-AMP Sepharose affinity chromatography step. None of the substitutions altered the oligomeric state of the recombinant protein, as all forms exhibited elution patterns identical to the wild-type enzyme on a Sephacryl-200 column with molecular masses approximating 200 kD (data not shown).
2.5. Kinetic assays
Enzyme activities were monitored by following the increase in NADH fluorescence at 450 nm, as previously described [29]. Comparison of activities with propionaldehyde versus chloroacetaldehyde were measured in 25 mM sodium Hepes buffer (pH 7.4) at fixed single concentrations of NAD+ and substrate, with and without 5 mM magnesium (Table 1).
Table 1.
Effect of Mg2+ ions on activity of native and T244-substituted ALDH2 against propionaldehyde and chloroacetaldehyde.
| Propionaldehydea | Chloroacetaldehyde | |||
|---|---|---|---|---|
|
|
||||
| No Mg2+ | + 5 mM Mg2+ | No Mg2+ | + 5 mM Mg2+ | |
| Enzyme | V (min−1) | V (min−1) | V (min−1) | V (min−1) |
| ALDH2 | 190 ± 20 | 470 ± 38 | 520 ± 65 | 670 ± 52 |
| T244V | 67 ± 9 | 170 ± 48 | 160 ± 15 | 250 ± 63 |
| T244A | 35 ± 2 | 120 ± 14 | 280 ± 47 | 560 ± 127 |
| T244G | 6 ± 1 | 54 ± 5 | 40 ± 5 | 80 ± 12 |
| T244S | 7 ± 1 | 67 ± 9 | 32 ± 4 | 99 ± 10 |
Reaction velocities were determined in 50 mM sodium Hepes buffer at pH 7.4 in the absence or presence of Mg2+ with 1.2 mM NAD+ and 50 μM propionaldehyde or 200 μM chloroacetaldehyde (native, T224G, and T244S) or with 4 mM NAD+ and 1.5 mM propionaldehyde or 0.8 mM chloroacetaldehyde (T244A and T244V).
Co-variation assays were conducted in 50 mM sodium phosphate buffer (pH 7.4) with the ranges of chloroacetaldehyde and NAD+ adjusted for each enzyme (Table 2). The kinetic constants, KM, Vmax and Kia shown in Table 2 were obtained by bisubstrate kinetic analyses. The velocity data were fit to the following bi-bi ordered sequential equation in the absence of products: v = Vmax [A][B] / (KiaKB + KA[B] + KB[A] + [A][B]) (Equation 1) where [A] and [B] correspond to the concentrations of NAD+ and aldehyde, respectively. The kcat values were derived from the Vmax values and the catalytic efficiency, kcat/KM for substrate was derived from Scheme 1 and is equivalent to the following collection of rate constants, k3k5 / (k4 + k5). Competitive inhibition assays were used to measure Kiq and were conducted in 25 mM sodium Hepes (pH 7.4) using 40–150 μM NADH as a competitive inhibitor against NAD+, as previously described [21]. Protein concentrations were determined using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA) with bovine serum albumin as a standard.
Table 2.
Kinetic constants for native and T244-substituted ALDH2 with chloroacetaldehyde as substrate.
| Kinetic Constant | ALDH2a | T244V | T244A | T244G | T244S |
|---|---|---|---|---|---|
| kcat (min−1) | 460 ± 50 | 150 ± 15 | 240 ± 20 | 38 ± 5 | 28 ± 4 |
| KM(NAD+) (μM) | 48 ± 8 | 390 ± 60 | 2100 ± 300 | 190 ± 20 | 94 ± 20 |
| KM(chloroacetaldehyde) (μM) | 39 ± 6 | 460 ± 50 | 300 ± 40 | 52 ± 9 | 13 ± 5 |
| Kia (μM) | 41 ± 6 | 140 ± 30 | 490 ± 80 | 240 ± 55 | 200 ± 40 |
| Kiq (μM) b | 89 ± 9 | 200 ± 30 | 150 ± 10 | 150 ± 20 | 110 ± 10 |
| kcat/KM(NAD+) (min−1mM−1) c | 9,500 | 390 | 110 | 200 | 300 |
| kcat/KM(chloroacetaldehyde) (min−1mM−1) | 12,000 | 330 | 790 | 730 | 2,000 |
| kcat/(Kia • KM(chloroacetaldehyde)) (min−1mM−2) | 290,000 | 2,400 | 1,600 | 3,100 | 11,000 |
Kinetic constants were determined by co-variation kinetics in 50 mM sodium phosphate buffer at pH 7.4 with 20–1200 μM NAD+ and 2–200 μM chloroacetaldehyde (native, T224G, and T244S) or with 0.5–4 mM NAD+ and 50–800 μM chloroacetaldehyde (T244A and T244V).
Kiq was obtained by competitive inhibition against 40–150 μM NAD+ in 25 mM sodium Hepes buffer at pH 7.4.
Kinetic constants obtained from fits were used to calculate values in italics (see Materials and Methods).
2.6. Crystallization and structure determination
Crystals of T244A ALDH2 were grown under conditions similar to that for the wild-type enzyme [17]. Briefly, the apo-enzyme form of T244A was concentrated to 7.5 mg/ml in 10 mM ACES, pH 6.6 containing 1 mM DTT and equilibrated against a crystallization solutions that contained 100 mM ACES, pH 6.4, 100–200 mM guanidine-HCl, 1–10 mM MgCl2 and 16–17% (w/v) PEG 6000. The complexes with NAD+ and NADH were prepared through direct soaking experiments. The complex with NADH was prepared by soaking an apo-enzyme crystal overnight against 0.5 mM NADH in 100 mM ACES, pH 6.4, 100 mM guanidine-HCl, 10 mM MgCl2 and 18% (w/v) PEG 6000. Due to a propensity for crystal cracking, the complex with NAD+ required a step-wise introduction of NAD+ into the crystal using the following protocol, 0.1 mM, 0.2 mM, 0.5 mM, 1 mM, 2 mM and 5 mM with 15 minutes of soaking at each concentration step in 100 mM sodium ACES, pH 6.4, 150 mM guanidine-HCl, 10 mM MgCl2 and 17% (w/v) PEG 6000. All crystals were flash cooled using a two-step protocol to introduce 18% (v/v) ethylene glycol into the crystallization/soaking solutions for cryoprotection. Diffraction data for the T244A-NADH complex were collected in our laboratory X-ray facility (Raxis IV++). Data for the apo-T244A, T244A-NAD+ complex, and apo-ALDH2 structures were collected at beamline 19-ID operated by the Structural Biology Consortium, located within the Advanced Photon Source at Argonne National Laboratory. All diffraction data were indexed, integrated and scaled using the HKL2000/HKL3000 program suite [41]. As all structures are essentially isomorphous with the wild-type ALDH2 orthorhombic data sets [17], each structure was solved by direct refinement using the wild-type apo-ALDH2 structure (with solvent removed) as the starting model. Confirmation of the substitution at position 244, as well as the binding of coenzyme was evaluated through inspection of the initial Fo-Fc electron density maps. Modeling of solvent molecules and bound ligands was performed independently for each structure. All structures were refined using Refmac [42] and visually inspected and adjusted using the visualization program Coot [43].
3. Results
3.1. Conservation of Thr-244 among aldehyde dehydrogenases
We generated a multiple sequence alignment of proteins which share greater than 20% sequence identity to human ALDH2. Among the 539 hits from diverse organisms, we find position 244 is a threonine in 483 sequences, a valine in 50, a cysteine in 4, and an isoleucine in 2. The four sequences with Cys-244 belong to ALDH family 16 (sequence identity ~30% to human ALDH2). With the additional substitutions of alanine at position 268 and glycine at 302, the function of the ALDH16 family members is unclear [1]. The two enzymes with Ile-244 are ALDH-like proteins from lower organisms, and are also of undetermined function. Forty-nine of the Val-244 containing enzymes belong to the CoA-dependent subfamily of ALDHs (also known as the MMSDH family [44]. The other sequence with valine at position 244 is a poorly characterized member of ALDH family 22 from Arabidopsis [45]. Thus, among the non-phosphorylating ALDHs, Thr-244 is conserved in more than 98% of CoA-independent sequences and Val-244 is conserved in all CoA-dependent enzymes. An interesting potential hydrogen bond partner for the Thr-244 side chain is lysine 178 (Fig. 2). This residue is moderately conserved, with a lysine appearing in 50% and a histidine in 22% of the non-phosphorylating ALDHs in our alignment. Additionally, in 68 of the 70 ALDH crystal structures that have a lysine at this position, the side chain amino group is within 3.4 Å of the side chain hydroxyl of Thr-244.
3.2. Kinetics of enzymes with substitutions ofThr-244
We made four Thr-244 substitutions in the human ALDH2 isozyme. The substitutions of glycine and alanine were chosen to study the effect of side chain deletion at this position, serine was selected for comparison with published results for GAPN and ALDH1A1, and valine is a naturally occurring variation appearing in CoA-dependent ALDHs and accounts for approximately 90% of the variance at this position.
We have previously established that the deacylation step is the major contributor to kcat for wild-type ALDH2 [7,46] and that that aldehydes substituted with an electron withdrawing group are oxidized more rapidly than propionaldehyde [7,22]. Hence we measured reaction velocities for propionaldehyde and chloroacetaldehyde for each enzyme (Table 1). All substituted enzymes exhibited lower catalytic rates than wild-type toward both substrates, with the T244G and T244S substitutions showing much larger decreases than the T244V and T244A enzymes (Table 1). Additionally, all reactions were 2.5- to 8-fold faster with chloroacetaldehyde than propionaldehyde as substrate. Since an electron withdrawing group adjacent to the aldehydic carbon would be expected to slow hydride transfer, the faster turnover indicates that hydride transfer does not significantly contribute to their observed maximal velocities in the Thr-244 substituted enzymes. Instead, the increase in Vmax is consistent with acceleration of either thiohemiacetal formation or acyl-enzyme hydrolysis. To distinguish between these possibilities, we investigated the effect of adding magnesium ions to the reaction. We and others have shown that addition of magnesium ions to ALDH2 catalyzed reactions increases the rate of acyl-enzyme hydrolysis without impacting thiohemiacetal formation or hydride transfer [29,47,48]. At low concentrations, magnesium ions result in an approximate 2-fold increase in Vmax with propionaldehyde as substrate but have little effect when chloroacetaldehyde is the substrate, presumably because the rate of chloroacetaldehyde oxidation cannot be further enhanced [29,47,48]. At the maximally activating concentrations used here, we found a 2-fold increase for propionaldehyde and no significant enhancement with chloroacetaldehyde for the wild-type enzyme, consistent with previous data [47], and obtained stronger effects for the Thr-244 substitutions, with a 2.5 to 9.5-fold increase in activity with propionaldehyde and 1.5 to 3.0-fold increase with chloroacetaldehyde (Table 1). Taken together, these data provide strong support that acyl-enzyme hydrolysis remains the major contributor to the turnover rate.
Because catalytic activity was higher for chloroacetaldehyde, steady-state co-variation assays were performed with this substrate and NAD+. The kinetic data obtained for the native and substituted enzymes were consistent with the sequential kinetic mechanism as described before for wild-type ALDH2 [22] (Scheme 1, Table 2). Consistent with the data shown in Table 1, we observe a split between the T244V(or A) and the T244G(or S) substitutions, where the latter two enzymes show the most drastic decreases in kcat. Interestingly, in the T244G and T244S substituted enzymes, KM(chloroacetaldehyde) values are quite similar to wild-type, instead it is the T244V and T244A substituted enzymes that show large increases in the KM for this substrate (Table 2). In the same way, the T244G and T244S substituted enzymes also show less drastic changes in KM(NAD+) than the T244V or T244A substituted enzymes, with a maximum KM(NAD+) of 2.1 mM for the T244A substituted enzyme. However, the variations in KM and kcat tended to offset one another such that kcat/KM(chloroacetaldehyde) varied 6-fold and kcat/KM(NAD+) varied only 3-fold across the substituted enzymes. The fit to the complete velocity equation for an ordered bi-bi reaction also provides an estimate of the dissociation constant for NAD+ [Kia ≈ KD(NAD+)] and this value increased 3- to 12-fold for the substituted enzymes. In contrast, the estimated dissociation constant, for NADH [Kiq ≈ KD(NADH)] was increased only slightly, from a nearly wild-type value for the T244S substituted enzyme to a just over 2-fold increase for the T244V substituted enzyme.
3.3. Structures of T244A ALDH2
The ALDH2 T244A enzyme was selected for structure determination because this substitution resulted in the largest changes in the overall catalytic efficiency when both substrates for the reaction were considered (Table 2, kcat/KiaKM(chloroacetaldehyde)). We solved the T244A substituted enzyme structure in the apo-enzyme form and as complexes with both oxidized and reduced cofactor to between 1.7 and 2.25 Å resolution (Table 3). All structures are essentially isomorphous with the wild-type and contain two independent copies of the tetramer in the asymmetric unit of the crystals.
Table 3.
Data collection and refinement statistics.
| Apo T244A | T244A-NAD+ | T244A-NADH | Apo ALDH2 | |
|---|---|---|---|---|
| PDB Code | 3n81 | 3n83 | 3n82 | 3n80 |
| Cell Dimensions (Å) | 141.8, 152.2, 177.3 | 145.1, 151.0, 177.7 | 142.2, 150.7, 177.3 | 141.7, 152.3, 177.3 |
| Resolution Range (Å) | 50.0 – 1.7 | 50.0 – 1.9 | 50.0 – 2.25 | 50.0 – 1.5 |
| Unique Reflections | 416,273 | 305,300 | 179,717 | 599,824 |
| Completeness (%) | 99.3 (98.4) | 99.8 (99.7) | 99.7 (98.1) | 98.9 (100) |
| Redundancy | 5.7 (4.0) | 6.0 (5.7) | 4.4 (3.3) | 3.1 (3.0) |
| <I>/σ<I> | 16.4 (2.4) | 20.6 (2.9) | 16.0 (4.8) | 20.1 (3.6) |
| Rmerge (%) | 9.2 (50.3) | 8.7 (53.9) | 7.3 (22.7) | 5.6 (26.7) |
| Refinement | ||||
| Rfree/Rwork (%) | 21.7/18.0 | 23.2/18.9 | 21.9/16.8 | 17.5/14.9 |
| Average B-value (Å2) | 18.1 | 21.2 | 25.6 | 13.1 |
| r.m.s.d bond lengths (Å)a | 0.012 | 0.011 | 0.008 | 0.013 |
| r.m.s.d bond angles (°)a | 1.27 | 1.26 | 1.07 | 1.54 |
Space group for all structures is orthorhombic (P212121). Values in parentheses correspond to the highest resolution shell.
Root-mean-square deviations (RMSDs) from ideal geometry of the final model.
3.3.1 Structural changes in the apo-enzyme due to T244A substitution
Overall, the structure of the apo-enzyme form of the T244A ALDH2 is similar to wild-type ALDH2 of similar resolution (Table 3), with an r.m.s.d. for all Cα atoms in the tetramer of 0.10 Å. The most obvious changes in the apo-T244A structure occur in the local environment surrounding residue 244. Here, we observe conformational heterogeneity of the Glu-268 and Lys-178 side chains, as well as a shift in the position of the Glu-476 side chain (Fig. 3A). In addition, when compared to wild-type, the Cα atoms of residues 245–248, which form the beginning of the αG helix, systematically shift in the direction of Cys-302 by between 0.3 and 0.7 Å (Fig. 3A). We also observe an increase in the thermal parameters for the whole αG helix (residues 246–260, Fig. 4). Lastly, we also see conformational heterogeneity of the side chain of Arg-264, at the C-terminal end of helix αG (Fig. 3A). Many of these structural changes are similar to those observed in the ALDH2 R475Q substituted enzyme [31].
Fig. 3.

Stereo views of the interactions N- and C-terminal to αG. (A) Apo T244A and wild-type ALDH2 (PDB codes 3n81 and 3n80) (B) NADH complexes of T244A and wild-type (PDB codes 3n82 and 1o02). (C) NAD complexes of T244A and C302S ALDH2 (PDB codes 3n83 and 1o04). 2Fo-Fc electron density maps contoured at 1.0 σ. Legend refers to color of the electron density (hash-mark color) and corresponding model (text color).
Fig. 4.

Relative variation in average B-factor as a function of residue number. Delta-B values were calculated as the difference between the refined values in each of the T244A structures and the refined values in the wild-type apo ALDH2 structure, after baseline correction using the overall B for each structure.
3.3.2 Restoration of the T244A ALDH2 structure upon NADH binding
The overall structure of the T244A-NADH complex is similar to both the apo-T244A and the wild-type NADH binary complex (PDB code 1o02) [17] with r.m.s.d.s for all Cα atoms in the tetramer of 0.22 and 0.13 Å, respectively. The coenzyme in the T244A-NADH structure displays a single well-ordered conformation in the ‘hydrolysis’ position with clear density for the Mg2+ ion bound to the pyrophosphates, both typical for NADH binding to ALDH2 [17]. The majority of the active site residues are also restored to positions observed in the wild-type NADH-bound structure (PDB code 1o02) [17], including residues 245–248 and the side chains of Arg-264 and Lys-178 (Fig. 3B). Additionally, the thermal parameters for αG are reduced to baseline values (Fig. 4). Interestingly, although Glu-268 is ordered in this structure, it is positioned with the carboxyl group close (dave = 3.5 Å) to both atoms of the carboxamide group of NADH (Fig. 3B). In wild-type structures, Glu-268 is found to occupy one of two preferred conformations; (a) with one side chain carboxyl oxygen at 3.4 Å from Cys-302 and the other at 3.4 Å from the amide nitrogen of the carboxamide moiety of NADH, or (b) with both side chain carboxyl oxygens adjacent to the main-chain atoms of residues 465 and 476 [17]. The conformation observed for Glu-268 in the T244A-NADH complex is similar to conformation (a), but the interaction with Cys-302 is lost in favor of the second interaction with the carboxamide group (Fig. 3B).
3.3.3 Partial restoration of the T244A ALDH2 structure upon NAD+ binding
In contrast to the well-ordered conformation of NADH in the T244A enzyme, the complex with NAD+ lacks a single well-defined position for the nicotinamide mononucleotide (NMN) portion of the coenzyme (Fig. 3C). Electron density for the adenosine is clear as are two distinct positions for the pyrophosphates, however the electron density for the NMN is discontinuous, making an accurate fit difficult. Hence, we chose to model only the ADP portion of NAD+ for this structure. Nevertheless, NAD+ binding does result in partial restoration of the structural changes observed in the apo-T244A enzyme. Residues 245–248 and residue Lys-178 in the T244A-NAD+ complex structure are restored to positions similar to those observed in the wild-type apo-enzyme structure as well as the structure of the C302S substituted enzyme with bound NAD+ (PDB code 1o04) [17]. As observed in the NADH complex, the B-factors for αG in the NAD+ complex are also reduced to baseline values (Fig. 4). On the other hand, the side chain of Arg-264 retains the conformational disorder observed in the apo-form of the T244A substituted enzyme structure, as does the side chain of Glu-268 (Fig. 3C). The overall structural similarity of the T244A-NAD+ complex to the apo-T244A, NAD-bound wild-type, and NAD-bound C302S ALDH2 is 0.24, 0.18, and 0.20 A r.m.s.d. for all Cα atoms in the tetramers, respectively.
4. Discussion
One of the unique aspects of catalysis in ALDH isozymes is the apparent requirement for coenzyme isomerization within the active site in order to facilitate distinct steps of the catalytic cycle. Structural studies on multiple ALDH family members have demonstrated the mobility of the coenzyme, and some isozymes show distinct coenzyme conformation preferences (Figs. 1B and 2) [16,49–51]. The most obvious conclusion from these studies is that while catalysis must proceed through a common subset of these conformations, each isoenzyme would appear to adopt different stable ground state conformations that are accessible in crystal structures. For ALDH2, there appear to be two relatively stable conformations of the coenzyme, the so-called ‘hydride transfer’ and ‘hydrolysis’ positions both of which are applicable to particular steps in the catalytic cycle; namely, hydride transfer and acyl-enzyme hydrolysis (Fig. 2) [17]. Given the structural precision necessary to accommodate at least two catalytically competent coenzyme conformations, it is not surprising that substitutions within the coenzyme binding cleft affect not only the catalytic efficiency for coenzyme, but also the catalytic efficiency for substrate aldehydes and the rate-limiting step. For instance, substitution of either Lys-192 or Glu-399, which interact with the coenzyme ribose moieties, change the major rate-limiting step in ALDH2 from acyl-enzyme hydrolysis to hydride transfer [52]. This result is consistent with important stabilizing interactions between the coenzyme and enzyme that affect the ability to precisely position the nicotinamide ring for hydride transfer.
In contrast to Lys-192 or Glu-399, the side chain of Thr-244 does not make any direct hydrogen bonds to the coenzyme [17,29]. Instead, the methyl substituent of Thr-244 is within van der Waals contact distance of the B-face of the nicotinamide ring when the coenzyme occupies the ‘hydride transfer’ position (Fig. 2A) [17]. Proper positioning of the nicotinamide ring is essential for efficient hydride transfer to the C4N carbon and may also be important in activating the catalytic cysteine in preparation for thiohemiacetal formation [53]. Furthermore, when the nicotinamide ring is in the contracted conformation ideal for the deacylation reaction, the hydroxyl group of the Thr-244 side-chain may indirectly stabilize the deacylating water molecule through an intervening ordered water molecule [17,18]. These observations suggest that Thr-244 may also have a role in facilitating the deacylation reaction.
Our data for the Thr-244 substitutions in ALDH2 show that the kcat/KM for chloroacetaldehyde was reduced by at least a factor of six and that kcat/KM for NAD+ was reduced by a factor of more than 25 (Table 2). We previously found catalytic efficiencies for substrate and cofactor to be reduced by similar amounts in T244S ALDH1A1, where additionally, the rate-limiting step was altered from NADH release to deacylation [29]. The T244S substitution in GAPN also resulted in decreased catalytic efficiency for substrate and coenzyme of about 700- and 1,100-fold, respectively [19]. A change in the rate-limiting step was also observed in GAPN, in this case the shift was from deacylation to hydride-transfer [19]. In contrast, when Thr-244 was substituted in ALDH2, deacylation remained the major rate-limiting step. However, in each of these other two enzymes (ALDH1A1 and GAPN) substitution at position 244 reduced catalytic efficiency by slowing chemical steps that are associated with the positioning of the cofactor, either just prior (slower hydride transfer in GAPN) or just after (slower acyl-enzyme hydrolysis in ALDH1A1) the generally accepted coenzyme isomerization. Another interesting point of difference between our results and those obtained for GAPN is in the magnitude of the changes in kcat resulting from, in particular, the T244V substitution. In ALDH2 all Thr-244 substituted enzymes were catalytically competent (Tables 1, 2). However, the kcat for the T244V substituted GAPN enzyme was reduced by almost 250,000-fold, resulting in a practically inactive enzyme [19]
These differences are reminiscent of those obtained for substitutions of another highly conserved active site residue, Glu-268. There have been two roles ascribed to Glu-268: activation of Cys-302 and activation of the hydrolytic water molecule [25]. However, it is fair to say that the relative contribution of Glu-268 to either or both of those functions appears to vary amongst different ALDH isozymes [54–56]. Similarly, the data presented here support a role for Thr-244 in both thiohemiacetal formation and in acyl-enzyme hydrolysis, with different substitutions at 244 impacting each process slightly differently. The T244S enzyme has the lowest kcat, but the highest catalytic efficiency toward chloroacetaldehyde and the best overall catalytic efficiency (kcat/KiAKMB, Table 2). For the T244S substituted enzyme, decreased kcat primarily reflects decreased acyl-enzyme hydrolysis, implying a slower rate of deacylation for this enzyme. On the other hand, the enzymes with more hydrophobic residues at position 244, T244A and T244V, showed less impact on kcat, but larger decreases in catalytic efficiency toward chloroacetaldehyde (Table 2). Thus, deacylation was least impaired for these substitutions, but thiohemiacetal formation appeared diminished. The T244G enzyme was intermediate, with effects both on kcat and kcat/KM, but not as severe for either measurement. What these results suggest is that the more hydrophilic environment promoted by the substitution of Thr-244 to Ser, and less so Gly, does not dramatically impact thiohemiacetal formation, nor the KM(NAD+). Instead, the smaller size of the Ser and Gly side chains likely increases the number of non-productive interactions between Glu-268, the nicotinamide ring, and the activated water molecule thereby negatively impacting acyl-enzyme hydrolysis and, ultimately, kcat. On the other hand, the more hydrophobic substitutions of Ala and Val appear to impact thiohemiacetal formation, while having a smaller impact on kcat. This effect might be ascribed to the charged nicotinamide ring, which would interact less favorably in the more hydrophobic environment of the T244A and T244V substituted enzymes. In this context, the Ala and Val substitutions could simply provide a less stable environment to support the precise positioning of the positively charged nicotinamide for efficient thiohemiacetal formation, as was suggested for the GAPDH isozyme [56]. Once the reaction has proceeded through hydride transfer and coenzyme isomerization, the impact of the hydrophobic substitutions upon acyl-enzyme hydrolysis in ALDH2 may be reduced by decreasing the number of non-productive encounters between the activated water and the surrounding active site residues and minimizing any impact on kcat. Thus, we conclude that our kinetic data for ALDH2 are in agreement with results obtained for ALDH1A1 [29] and GAPN [19] in supporting a role for Thr-244 in the stable positioning of the nicotinamide ring, with potential impact on the catalytic steps between thiohemiacetal formation and acyl-enzyme hydrolysis.
The present study also identifies a new role for Thr-244, specifically in anchoring the N-terminal end of the key NAD binding helix, αG. Rather than leading to the large-scale disordering observed when the underlying structural scaffold is disrupted, such as those observed in the ALDH2*2 enzyme structure [20], these effects are more subtle and are only visualized by inspection of the local thermal parameters for these regions of protein structure in our apo-T244A ALDH2 crystal structure (Figs. 3A, 4). However, similar to the changes in ALDH2*2 and E475Q ALDH2, these perturbations are largely restored with coenzyme binding, provided sufficient coenzyme is present [31] (Figs. 3B, 3C, 4). This is also consistent with the 3- to 11-fold increase in Kia for the T244 substitutions (Table 2) and with our prior data for the T244S substitution in ALDH1A1 where the estimated KD for NAD+ was increased about 9-fold [19]. Presumably the descreased affinity for coenzyme reflects the extent to which restoration of a stable structural scaffold to support the nicotinamide ring impacts binding. The smaller changes in the NADH inhibition constants (estimated KD) seen in ALDH1A1 [29], GAPN [19], and ALDH2 (1.2- to 2.2-fold, Table 2) probably reflect the lack of interaction between residue 244 and the reduced nicotinamide ring when the coenzyme is positioned away from Cys302 in the hydrolysis position. In addition to a role in positioning the nicotinamide ring, our data suggests that Thr-244 also plays a role in facilitating coenzyme binding by stabilizing the N-terminus of helix αG. We further propose that the potential hydrogen bond between the Thr-244 delta hydroxyl and the zeta nitrogen of Lys-178 could support both functions of Thr-244 by orienting the side chain, as has been previously shown for GAPN [29]. Indeed, Thr-244 is observed to be within hydrogen bonding distance to Lys-178 in the majority of the ALDH entries in the PDB and a lysine or histidine is observed at this position in nearly 75% of CoA-independent ALDHs. In ALDH2, we suggest that the substitution of Thr-244 to Ala breaks the link with Lys-178, releasing a conformational constraint that limited the mobility of the αG helix. Although this hypothesis does not fully explain the kinetic data in Table 2, the role of Lys-178 in tethering Thr-244 is an interesting avenue for further investigation.
In conclusion, the data presented here support the idea that ALDH family members all utilize similar catalytic strategies. Clearly each enzyme active site presents a unique context in which the conserved residues serve their respective roles and this will impact the magnitude of the changes that are observed when substitutions are introduced.
Highlights.
The function of Threonine 244 in aldehyde dehydrogenase family members is enigmatic.
We characterized four different mutants of ALDH2 substituted at position 244.
All mutants showed decreased activity and reduced efficiency toward both substrates.
The crystal structure of the T244A mutant showed reduced stability of the αG helix.
Our data supports the notion that Thr244 is critical for proper coenzyme positioning.
Acknowledgments
T.D.H. wishes to thank the staff at the 19-ID beamline facility, especially M. Cuff, N. Duke and S. Ginell. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by U Chicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. This work was supported in part by US National Institutes of Health grants AA011982 and AA018123 to T.D.H. S.P.-M. was supported by US National Institutes of Health training fellowship T32-DK064466.
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vasiliou V, Nebert DW. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum Genomics. 2005;2:138–143. doi: 10.1186/1479-7364-2-2-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hurley TD, Edenberg HJ, Li TK. Pharmacogenomics of alcoholism. In: Licinio J, Wong M, editors. Pharmacogenomics: The search for individualized therapies. 2002. pp. 417–441. [Google Scholar]
- 3.Brooks PJ, Enoch MA, Goldman D, Li TK, Yokoyama A. The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLoS Med. 2009;6:e50. doi: 10.1371/journal.pmed.1000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higuchi S, Matsushita S, Imazeki H, Kinoshita T, Takagi S, Kono H. Aldehyde dehydrogenase genotypes in Japanese alcoholics. Lancet. 1994;343:741–742. doi: 10.1016/s0140-6736(94)91629-2. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Foster MW, Zhang J, Mao L, Rockman HA, Kawamoto T, Kitagawa K, Nakayama KI, Hess DT, Stamler JS. An essential role for mitochondrial aldehyde dehydrogenase in nitroglycerin bioactivation. Proc Natl Acad Sci USA. 2005;102:12159–12164. doi: 10.1073/pnas.0503723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beretta M, Gruber K, Kollau A, Russwurm M, Koesling D, Goessler W, Keung WM, Schmidt K, Mayer B. Bioactivation of nitroglycerin by purified mitochondrial and cytosolic aldehyde dehydrogenases. J Biol Chem. 2008;283:17873–17880. doi: 10.1074/jbc.M801182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman RI, Weiner H. Horse liver aldehyde dehydrogenase. II. Kinetics and mechanistic implications of the dehydrogenase and esterase activity. J Biol Chem. 1972;247:267–272. [PubMed] [Google Scholar]
- 8.Mukerjee N, Pietruszko R. Human mitochondrial aldehyde dehydrogenase substrate specificity: Comparison of esterase with dehydrogenase reaction. Arch Biochem Biophys. 1992;299:23–29. doi: 10.1016/0003-9861(92)90239-s. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi K, Weiner H. Nicotinamide adenine dinucleotide activation of the esterase reaction of horse liver aldehyde dehydrogenase. Biochemistry. 1981;20:2720–2726. doi: 10.1021/bi00513a003. [DOI] [PubMed] [Google Scholar]
- 10.Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, Mochly-Rosen D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science. 2008;321:1493–1495. doi: 10.1126/science.1158554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brichac J, Ho KK, Honzatko A, Wang R, Lu X, Weiner H, Picklo MJ., Sr Enantioselective oxidation of trans-4-hydroxy-2-nonenal is aldehyde dehydrogenase isozyme and Mg2+ dependent. Chem Res Toxicol. 2007;20:887–895. doi: 10.1021/tx7000509. [DOI] [PubMed] [Google Scholar]
- 12.Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol. 2006;19:102–110. doi: 10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- 13.Beretta M, Gorren AC, Wenzl MV, Weis R, Russwurm M, Koesling D, Schmidt K, Mayer B. Characterization of the east asian variant of aldehyde dehydrogenase-2: Bioactivation of nitroglycerin and effects of alda-1. J Biol Chem. 2010;285:943–952. doi: 10.1074/jbc.M109.014548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Budas GR, Disatnik MH, Chen CH, Mochly-Rosen D. Activation of aldehyde dehydrogenase 2 (ALDH2) confers cardioprotection in protein kinase C epsilon (PKCvarepsilon) knockout mice. J Mol Cell Cardiol. 2009;48:757–764. doi: 10.1016/j.yjmcc.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perez-Miller S, Younus H, Vanam R, Chen CH, Mochly-Rosen D, Hurley TD. Alda-1 is an agonist and chemical chaperone for the common human aldehyde dehydrogenase 2 variant. Nat Struct Mol Biol. 2010;17:159–164. doi: 10.1038/nsmb.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Ambrosio K, Pailot A, Talfournier F, Didierjean C, Benedetti E, Aubry A, Branlant G, Corbier C. The first crystal structure of a thioacylenzyme intermediate in the ALDH family: New coenzyme conformation and relevance to catalysis. Biochemistry. 2006;45:2978–2986. doi: 10.1021/bi0515117. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Miller SJ, Hurley TD. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry. 2003;42:7100–7109. doi: 10.1021/bi034182w. [DOI] [PubMed] [Google Scholar]
- 18.Steinmetz CG, Xie P, Weiner H, Hurley TD. Structure of mitochondrial aldehyde dehydrogenase: The genetic component of ethanol aversion. Structure. 1997;5:701–711. doi: 10.1016/s0969-2126(97)00224-4. [DOI] [PubMed] [Google Scholar]
- 19.Pailot A, D’Ambrosio K, Corbier C, Talfournier F, Branlant G. Invariant Thr244 is essential for the efficient acylation step of the non-phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutans. Biochem J. 2006;400:521–530. doi: 10.1042/BJ20060843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larson HN, Weiner H, Hurley TD. Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” Variant. J Biol Chem. 2005;280:30550–30556. doi: 10.1074/jbc.M502345200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farres J, Wang X, Takahashi K, Cunningham SJ, Wang TT, Weiner H. Effects of changing glutamate 487 to lysine in rat and human liver mitochondrial aldehyde dehydrogenase. A model to study human (oriental type) class 2 aldehyde dehydrogenase. J Biol Chem. 1994;269:13854–13860. [PubMed] [Google Scholar]
- 22.Farres J, Wang TT, Cunningham SJ, Weiner H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry. 1995;34:2592–2598. doi: 10.1021/bi00008a025. [DOI] [PubMed] [Google Scholar]
- 23.Sheikh S, Ni L, Hurley TD, Weiner H. The potential roles of the conserved amino acids in human liver mitochondrial aldehyde dehydrogenase. J Biol Chem. 1997;272:18817–18822. doi: 10.1074/jbc.272.30.18817. [DOI] [PubMed] [Google Scholar]
- 24.Jones KH, Lindahl R, Baker DC, Timkovich R. Hydride transfer stereospecificity of rat liver aldehyde dehydrogenases. J Biol Chem. 1987;262:10911–10913. [PubMed] [Google Scholar]
- 25.Wang X, Weiner H. Involvement of glutamate 268 in the active site of human liver mitochondrial (class 2) aldehyde dehydrogenase as probed by site-directed mutagenesis. Biochemistry. 1995;34:237–243. doi: 10.1021/bi00001a028. [DOI] [PubMed] [Google Scholar]
- 26.Hammen PK, Allali-Hassani A, Hallenga K, Hurley TD, Weiner H. Multiple conformations of nad and nadh when bound to human cytosolic and mitochondrial aldehyde dehydrogenase. Biochemistry. 2002;41:7156–7168. doi: 10.1021/bi012197t. [DOI] [PubMed] [Google Scholar]
- 27.Shone CC, Fromm HJ. Steady-state and pre-steady-state kinetics of coenzyme a linked aldehyde dehydrogenase from Escherichia coli. Biochemistry. 1981;20:7494–7501. doi: 10.1021/bi00529a026. [DOI] [PubMed] [Google Scholar]
- 28.Sohling B, Gottschalk G. Purification and characterization of a coenzyme-a-dependent succinate-semialdehyde dehydrogenase from Clostridium kluyveri. Eur J Biochem. 1993;212:121–127. doi: 10.1111/j.1432-1033.1993.tb17641.x. [DOI] [PubMed] [Google Scholar]
- 29.Ho KK, Hurley TD, Weiner H. Selective alteration of the rate-limiting step in cytosolic aldehyde dehydrogenase through random mutagenesis. Biochemistry. 2006;45:9445–9453. doi: 10.1021/bi060718c. [DOI] [PubMed] [Google Scholar]
- 30.Ho KK, Mukhopadhyay A, Li YF, Mukhopadhyay S, Weiner H. A point mutation produced a class 3 aldehyde dehydrogenase with increased protective ability against the killing effect of cyclophosphamide. Biochem Pharmacol. 2008;76:690–696. doi: 10.1016/j.bcp.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Larson HN, Zhou J, Chen Z, Stamler JS, Weiner H, Hurley TD. Structural and functional consequences of coenzyme binding to the inactive asian variant of mitochondrial aldehyde dehydrogenase: Roles of residues 475 and 487. J Biol Chem. 2007;282:12940–12950. doi: 10.1074/jbc.M607959200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaffer AA, Aravind L, Madden TL, Shavirin S, Spouge JL, Wolf YI, Koonin EV, Altschul SF. Improving the accuracy of psi-blast protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 2001;29:2994–3005. doi: 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The Uniprot Consortium, The universal protein resource (uniprot) Nucleic Acids Res. 2007;35:D193–197. doi: 10.1093/nar/gkl929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papadopoulos JS, Agarwala R. COBALT: Constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23:1073–1079. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- 36.Nicholas KB, Nicholas HB, Jr, Deerfield DW., II GeneDoc: Analysis and visualization of genetic variation. EMBNEW NEWS. 1997;4:14. ( http://www.Psc.Edu/biomed/genedoc) [Google Scholar]
- 37.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.The PyMol Molecular Graphics System, Version 0.99. Schrödinger, LLC; [Google Scholar]
- 39.Zheng CF, Wang TT, Weiner H. Cloning and expression of the full-length cDNAs encoding human liver class 1 and class 2 aldehyde dehydrogenase. Alcohol Clin Exp Res. 1993;17:828–831. doi: 10.1111/j.1530-0277.1993.tb00849.x. [DOI] [PubMed] [Google Scholar]
- 40.Ghenbot G, Weiner H. Purification of liver aldehyde dehydrogenase by p-hydroxyacetophenone-sepharose affinity matrix and the coelution of chloramphenicol acetyl transferase from the same matrix with recombinantly expressed aldehyde dehydrogenase. Protein Expres Purif. 1992;3:470–478. doi: 10.1016/1046-5928(92)90064-4. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Kedishvili NY, Popov KM, Rougraff PM, Zhao Y, Crabb DW, Harris RA. CoA-dependent methylmalonate-semialdehyde dehydrogenase, a unique member of the aldehyde dehydrogenase superfamily. cDNA cloning, evolutionary relationships, and tissue distribution. J Biol Chem. 1992;267:19724–19729. [PubMed] [Google Scholar]
- 45.Kirch HH, Schlingensiepen S, Kotchoni S, Sunkar R, Bartels D. Detailed expression analysis of selected genes of the aldehyde dehydrogenase (ALDH) gene superfamily in Arabidopsis thaliana. Plant Mol Biol. 2005;57:315–332. doi: 10.1007/s11103-004-7796-6. [DOI] [PubMed] [Google Scholar]
- 46.Weiner H, Hu JHJ, Sanny CG. Rate-limiting steps for the esterase and dehydrogenase reaction catalyzed by horse liver aldehyde dehydrogenase. J Biol Chem. 1976;251:3853–3855. [PubMed] [Google Scholar]
- 47.Ho KK, Allali-Hassani A, Hurley TD, Weiner H. Differential effects of Mg2+ ions on the individual kinetic steps of human cytosolic and mitochondrial aldehyde dehydrogenases. Biochemistry. 2005;44:8022–8029. doi: 10.1021/bi050038u. [DOI] [PubMed] [Google Scholar]
- 48.Vallari RC, Pietruszko R. Interaction of Mg2+ with human liver aldehyde dehydrogenase. J Biol Chem. 1984;259:4922–4926. [PubMed] [Google Scholar]
- 49.Liu ZJ, Sun YJ, Rose J, Chung YJ, Hsiao CD, Chang WR, Kuo I, Perozich J, Lindahl R, Hempel J, Wang BC. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat Struct Biol. 1997;4:317–326. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 50.Cobessi D, Tete-Favier F, Marchal S, Azza S, Branlant G, Aubry A. Apo and holo crystal structures of an NADP-dependent aldehyde dehydrogenase from Streptococcus mutans. J Mol Biol. 1999;290:161–173. doi: 10.1006/jmbi.1999.2853. [DOI] [PubMed] [Google Scholar]
- 51.Tsybovsky Y, Donato H, Krupenko NI, Davies C, Krupenko SA. Crystal structures of the carboxyl terminal domain of rat 10-formyltetrahydrofolate dehydrogenase: Implications for the catalytic mechanism of aldehyde dehydrogenases. Biochemistry. 2007;46:2917–2929. doi: 10.1021/bi0619573. [DOI] [PubMed] [Google Scholar]
- 52.Ni L, Sheikh S, Weiner H. Involvement of glutamate 399 and lysine 192 in the mechanism of human liver mitochondrial aldehyde dehydrogenase. J Biol Chem. 1997;272:18823–18826. doi: 10.1074/jbc.272.30.18823. [DOI] [PubMed] [Google Scholar]
- 53.Marchal S, Branlant G. Evidence for the chemical activation of essential Cys-302 upon cofactor binding to nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 1999;38:12950–12958. doi: 10.1021/bi990453k. [DOI] [PubMed] [Google Scholar]
- 54.Vedadi M, Meighen E. Critical glutamic acid residues affecting the mechanism and nucleotide specificity of Vibrio harveyi aldehyde dehydrogenase. Eur J Biochem. 1997;246:698–704. doi: 10.1111/j.1432-1033.1997.t01-1-00698.x. [DOI] [PubMed] [Google Scholar]
- 55.Mann CJ, Weiner H. Differences in the roles of conserved glutamic acid residues in the active site of human class 3 and class 2 aldehyde dehydrogenases. Protein Sci. 1999;8:1922–1929. doi: 10.1110/ps.8.10.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marchal S, Rahuel-Clermont S, Branlant G. Role of glutamate-268 in the catalytic mechanism of nonphosphorylating glyceraldehyde-3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 2000;39:3327–3335. doi: 10.1021/bi9914208. [DOI] [PubMed] [Google Scholar]