

Abstract
The conformational preferences of polyglutamine (polyQ) sequences are of major interest because of their central importance in the expanded CAG repeat diseases that include Huntington’s disease (HD). Here we explore the response of various biophysical parameters to the introduction of β-hairpin motifs within polyQ sequences. These motifs (trpzip, disulfide, D-Pro-Gly, Coulombic attraction, L-Pro-Gly) enhance formation rates and stabilities of amyloid fibrils with degrees of effectiveness well-correlated with their known abilities to enhance β-hairpin formation in other peptides. These changes led to decreases in the critical nucleus for amyloid formation from a value of n* = 4 for a simple, unbroken Q23 sequence to approximate unitary n* values for similar length polyQs containing β-hairpin motifs. At the same time, the morphologies, secondary structures, and bioactivities of the resulting fibrils were essentially unchanged from simple polyQ aggregates. In particular, the signature pattern of SSNMR 13C Gln resonances that appears to be unique to polyQ amyloid is replicated exactly in fibrils from a β-hairpin polyQ. Importantly, while β-hairpin motifs do produce enhancements in the equilibrium constant for nucleation in aggregation reactions, these Kn* values remain quite low (~ 10−10) and there is no evidence for significant embellishment of β-structure within the monomer ensemble. The results indicate an important role for β-turns in the nucleation mechanism and structure of polyQ amyloid and have implications for the nature of the toxic species in expanded CAG repeat diseases.
In Huntington’s disease and nine other expanded polyglutamine (polyQ) diseases, a genetic expansion of the polyQ sequence in a disease protein into a pathological repeat length range typically above 35 residues increases disease risk and decreases age-of-onset 1. One hypothesized biophysical explanation for this dramatic repeat length effect is that expanded polyQ sequences populate an alternative monomer conformation that triggers a dysfunctional and/or toxic response in the cell 2. However, the observations of polyQ aggregates in disease brain tissue 3 and of a repeat length dependence of aggregation both in vitro 4, 5 and in cell and animal models 6 suggest an alternative hypothesis featuring a strong role for polyQ aggregation 7. For this reason, particular emphasis has been placed on understanding the mechanisms of polyQ amyloid nucleation and how this might be affected by repeat length, sequence context, and cellular environment 8. Although flanking sequences can clearly have a major impact on aggregation rates and mechanisms 8–10, it is likely that some fundamental aspects of polyQ amyloid formation deduced from studies on simple polyQ sequences will apply to polyQ behavior in disease proteins.
Mature aggregates of polyQ disease proteins and model peptides exhibit many features of amyloid structure, including a filamentous architecture in electron micrographs 4 and β-rich secondary structure by CD 11, FTIR 10, X-ray diffraction 12–14, and SSNMR 15, 16. In contrast to a number of relatively well-characterized polypeptide amyloids, which exhibit secondary structures dominated by in-register, parallel β-sheet 17–19, polyQ amyloid appears more likely to possess anti-parallel β-sheet architecture 13–16, 20, 21. Two basic models for how polypeptide sequences might be accommodated into such anti-parallel β-sheet structures have been delineated by Kajava and Steven 22. In the first, the chain remains within a single sheet by undergoing a series of intramolecularly H-bonded β-turn/β-hairpin chain reversals (Fig. 1). In the second, the chain undergoes reverse turns (“β-arcs”) that connect adjacent β-sheets in a conformation that has been referred to as a β-arch 22 (Fig. 1). Such chain reversals are reminiscent of polypeptide conformations found in a number of parallel, in-register β-sheet amyloids, including fibrils of Aβ 22.
Figure 1.

Reverse turn models of polyQ peptides and their amyloid fibrils. Schematic anti-parallel β-sheet models for a segment of amyloid fibril showing two fundamental ways that fibrils might accommodate longer peptides requiring reverse turns for optimal involvement in fibril structure. In the left panel, monomers are folded into β-hairpins mediated by β-turn chain reversals. In the right panel, monomers are folded into β-arches mediated by β-arc chain reversals. Red dotted lines denote backbone intra-strand H-bonding.
Previously, our lab found that all simple polyQ peptides tested with the sequence format K2QNK2 spontaneously form amyloid via a classical nucleated growth polymerization mechanism without forming any required, on-pathway non-amyloid intermediates 11, 23. At the same time, nucleation efficiencies varied considerably within this series, such that peptides with polyQ repeat lengths of 23 or lower exhibited a critical nucleus (n*) of 4, while those with repeat lengths of 26 or above had n* = 1 23. We interpreted these data to be consistent with β-hairpin formation playing a critical role in nucleus structure, based on the hypothesis that longer polyQ sequences can form more stable β-hairpins 23. It seems also possible, however, that the chain reversal required for enhancing polyQ amyloid nucleation is the β-arc 22 in which the reverse-turn conformation is stabilized by side chain interactions rather than main-chain H-bonding (Fig. 1a). Indeed, some have argued that the anti-parallel β-sheet architecture of polyQ amyloid may feature such β-arc connectivity 16.
A number of recent studies have interpreted a variety of biochemical and biophysical data to indicate that longer, monomeric polyQ sequences in solution are enriched in β-hairpin like structures (compared with shorter polyQ sequences), which would thus be prime candidates for the toxic species in expanded polyQ disorders 24–27. In contrast, other experimental and computational studies have indicated that monomeric polyQ sequences of all repeat lengths longer than ~ 10 Gln residues exhibit similar conformations that are compact but at the same time disordered 5, 8, 11, 28–31.
To address some of these fundamental issues concerning polyQ folding and cytotoxicity, we designed and analyzed a series of mutated polyQ sequences containing various well-characterized β-hairpin encouraging sequence elements. Our results provide a number of insights into nucleation mechanism and amyloid structure for the polyQ homopolymer, and place stringent limits on the ability of polyQ monomers in water to populate β-hairpin conformations.
RESULTS
A number of sequence motifs have been described that highly favor β-hairpin formation 32–42. One class of motifs encourages β-hairpin formation from the center of the peptide (e.g., L-Pro-X 32 and D-Pro-Gly 32, 35). Another class encourages the close association of chain termini in orientations compatible with β-hairpin formation (e.g., disulfide crosslinks 41, “tryptophan zippers” (trpzips) 42, and favorable Coulombic interactions 36, 38). We set out to explore the effects of both classes of motif on the aggregation kinetics of sequences in the Q22–Q23 repeat length range.
A survey of this series of mutated polyQ peptides (Table 1) shows that β-hairpin encouraging mutations consistently enhance aggregation kinetics (Fig. 2a). The L-Pro-Gly motif, which is compatible with β-hairpin conformation without greatly favoring it 32, modestly enhances aggregation kinetics under these conditions (Fig. 2a, ◆) compared with a K2Q23K2 peptide (Fig. 2a, ■). In contrast, the D-Pro-Gly motif, known to be more effective at encouraging β-hairpin formations than L-Pro-Gly in monomeric peptide model studies 32, 35, dramatically enhances aggregation kinetics (Fig. 2a, ▼). Peptides containing either an intramolecular disulfide bond 41 (Fig. 2a, ▲) or a “trpzip” motif (Fig. 2a, ■) consisting of N-terminal Nα-Ac-Trp and C-terminal WTG 42 also exhibit dramatic rate increases. In addition, a D2Q23K2 peptide, engineered for the potential to undergo intramolecular Coulombic attraction between Asp and Lys residues at chain termini, exhibits a relatively modest rate enhancement (Fig. 2a, ●).
Table 1.
Structures of peptides
| Name | Sequence | ||||
|---|---|---|---|---|---|
| K2Q23K2 | KKQQQQQQQQ | QQQQQQQQQQ | QQQQQKK | ||
| K2Q10PGQ11K2 | KKQQQQQQQQ | QQPGQQQQQQ | QQQQQKK | ||
| K2Q10pGQ11K2 | KKQQQQQQQQ | QQpGQQQQQQ | QQQQQKK | ||
| D2Q23K2 | DDQQQQQQQQ | QQQQQQQQQQ | QQQQQKK | ||
| K2CQ22CK2 (ox.) | KKCQQQQQQQ | QQQQQQQQQQ | QQQQQCKK | ||
| K2Q11PGQ11D2 | KKQQQQQQQQ | QQQPGQQQQQ | QQQQQQDD | ||
| AcWQ22WTGK2 | AcWQQQQQQQ | QQQQQQQQQQ | QQQQQWTGKK | ||
| AcWQ11pGQ11WTGK2 | AcWQQQQQQQ | QQQQpGQQQQ | QQQQQQQWTG | KK | |
| K2Q25K2 | KKQQQQQQQQ | QQQQQQQQQQ | QQQQQQQKK | ||
| K2Q41K2 | KKQQQQQQQQ | QQQQQQQQQQ | QQQQQQQQQQ | QQQQQQQQQQ | QQQKK |
| Biotinyl-K2Q30K2 | B*KKQQQQQQ | QQQQQQQQQQ | QQQQQQQQQQ | QQQQKK | |
| NLS-GGQ11PGQ12CK2 | PKKKRKVGGQ | QQQQQQQQQQ | PGQQQQQQQQ | QQQQCKK | |
| NLS-GGQ25CK2 | PKKKRKVGGQ | QQQQQQQQQQ | QQQQQQQQQQ | QQQQCKK | |
Lower case p is the D-enantiomer of Pro. In K2CQ22CK2 (ox.) the Cys residues are intramolecularly disulfide-bonded. “Ac” represents an Nα-acetyl group. “NLS” is the nuclear localization sequence PKKKRKV. B* is Glu(biotinyl-PEG) (see Materials and Methods).
Figure 2.

Aggregation kinetics of polyQ peptides with varying β-hairpin propensity. (a) Aggregation kinetics (HPLC sedimentation assay) showing rates of monomer disappearance for approximately 100 μM solutions of different polyQ peptides; (b) aggregation of various polyQ peptides incubated in the 10–20 μM range in PBS at 37 °C; (c) aggregation of AcWQ11pGQ11WTGK2 monitored by CD spectroscopy; (d) circular dichroism spectra of 20–30 μM freshly disaggregated monomers: K2Q10pGQ11K2 (red); K2Q25K2 (black); K2Q10pGQ11K2 – K2Q25K2 (green).
We also found that the aggregation enhancing abilities of these β-hairpin mimetics are additive. The peptide AcWQ11pGQ11WTGK2 (Fig. 2a, ★) combines two motifs, one that encourages β-hairpin formation from the chain termini and one that facilitates β-turn formation from the center of the sequence. (The design of this peptide has conformational features that are conceptually similar to that of a Gln-rich, D-Pro-Gly/disulfide peptide previously described by the Fairman group 43.) We found that this peptide aggregates so rapidly at ~100 μM that the kinetics could not be accurately determined by our methods. We therefore repeated the analysis at lower concentration. Figure 2a shows that even at a mere ~ 15 μM, AcWQ11pGQ11WTGK2 aggregates more rapidly than the disulfide or trpzip mutants at ~100 μM. We then compared this peptide to the two corresponding indvidual β-hairpin mutants at this lower concentration of ~15 μM (Fig. 2b, ■). The results show clearly that the double β-hairpin mutant AcWQ11pGQ11WTGK2 (■) aggregates more rapidly than both the single D-Pro-Gly (▼) and trpzip (▲) mutants. Moreover,, the figure also shows that all of these β-hairpin mutated versions of short (Q23) polyQ peptides aggregate nearly as fast or faster than an equivalent concentration of a much longer simple polyQ peptide with a pathological repeat length, K2Q37K2 (●). The K2Q23K2 peptide, at this concentration, does not detectibly aggregate (not shown). We monitored aggregation of the double mutant AcWQ11pGQ11WTGK2 by CD and obtained a random coil to β-sheet transition (Fig. 2c) very similar to data previously reported for simple polyQ aggregation 11.
Detailed nucleation kinetics analysis of K2Q10pGQ11K2
To investigate how these mutations lead to aggregation rate enhancements, we carried out nucleation kinetics analysis 11, 23, 44. For example, we studied the concentration dependence of the initial aggregation of the D-Pro-Gly peptide K2Q10pGQ11K2 (Fig. 3a). As previously carried out for other polyQ peptides 11, 23, 44, these data were plotted vs. time2 (Fig. 3b) and the resulting rates plotted vs. starting concentration in a log-log plot (Fig. 3c, ■). From the resulting slope of ~ 2.7, we obtain a value for the critical nucleus (n*, the number of molecules involved in nucleus formation) of 0.7 using the relationship n* = slope - 2 11, 23, 44. This contrasts strongly with the n* of 3.9, based on a log-log slope of 5.9 (Fig. 3c, ●), previously reported for K2Q23K2 23. Thus, the rate enhancement effect of replacing two Gln residues in the center of a Q23 sequence with D-Pro-Gly is due at least in part to a dramatic reduction in the size of the critical nucleus from n* ≈ 4 to n* ≈ 1.
Figure 3.

Detailed nucleation kinetics analysis for K2Q10pGQ11K2. (a) Concentration dependent aggregation kinetics. (b) Time2 plots of the data from panel a. (c) Log-log plot of the rates from panel b vs. starting concentration for K2Q10pGQ11K2 compared to similarly obtained data 23 for K2Q23K2. (d) Determination of the equilibrium position for fibril elongation (i.e., the Cr value) from fibril association (i.e., growth) and dissociation curves. (e) Pseudo-first order elongation kinetics from a solution of 59 μM monomer seeded with ~ 12 % weight/weight amyloid. (f) Titration of the growth points on the amyloid preparation used in the panel e experiment, using biotinyl-K2Q30K2 (Methods); the plateau (~ 60 fmol) indicates the molar amount of growth sites in the sample tested.
To complement the nucleation kinetics analysis, we also determined the second order elongation rate constant, k+. From the measurement of an aggregate preparation’s pseudo-first order elongation rate constant (Fig. 3e) and its molar concentration of fibril growth points (Fig. 3f) we determined a k+ of 0.17 × 104 M−1s−1 for K2Q10pGQ11K2. In contrast, we previously determined k+ to be 1.24 × 104 M−1s−1 for K2Q23K2 23. Thus, while nucleation of amyloid formation is greatly facilitated by the β-hairpin encouraging D-Pro-Gly insertion, we find that amyloid elongation is actually somewhat retarded by this change.
Two measures of the energetics of fibril assembly were determined for this peptide from the aggregation data. First, the equilibrium constant for nucleation, Kn*, was calculated from the y-intercept of the log-log plot (Fig. 3c) and the k+ value 45. This yields a value of Kn* = 0.93 × 10−10 for the monomeric folding reaction responsible for nucleus formation, dramatically illustrating the high energetic barrier to nucleus formation for even a rapidly aggregating peptide containing a β-hairpin enhancing mutation. (Unfortunately, a comparable analysis cannot be conducted for K2Q23K2 because its critical nucleus is not monomeric.) Second, the thermodynamic contribution of the D-Pro-Gly to the stability of the fibril product was obtained by determining Cr, the concentration of monomer when fibril association/dissociation reaches equilibrium 46, 47. The Cr was obtained by separately monitoring both fibril assembly and disassembly (Fig. 3d), and the corresponding free energy change calculated (see Methods). For K2Q10pGQ11K2, we obtained Cr = 0.35 ± 0.02 μM (Fig. 3d; Table 2), corresponding 46, 47 to a ΔGelong of −38.3 kJ/mol (Table 2). In contrast, the Cr and ΔGelong for K2Q23K2 are 2.9 ± 0.5 μM and −32.9 kJ/mol (Table 2). Thus, fibrils are stabilized relative to the monomer state by 5.4 kJ/mol when Gln-Gln is replaced with D-Pro-Gly in a Q23 peptide.
Table 2.
Summary of nucleation kinetics and fibril stability analysis
| Peptides Studied | Data pointsa | Slopeb | R2 of fitc | n*d | Cre (μM) | ΔGelongf (kJ/mol) |
|---|---|---|---|---|---|---|
| K2Q23K2g | 8 | 5.9 | 0.9875 | 3.9 | 2.9 ± 0.5 | −32.9 |
| K2Q10PGQ11K2 | 7 | 3.4 | 0.9197 | 1.4 | 1.7 ± 0.4 | −34.3 |
| D2Q23K2 | 7 | 3.6 | 0.9671 | 1.6 | 1.2 ± 0.3 | −35.3 |
| K2Q10pGQ11K2 | 7 | 2.7 | 0.9864 | 0.7 | 0.35 ± 0.02 | −38.3 |
| K2CQ22CK2h | 5 | 2.8 | 0.9557 | 0.8 | 0.36 ± 0.03 | −38.3 |
| AcWQ22WTGK2 | 6 | 2.7 | 0.9593 | 0.7 | 0.21 ± 0.02 | −39.6 |
| AcWQ11pGQ11WTGK2 | 5 | 2.9 | 0.9664 | 0.9 | 0.11 ± 0.01 | −41.3 |
Number of data points in the log-log plot;
Slope of log-log plot;
Fit of log-log plot;
n* from log-log slope;
Equilibrium concentration of peptide from average of final positions of fibril growth and dissociation curves;
Calculated free energy change on elongation calculated from the Cr value 46;
Data for K2Q23K2 from 23;
This is the oxidized, disulfide cross-linked form of this molecule.
Kinetics analysis of other mutated polyQ peptides
Similar analyses were conducted for the other peptides shown in Figure 2a (Fig. 4; Table 2). The results show that, in every case, β-hairpin favoring mutations in a Q22 or Q23 background reduce n* from ~4 to ~1 (Fig. 4, a–e; Table 2). Consistent with the trends in the aggregation kinetics at 100 μM, n* for the moderately effective L-Pro-Gly mutation and for the D2/K2 Coulombic attraction mutation give n* values perched between 1 and 2, which may indicate a mixed mechanism for nucleation for these peptides. At the same time, strong β-hairpin favoring mutations such as the trpzip motif and the covalent disulfide, both of which at 100 μM produce more rapid spontaneous aggregation than the D-Pro-Gly mutant, yield n* values of 0.7 to 0.8, in the same range as the D-Pro-Gly peptide (Table 2). The variations in aggregation kinetics within those peptides that exhibit similar n* values are presumably due to an interplay between Kn* and/or k+ values.
Figure 4.

Kinetics analysis of other β-hairpin mimetic peptides. Shown are the log-log plots (a–e) and the Cr plots (f–j) for the peptides K2Q10PGQ11K2 (a,f); oxidized K2CQ22CK2 (b,g); D2Q23K2 (c,h); AcWQ22WTGK2 (d,i); AcWQ11pGQ11WTGK2 (e,j). For values resulting from these plots see Table 2.
Displaying a similar correlation, mutations known to provide modest enhancement of β-hairpin stability provide relatively low stabilization to the amyloid fibril, as assessed by Cr value, while motifs that strongly enhance β-hairpin formation provide greater fibril stabilization. Thus, the L-Pro-Gly mutation yields a ΔGelong value of −34.3 kJ/mol, only 1.4 kJ/mol more stable than the K2Q23K2 peptide, and the D2/K2 pair yields a ΔGelong of −35.3 kJ/mol for a stabilization, compared to the K2Q23K2 sequence context, of only 2.4 kJ/mol (Table 2). In contrast, the disulfide and trpzip mutations give ΔGelong values of −38.3 and −39.6 kJ/mol, for ΔΔGelong values, compared with K2Q23K2, of 5.4 and 6.4 kJ/mol, respectively.
Analysis of the double β-hairpin mutant AcWQ11pGQ11WTGK2 yielded data consistent with the comparative analysis shown in Figures 2a and b. The log-log plot of this peptide (Fig. 4e) yields a value for n* of 0.9 (Table 2), and the Cr determination (Fig. 4j) led to a value of ~ 0.11 μM (Table 2), i.e., more stabilizing than either of the “individual β-hairpin” mutations.
Aggregate structure and properties in vitro
The above results suggest that β-hairpin formation is not only easily tolerated as part of the nucleation mechanism, but also actually enhances the efficiency of nucleated growth by reducing the value of n*. Nonetheless, the results do not formally prove that amyloid nucleation within simple, unbroken polyQ sequences also occurs via β-hairpin formation. To gain insight into this important question, we compared the aggregates that are obtained from β-hairpin mutants to the amyloid fibrils generated by normal polyQ peptides. We found that the EM morphologies of the aggregates formed by the above mutated polyQ sequences (Fig. 5b–g) are very similar to amyloid-like aggregates from simple polyQ peptides in both the Q23 (Fig. 5a) and Q41 (Fig. 5h) ranges. Thus, all these aggregates exhibit morphologies built upon unbranched monofilaments of diameter 3.2–4.5 nm that appear to associate into rigid-looking non-twisted ribbons or tapes consisting of multiple filaments. For some peptides these ribbons tend to be relatively short, in the 200–400 nm range (Fig. 5a, c–g), while for other peptides the ribbons are over 0.5 μm in length (Fig. 5b,h). These aggregate morphologies are distinct from more typical amyloid fibrils that exhibit more uniform diameters in the 8–12 nm range. We also conducted FTIR analysis of the aggregates (Fig. 6). In all cases we obtained spectra dominated by a triplet of strong peaks reporting on β-sheet and on ordered Gln side chains 48 that is characteristic of polyQ amyloid and identical to a sample of K2Q23K2 amyloid.
Figure 5.

Electron micrographs of final aggregates. (a) K2Q23K2 (from reference 23); (b) K2Q10PGQ11K2; (c) D2Q23K2; (d) oxidized K2CQ22CK2; (e) K2Q10pGQ11K2; (f)AcWQ11pGQ11WTGK2; (g) AcWQ22WTGK2; (h) K2Q41K2. The 50 nm scale bar in part a applies to all panels.
Figure 6.

Secondary structures of amyloid fibrils. Second derivative Fourier transform infrared spectra of isolated amyloid fibrils. The three major bands in these spectra are typical for polyQ aggregates and are assigned to NH2 deformations in the Gln side chains (~ 1605 cm−1), β-sheet (1625–30 cm−1), and C = O stretching in the Gln side chains (1655–1660 cm−1) 48.
As a more detailed probe for changes in the core structure of the resulting amyloid fibrils we also conducted magic-angle-spinning (MAS) ssNMR experiments on these fibrils. Previous reports from our labs and others 15, 16 have established that Gln residues within the core of normal polyQ fibrils feature two sets of reproducible NMR signatures. Such 13C chemical shifts are sensitive to the local structure as well as dynamics 49, and are therefore sensitive probes of the structure within the amyloid fibril core. Indeed, amyloid fibrils in different polymorphic forms are often identified based on their chemical shifts, which in turn reflect variations in their internal structure 50–53. To probe for any disturbance of the amyloid core structure in mutant fibrils, we introduced a specifically 13C,15N-labeled Gln into K2Q11PGQ11D2, a peptide that combines two sequence motifs (PG and K2/D2) found to independently enhance aggregation and decrease n* (Table 2). The label was placed in the eighth Gln in the first Q11 segment (i.e., residue Q10). Using 2D 13C-13C MAS ssNMR spectroscopy, we determined this Gln’s NMR signals, resulting in the 13C NMR data in Fig. 7a. Again, this single labeled Gln residue yields a doubled set of resonances, indicating two distinct conformations. These are the same doubled resonances, populated in approximately equal amounts, previously found in amyloid fibrils of both simple polyQ and the polyQ portion of huntingtin fragments 15, 16. The 13C line widths for the labeled Q10 vary from ~180–225 Hz, depending on the atomic site (1.2–1.5 ppm at 600MHz 1H frequency). These line widths are the same as those in our previously published 15 data on K2Q30K2 fibrils in which the fourth Gln of the polyQ track was isotopically labeled (Fig. 7b). Although these line widths exceed those recently observed for certain amyloid fibrils with particularly high structural homogeneity, they are typical of the range of line widths seen for the variety of amyloids studied by MAS ssNMR (e.g., 51, 54–56).
Figure 7.

Solid-state NMR on fibrils. (a) Doubled cross-peak patterns of a single labeled Gln in fibrils prepared from [U-13C, 15N-Q10] K2Q11PGQ11D2 reproduce the Gln peaks in fibrils (b) from [U-13C, 15N-Q6] K2Q30K2 peptide that lacks β-hairpin encouraging motifs 15. Red and blue lines mark peaks of the two distinct conformers. Asterisks mark spinning side bands. Spectra were obtained at 600MHz (for 1H), using 10 kHz MAS and 8ms or 25ms DARR mixing, respectively. (c) For the >10,000 Gln in the Biomolecular NMR database (bar graphs) the Gln Cβ (light bars) and Cγ shifts (dark bars) are generally well-separated. In each of the two polyQ conformers ‘form a’ (red bracket) and ‘form b’ (blue) there is instead small shift difference between Cβ and Cγ. The Cβ shift of ‘form a’ is highly unusual, and the Cβ and Cγ shifts of ‘form b’ are both atypical. (d) The average shifts of the few BMRB Gln that feature Cβ/Cγ shifts very close together (within 1 ppm). The particular shifts of conformer a (red) or b (blue) are seen in 0.5% and 0.4% of all Gln, respectively.
To examine the likelihood that these chemical shifts are reproduced by chance, without preservation of structure, we extracted the 13C shifts of >10,000 Gln residues reported in the Biological Magnetic Resonance Data Bank (BMRB; http://www.bmrb.wisc.edu) of proteins studied by NMR. Protein NMR signals for the Cβ and Cγ of Gln are typically well separated in chemical shift (Fig. 7c). In contrast, in each of the two observed polyQ Gln conformers ‘form a’ (indicated in red) and ‘form b’ (marked in blue) there is an unusually small shift difference between Cβ and Cγ (see Fig. 7 and refs 15, 16). Figure 7d summarizes the (small) subset of Gln in the BMRB that do have a similarly small Cβ/Cγ shift difference, and reveals that only 0.5% (form a) and 0.4% (form b) of Gln match either of the observed chemical shift values. The unlikelihood of the observed shifts is also apparent from inspection of the actual shift values in Figure 7c, as the Cβ shift of ‘form a’ is in itself very unusual, and both the Cβ and Cγ shifts of ‘form b’ are atypical. Thus, the consistent and reproducible observation of both sets of resonances in polyQ amyloids (here and elsewhere 15, 16, 57) seems exceedingly unlikely to occur by chance. We also note that both the separation between the two sets of peaks (Fig. 7), and the differences between the observed and ‘typical’ Gln shifts (Fig. 7c) significantly exceed the width of the peaks. Further studies of this seemingly unique polyQ structure will be required to fully characterize the nature of the two co-existing forms, necessitating further MAS ssNMR experiments that provide site-specific structural insights into these complex and composite amyloid fibril structures 52, 53, 56, 58. Nonetheless, it is clear that the incorporation of these β-hairpin-promoting mutations does not appreciably modulate these key indicators of the internal amyloid core structure.
Aggregate structure and properties in cells
Another measure of the relation of our β-hairpin-peptide amyloid fibrils to fibrils made of simple polyQ sequences is their respective behavior in cell assays. Previous work with simple polyQ aggregates showed that finely dispersed aggregates can enter mammalian cells in culture, and that, if the constituent peptides are fitted with a nuclear localization signal (NLS), the aggregates are both extremely toxic 59 and can recruit GFP-tagged polyQ sequences within the cell 60. We obtained the peptide NLS-GGQ11PGQ12CK2, modified the Cys residue with the fluorophore Cy5, prepared aggregates from the labeled peptide, and exposed PC12 cells in culture to these aggregates in growth media (Methods).
Similar to normal polyQ counterparts, these mutant aggregates are taken up by PC12 cells in culture (Fig. 8b) and, in cells producing a huntingtin (htt) exon1 fragment with Q25 fused to EGFP, the aggregates stimulate the formation of GFP puncta (Fig. 8d), many of which co-localize with the Cy5 labeled internalized aggregates (Fig. 8e). Quantitation of the number of cells with puncta shows that PC12 cells without internalized aggregates exhibit only diffuse GFP fluorescence consistent with low molecular weight species, and that GFP puncta formation is equivalent in cells taking up either simple polyQ aggregates or β-hairpin mutant aggregates (Fig. 8f). In addition, we find that the cytotoxicity of the NLS-β-hairpin aggregates is equivalent to similar, simple polyQ aggregates 59 (Fig. 8g).
Figure 8.
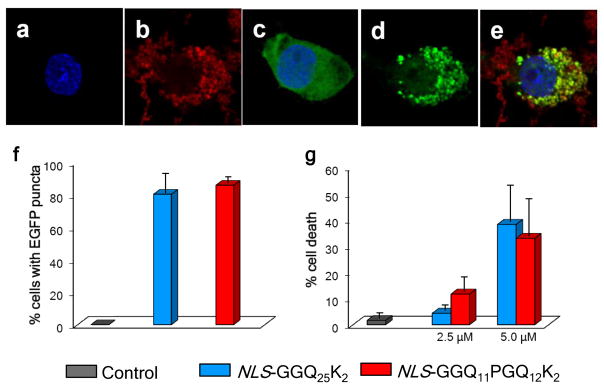
Cell activities of aggregates. (a–e) Confocal fluorescence microscope images of PC12 cells stably transfected to express a Huntingtin exon-1 fragment containing a Q25 repeat and fused to green fluorescent protein, 24 hrs after induction with 1 μM ponasterone with (a,b,d,e) or without (c) treatment with tagged amyloid fibrils. (a) Blue only, Hoechst 33342 (Invitrogen) stained nuclei; (b) red only, Cy5-labelled NLS-GGQ11PGQ12CKK amyloid; (c) blue-stained nucleus and diffuse green showing non-aggregated exon1 in cells not treated with exogenous aggregates; (d) green only, showing aggregate formation from exon1 protein in cells treated with exogenous aggregates; (e) merge colors in aggregate-treated cells showing that aggregates outside the cell remain red, while aggregates within the exon1-producing cell are mostly overlapping red + green. (f) cellular recruitment, as percent PC12 cells with EGFP inclusions, after 16 hrs of expressed (1 μM ponasterone induction) Q25 exon1-EGFP by polyQ fibrils internalized from a 2.5 μM (monomer equivalent) suspension in the growth medium; control data is for similarly induced cells not treated with aggregates. (g) cell death, assessed after 24 hrs by LDH release assay, in PC12 cells treated with different concentrations of aggregates in the growth medium; control data is for cells grown without aggregates.
Solution structure of β-hairpin polyQ monomers
Since the conformational flexibility and preferences of monomeric polyQ peptides are of great interest to the HD field, we wanted to investigate the monomeric state of peptides with installed β-hairpin motifs. We employed CD to assess the extent to which β-hairpin encouraging mutations force measureable levels of intact β-hairpin structure in monomers in solution. In contrast to their β-hairpin enhancing effects on other peptides 32, 36, 42, we found no indication of a substantial amount of β-hairpin structure in the native ensemble of these β-hairpin motif polyQ peptides. Thus, the CD spectrum of monomeric K2Q10pGQ11K2 (Fig. 2d, red) is similar to the spectrum of K2Q25K2 (Fig. 2d, black), both spectra exhibiting random coil characteristics. The inability of the installed β-hairpin encouraging mutations to enhance β-sheet content is emphasized in the difference spectrum generated by subtracting the spectrum of the K2Q25K2 peptide from that of K2Q10pGQ11K2. This difference spectrum is weak and without typical β-strand or turn features (Fig. 2d, green).
DISCUSSION
The results reported here support and extend our model for the nucleation of amyloid formation by simple polyQ sequences. We previously hypothesized that polyQ sequences in the Q18–Q23 length range are precluded from operating via a monomeric nucleus due to their energetically restricted abilities to fold, even transiently, into a monomeric nucleus, which we modeled as a β-hairpin type structure 23. In our studies described here, we attempted to overcome this hypothetical energy barrier by introducing mutations that are well-known to generally enhance β-hairpin formation. The success of this approach, in generating short polyQ sequences with greatly enhanced aggregation rates (Fig. 2a) controlled by a monomeric nucleus (Table 2) provides additional support for our models for the structure of the critical nucleus and resulting amyloid fibril, and also places severe restrictions on the concentration of β-hairpin-like conformations that are normally populated within the monomer ensemble.
Detailed kinetics analysis of one of these peptides, K2Q10pGQ11K2, shows that this β-hairpin mutation not only confers an n* ~ 1 but also a nucleation equilibrium constant (Kn*; see Methods) in the range of 10−10, similar to values obtained for other rapidly aggregating polyQ peptides 8, 23, 45. Somewhat surprisingly, the second-order elongation rate constant (k+) for the D-Pro-Gly peptide is about an order of magnitude smaller than the rate constant we obtained previously for K2Q23K2 23. Although our model predicts that facilitating β-turn formation should favor both nucleation and elongation, it is conceivable that the D-Pro-Gly sequence provides an unfavorable configurational constraint onto the multi-step fibril elongation mechanism 61–64. The fact that K2Q10pGQ11K2 undergoes amyloid formation much more rapidly than K2Q23K2, in spite of a less favorable elongation rate constant, is a dramatic illustration of the large benefit of a small nucleus. Using other methods, similar overall aggregation rate enhancement, albeit with different underlying parameters, was recently reported for a D-Pro-Gly containing short polyQ sequence 65, 66. Introduction of regularly spaced D-Pro-Gly insertions was previously reported to also greatly enhance aggregation rates for longer polyQ sequences 20, and introduction of D-Pro-Gly into a predicted turn location in the Aβ sequence greatly enhances its aggregation rate 67.
Several lines of experimental evidence suggest that amyloid fibrils from simple, unbroken polyQ contain chain reversals that are predominantly β-turns and not β-arcs. If the normal aggregation pathway did not involve β-hairpin structures, then the introduction of such mutations into polyQ would be expected to disrupt the normal aggregation process, reducing aggregation kinetics and potentially significantly altering aggregate morphology in an example of mutation-dependent amyloid polymorphism 68, 69. However, we found that β-hairpin motifs uniformly enhance aggregation kinetics, and EM, FTIR and SSNMR data all suggest that the structures of the amyloid fibrils formed by β-hairpin motif-containing polyQ peptides are very similar to aggregates of unbroken polyQ. In particular, the doublet of 13C resonances observed for Gln in simple polyQ amyloid fibrils 15, 16, which are shown here to be at chemical shift positions that are highly unusual for Gln residues in other proteins, are replicated in amyloid fibrils of a polyQ containing β-hairpin enhancing motifs. In addition, data from cell studies show that the aggregates from these modified polyQ peptides behave analogously to normal unbroken polyQ molecules in both their cytotoxicities and intracellular polyQ recruitment activities (Fig. 8).
Finally, the Cr data and derived ΔG values (Table 1) suggest modest stabilizations of fibril structure in β-turn motif polyQ peptides that are are quantitatively consistent with their abilities to stabilize β-hairpin structures in non-polyQ monomeric peptides 42. For example, a disulfide bond between chain termini was previously shown to provide about 4 kJ/mol stabilization in the folding of a monomeric β-hairpin 41, while we found a 4.4 kJ/mol contribution to polyQ amyloid stability (Table 2, compare K2CQ22CK2 to K2Q23K2). Similarly, Coulombic effects at chain termini provide 1.5 – 2.5 kJ/mol in the folding of monomeric β-hairpins 42 and we found a 2.4 kJ/mol effect on polyQ amyloid stability (Table 2, compare D2Q23K2 to K2Q23K2).
Thus, our preliminary conclusions are that (a) polyQ peptides containing β-hairpin encouraging mutations aggregate in such a way that the structures of both the nucleus and final amyloid involve β-hairpin structures, and (b) that simple polyQ peptide aggregation involves similar structures except where energetically unfavorable (i.e., short polyQ peptides like K2Q23K2). Further studies, including more detailed SSNMR evaluations and comparisons, should provide a deeper look into the structures of these β-hairpin polyQ amyloid fibrils.
Although these β-hairpin encouraging mutations make monomeric nucleation viable, CD analysis (Fig. 2d) shows that they do not work well enough to allow spectroscopic detection of enhanced β-sheet/β-turn in the monomer ensemble. This appears counter-intuitive but in fact is consistent both with the nucleation data and with what we know about polyQ structure in water. Monomeric polyQ in water exists as a compact coil 30, 70, 71 in which backbone amides appear to favor H-bonding to Gln side chains rather than being exclusively H-bonded to solvent water 29. This may place a significant barrier on β-hairpin formation, since the monomer already possesses stabilizing intramolecular H-bonds, and the entropic cost of ordering the backbone into stable β-turn structures is unlikely to be effectively compensated by “zero-sum” rearrangements within this H-bonding network 29. These considerations are consistent with our model that the β-hairpin conformation serves as a nucleus for aggregation. Our Kn* measurements show that the fraction of monomers in the ensemble that is present as (β-hairpin) nuclei is expected to be well below the levels of spectroscopic detection, in the range of 10−10, even for the efficiently aggregating D-Pro-Gly mutant.
This analysis may have implications for recent suggestions that the toxic species responsible for expanded polyQ pathology are monomers that are “misfolded” into β-hairpin conformations 24–27. (Such “toxic conformation” models are distinctly different from one in which the intrinsically disordered polyQ molecule engages a specific folding pattern in the process of forming a “toxic complex” with some cellular target 8.) Our inability to detect even in difference spectra any enhanced β-structure in a polyQ containing a strong β-hairpin encouraging motif (Fig. 2d) suggests a very low propensity for simple polyQ peptides to adopt such conformations as isolated monomers in solution. If we are correctly interpreting our data to suggest that the critical nucleus for amyloid formation consists of just such β-hairpin structures, we can put a value on the extent of β-hairpin formation within the monomer ensemble. Thus, at 1 μM (a liberal estimate for the steady-state concentration of disease-related polyQ fragments in the cell) and at a Keq of 10−10, the concentration of monomeric polyQ with β-hairpin conformations in the steady state is calculated to be in the range of 10−16 M. It is not clear how such a vanishingly low concentration of β-hairpin polyQ molecules could elicit a toxic cellular response, other than perhaps by nucleating amyloid growth.
MATERIALS AND METHODS
Most peptides were synthesized at the Small Scale Synthesis facility at the Keck Biotechnology Resource Laboratory of Yale University (http://keck.med.yale.edu/) and supplied crude. The peptide K2CQ22CK2 was oxidized by incubating a solution of the peptide in 6 M Gdn.HCl, 10 mM Tris.HCl, 10 μM CuCl2, pH 8.0 at 24 °C for 2 hrs, followed by purification of the crosslinked product.
Peptide AcWQ11pGQ11WTGKK was synthesized by microwave-assisted Fmoc solid-phase methods on a MARS microwave reactor (CEM) using H-Lys(Boc)-HMPB NovaPEG resin (Novabiochem) as the solid support. Couplings were carried out in N-Methyl-2-pyrrolidone with a 2 min ramp to 70 °C and a 4 min hold at that temperature using 4 equiv of Fmoc-protected amino acid, 4 equiv of HCTU [2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium hexafluorophosphate], and 6 equiv of diisopropylethylamine. Deprotections were performed with a 2 min ramp to 80 °C followed by a 2 min hold at that temperature using 20% 4-methylpiperidine in dimethylformamide. Resin was washed three times with dimethylformamide between each cycle. N-terminal acetylation was carried out on resin by treatment with 8:2:1 v/v/v dimethylformamide/diisopropylethylamine/acetic anhydride. Peptide was cleaved from resin by treatment with 94% trifluoroacetic acid, 2.5% water, 2.5% ethanedithiol, and 1% triisopropyl silane. The cleavage mixture was precipitated into cold ether, centrifuged, and the supernatant drained to afford the peptide as a crude pellet.
All peptides were purified and disaggregated as described 23, 46. Aggregation reactions were initiated, and monitored by an HPLC-based sedimentation assay, as described 23, 46. In particular, in addition to HFIP/TFA treatment and high speed centrifugation of aqueous stock solution, all PBS solutions of peptides were filtered through a 20 nm membrane filter (Anotop 10, Whatman) 23 before incubation for monitoring aggregation. Fibril dissociation reactions for confirming Cr were initiated by PBS dilution of a late-stage fibril formation reaction, as described 23. Electron micrographs and FTIR were carried out as described previously 23. Far-UV CD measurements were performed on 20 – 30 μM peptide solutions in 20 mM Tris.HCl, pH 7.4, on a JASCO J-810 spectropolarimeter using a 0.1 cm path length cuvette. CD spectra were analyzed using the CONTINLL program from the CDPro package (lamar.colostate.edu/~sreeram/CDPro) in which the SP37A reference set (ibasis 5) was used to estimate the amount of secondary structure 72.
Preparation of labeled aggregates for cell experiments
For the labeling, a solution of ~24 mM Cy5 maleimide Mono-Reactive dye (GE Healthcare, Life Sciences) in ~50 μl DMSO was added to a solution of ~ 200 μM peptide in 6 M GdnHCl, 20 mM Tris.HCl, pH 7.5 and 2 mM Tris-(2-carboxethyl) phosphine (TCEP), and the mixture stirred at 25 °C and monitored by analytical HPLC. When the reaction was 90% complete (~ 2 hrs) the labeled peptide was purified by HPLC. For aggregate preparation, monomer solutions (1 part labeled peptide to 15 parts of the same sequence but lacking Cys) of approximately 150 μM in PBS were snap frozen in liquid N2, then incubated at −20 °C until aggregation was at least 90% complete (~24 hrs) 46. Thawed, aggregated samples contained long, single filament fibrils which were then subjected to multiple rounds of sonication with the reduction in average particle size monitored by DLS. Typically, 10 rounds, each consisting of 30 secs sonication followed by 30 secs rest, were performed using a micro-probe sonicator (Sonic Dismembrator Model 500-Fisher Scientific (20kHz)) with amplitude control knob set at 40%, with the sample cooled on ice. The resulting aggregate suspension was then filtered using a 0.2 μm filter (VWR), yielding a final stock suspension with a 40–65 nm size range by DLS. The concentrations of the aggregate suspensions were determined as described 46.
Analysis of nucleation kinetics, fibril stability and simulated aggregation curves
Aggregation kinetics data were treated as described previously 11, 23, 46, using the equation , in which Δ is the molar concentration of monomers that has been converted to aggregates at time t, and k+ is the second order elongation rate constant for nucleus elongation and aggregate elongation, c is the molar concentration of monomers at the start of the reaction, n* is the critical nucleus size, and Kn* is the nucleation equilibrium constant. The slope of the log-log plot (Figure 3C) = n* + 2. The y-intercept of the plot = log [½(k+2)(Kn*)]. Calculation of Kn* and ΔhGn* from the y-intercept has been described 23, 45, 46.
Critical concentrations (Cr) were determined by the convergence of the fibril formation and fibril dissociation curves (Figs. 3d and 4f–j) 46, 47, 73. Amyloid formation reactions were followed until the residual monomer concentration after centrifugation remained constant. In parallel, late in the fibrillization reaction, a portion of the reaction mixture was removed and diluted with PBS in such a way that the total polyQ concentration (fibril plus monomer) after dilution remained above the forward plateau value. This reaction was incubated under identical conditions and aliquots removed to determine residual monomer concentration after centrifugation. Ideally, this value should converge with the value from the forward reaction (Figs. 3d and 4f–j). Where convergence did not occur (probably because of very slow rates), the mean value of the association and dissociation values was calculated as the Cr. Free energies of elongation (ΔGelong) were determined from these Cr values using the expression ΔGelong = −RTln(1/Cr) 47.
Solid-state NMR experiments
SSNMR was generally performed as described previously 15. Mature fibrils were pelleted into a 3.2 mm zirconia MAS rotor (Bruker Biospin, Billerica, MA) by centrifugation and were kept hydrated and unfrozen during and between experiments. SSNMR was done with a wide-bore Bruker Avance I spectrometer operating at 600 MHz 1H Larmor frequency (14.3 T) and a Bruker standard-bore 3.2 mm MAS EFree HCN probe (Bruker Biospin, Billerica, MA). A spinning rate of 10 kHz was maintained throughout all experiments while cooling with pre-cooled N2 gas at 2°C. Assignments were based on 2D 13C-13C experiments, which used 1H-13C cross polarization (CP) followed by dipolar-assisted rotational resonance (DARR) 74 mixing for the 13C-13C transfers and 83 kHz two-pulse phase modulation (TPPM) 1H decoupling 75 during acquisition and evolution. The data in Figure 7a (7 mg K2Q11PGQ11D2 fibrils) were acquired in approximately 11.5 hours, whilst the smaller sample (4 mg peptide) in Figure 7b necessitated an acquisition time of ~44 hours 15. The NMR data were referenced, processed and analyzed as described previously 15.
Cell culture experiments
Cells (WT or transfected PC12 “Schweitzer morph A cells” 76) were maintained in Dulbecco’s modified Eagle’s media (DMEM) containing 25mM HEPES (Cellgro), 5% supplemented calf serum (Hyclone), 5% horse serum (Hyclone), 2 mM L-glutamine, penicillin and streptomycin on collagen IV coated plates (Trevigen) at 37°C in 9.5% CO2. Cell media was changed every 3 days. Medium for transfected (Htt-exon1-Q25-EGFP) cells also included 0.5 mg/ml G418 (Mediatech) and 1 μM ponesterone. For aggregate internalization 59, freshly sonicated aggregates, prepared as described above, were diluted into OptiMEM (Gibco) medium supplemented with antibiotics. Cell toxicity was assessed by LDH release using the CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega).
For confocal microscopy analysis of cellular aggregates, cells were plated in collagen coated glass chamber slides (Nunc). Htt-exon1-Q25-EGFP PC12 cells were incubated with aggregates and simultaneously induced for exon1 expression with 1μM ponasterone. At specific times, cells were fixed with 4% paraformaldehyde (Cytofix, EB) and the nuclei stained with Hoechst 33342 (Invitrogen). Confocal images were collected using an Olympus Fluoview 1000 confocal microscope (×100 oil immersion lens) at room temperature. Random fields were scored (≥200 cells per condition over 3 experiments) for the percentage of cells presenting EGFP and Cy5 puncta using ImageJ software (NIH).
Data analysis and statistics
For the in vitro aggregation assays, error bars are standard deviations from analyses in duplicate. Data sets were fit in Origin 7.5 software (OriginLab). Most reaction profiles were fit to B-spline curves. Semi-log plots for heavily seeded elongation reactions used to determine elongation rate constants were fit by linear regression. Growing end titration data were fit to one site saturation ligand binding curves using Sigma Plot 10.0.
Cellular puncta counts and toxicity data were analyzed by GraphPad PRISM. Significance was determined using post-hoc analysis (Student’s t-tests with Bonferroni correction) using P < 0.05.
Highlights.
β-hairpin motifs added to short polyQ sequences greatly enhance aggregation rates
The enhancing effect is primarily due to a change in critical nucleus from 4 to 1
Amyloid structure of mutants is unchanged by SSNMR, EM, FTIR and bioactivities
Even with these added mutations, the monomeric polyQs are disordered in solution
The β-hairpin conformation is thus quite rare, consistent with a roll in nucleation
Acknowledgments
We gratefully acknowledge funding support from the University of Pittsburgh (W.S.H.) and NIH grants R01 AG019322 (to R.W. and P.v.d.W) and R01 GM099718 (to R.W.). We acknowledge Erik Schweitzer for providing the transfected PC12 cell line and Rakesh Mishra for suggestions and helpful discussions. EMs were collected in the Structural Biology Department’s EM facility administered by Drs. James Conway and Alexander Makhov.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bates GP, Benn C. The polyglutamine diseases. In: Bates GP, Harper PS, Jones L, editors. Huntington’s Disease. Oxford University Press; Oxford, U.K: 2002. pp. 429–472. [Google Scholar]
- 2.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 3.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 4.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci U S A. 1999;96:4604–9. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–82. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 6.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 7.Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–4. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 8.Wetzel R. Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J Mol Biol. 2012;421:466–90. doi: 10.1016/j.jmb.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellisdon AM, Thomas B, Bottomley SP. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem. 2006;281:16888–96. doi: 10.1074/jbc.M601470200. [DOI] [PubMed] [Google Scholar]
- 10.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–9. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S, Ferrone F, Wetzel R. Huntington’s Disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci U S A. 1994;91:5355–8. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma D, Shinchuk LM, Inouye H, Wetzel R, Kirschner DA. Polyglutamine homopolymers having 8–45 residues form slablike beta-crystallite assemblies. Proteins. 2005;61:398–411. doi: 10.1002/prot.20602. [DOI] [PubMed] [Google Scholar]
- 14.Sikorski P, Atkins E. New model for crystalline polyglutamine assemblies and their connection with amyloid fibrils. Biomacromolecules. 2005;6:425–32. doi: 10.1021/bm0494388. [DOI] [PubMed] [Google Scholar]
- 15.Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PC. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J Am Chem Soc. 2011;133:4558–66. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider R, Schumacher MC, Mueller H, Nand D, Klaukien V, Heise H, Riedel D, Wolf G, Behrmann E, Raunser S, Seidel R, Engelhard M, Baldus M. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J Mol Biol. 2011;412:121–36. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- 17.Benzinger TL, Gregory DM, Burkoth TS, Miller-Auer H, Lynn DG, Botto RE, Meredith SC. Propagating structure of Alzheimer’s beta-amyloid(10–35) is parallel beta-sheet with residues in exact register. Proc Natl Acad Sci U S A. 1998;95:13407–12. doi: 10.1073/pnas.95.23.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Supramolecular structural constraints on Alzheimer’s beta-amyloid fibrils from electron microscopy and solid-state nuclear magnetic resonance. Biochemistry. 2002;41:15436–50. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 19.Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q Rev Biophys. 2008;41:265–97. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 20.Thakur A, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci U S A. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R. Structural features and domain organization of huntingtin fibrils. J Biol Chem. 2012;287:31739–46. doi: 10.1074/jbc.M112.353839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kajava AV, Baxa U, Steven AC. Beta arcades: recurring motifs in naturally occurring and disease-related amyloid fibrils. FASEB J. 2010;24:1311–9. doi: 10.1096/fj.09-145979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328–36. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, Widjaja K, Shaby BA, Lotz GP, Newhouse Y, Mitchell EJ, Osmand A, Gray M, Thulasiramin V, Saudou F, Segal M, Yang XW, Masliah E, Thompson LM, Muchowski PJ, Weisgraber KH, Finkbeiner S. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7:925–34. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang QC, Yeh TL, Leyva A, Frank LG, Miller J, Kim YE, Langen R, Finkbeiner S, Amzel ML, Ross CA, Poirier MA. A compact beta model of huntingtin toxicity. J Biol Chem. 2011;286:8188–96. doi: 10.1074/jbc.M110.192013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, Yang G, Ratovitski T, Delannoy M, Muchowski PJ, Finkbeiner S, Legleiter J, Ross CA, Poirier MA. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J Biol Chem. 2012;287:16017–28. doi: 10.1074/jbc.M111.252577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters-Libeu C, Miller J, Rutenber E, Newhouse Y, Krishnan P, Cheung K, Hatters D, Brooks E, Widjaja K, Tran T, Mitra S, Arrasate M, Mosquera LA, Taylor D, Weisgraber KH, Finkbeiner S. Disease-Associated Polyglutamine Stretches in Monomeric Huntingtin Adopt a Compact Structure. J Mol Biol. 2012 doi: 10.1016/j.jmb.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–72. doi: 10.1016/s0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Vitalis A, Wyczalkowski MA, Pappu RV. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 30.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc Natl Acad Sci U S A. 2006;103:16764–9. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein FA, Pastore A, Masino L, Zeder-Lutz G, Nierengarten H, Oulad-Abdelghani M, Altschuh D, Mandel JL, Trottier Y. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: towards a length-dependent toxicity gradient. J Mol Biol. 2007;371:235–44. doi: 10.1016/j.jmb.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 32.Stanger HE, Gellman SH. Rules for antiparallel beta-sheet design: D-Pro-Gly Is superior to L-Asn-Gly for beta-hairpin nucleation. J Am Chem Soc. 1998;120:4236–37. [Google Scholar]
- 33.Blanco F, Ramirez-Alvarado M, Serrano L. Formation and stability of beta-hairpin structures in polypeptides. Curr Opin Struct Biol. 1998;8:107–11. doi: 10.1016/s0959-440x(98)80017-1. [DOI] [PubMed] [Google Scholar]
- 34.Lacroix E, Kortemme T, Lopez de la Paz M, Serrano L. The design of linear peptides that fold as monomeric beta-sheet structures. Curr Opin Struct Biol. 1999;9:487–93. doi: 10.1016/s0959-440x(99)80069-4. [DOI] [PubMed] [Google Scholar]
- 35.Espinosa JF, Syud FA, Gellman SH. Analysis of the factors that stabilize a designed two-stranded antiparallel beta-sheet. Protein Sci. 2002;11:1492–505. doi: 10.1110/ps.4140102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ciani B, Jourdan M, Searle MS. Stabilization of beta-hairpin peptides by salt bridges: role of preorganization in the energetic contribution of weak interactions. J Am Chem Soc. 2003;125:9038–47. doi: 10.1021/ja030074l. [DOI] [PubMed] [Google Scholar]
- 37.Searle MS, Ciani B. Design of beta-sheet systems for understanding the thermodynamics and kinetics of protein folding. Curr Opin Struct Biol. 2004;14:458–64. doi: 10.1016/j.sbi.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Huyghues-Despointes BM, Qu X, Tsai J, Scholtz JM. Terminal ion pairs stabilize the second beta-hairpin of the B1 domain of protein G. Proteins. 2006;63:1005–17. doi: 10.1002/prot.20916. [DOI] [PubMed] [Google Scholar]
- 39.Pantoja-Uceda D, Santiveri CM, Jimenez MA. De novo design of monomeric beta-hairpin and beta-sheet peptides. In: Guerois R, de la Paz LM, editors. Protein Design. 2006/09/08. Vol. 340. Springer; 2006. pp. 27–51. [DOI] [PubMed] [Google Scholar]
- 40.Hughes RM, Waters ML. Model systems for beta-hairpins and beta-sheets. Curr Opin Struct Biol. 2006;16:514–24. doi: 10.1016/j.sbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 41.Santiveri CM, Leon E, Rico M, Jimenez MA. Context-dependence of the contribution of disulfide bonds to beta-hairpin stability. Chemistry. 2008;14:488–99. doi: 10.1002/chem.200700845. [DOI] [PubMed] [Google Scholar]
- 42.Kier BL, Shu I, Eidenschink LA, Andersen NH. Stabilizing capping motif for beta-hairpins and sheets. Proc Natl Acad Sci U S A. 2010;107:10466–71. doi: 10.1073/pnas.0913534107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith MH, Miles TF, Sheehan M, Alfieri KN, Kokona B, Fairman R. Polyglutamine fibrils are formed using a simple designed beta-hairpin model. Proteins. 2010;78:1971–9. doi: 10.1002/prot.22713. [DOI] [PubMed] [Google Scholar]
- 44.Ferrone F. Analysis of protein aggregation kinetics. Meths Enzymol. 1999;309:256–274. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 45.Bhattacharyya AM, Thakur AK, Wetzel R. polyglutamine aggregation nucleation: thermodynamics of a highly unfavorable protein folding reaction. Proc Natl Acad Sci U S A. 2005;102:15400–5. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, Thiagarajan G, Wetzel R. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- 47.Williams AD, Shivaprasad S, Wetzel R. Alanine scanning mutagenesis of Aβ(1–40) amyloid fibril stability. J Mol Biol. 2006;357:1283–94. doi: 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- 48.Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R. Slow amyloid nucleation via alpha-helix-rich oligomeric intermediates in short polyglutamine-containing huntingtin fragments. J Mol Biol. 2012;415:881–99. doi: 10.1016/j.jmb.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Gortari I, Portella G, Salvatella X, Bajaj VS, van der Wel PC, Yates JR, Segall MD, Pickard CJ, Payne MC, Vendruscolo M. Time averaging of NMR chemical shifts in the MLF peptide in the solid state. J Am Chem Soc. 2010;132:5993–6000. doi: 10.1021/ja9062629. [DOI] [PubMed] [Google Scholar]
- 50.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A. 2005;102:15871–6. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van der Wel PCA, Lewandowski JR, Griffin RG. Structural characterization of GNNQQNY amyloid fibrils by magic angle spinning NMR. Biochemistry. 2010;49:9457–9469. doi: 10.1021/bi100077x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tycko R. Solid-state NMR studies of amyloid fibril structure. Ann Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel Œ≤-sheet structure. Proceedings of the National Academy of Sciences. 2006;103:19754–19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Wel PC, Lewandowski JR, Griffin RG. Solid-state NMR study of amyloid nanocrystals and fibrils formed by the peptide GNNQQNY from yeast prion protein Sup35p. J Am Chem Soc. 2007;129:5117–30. doi: 10.1021/ja068633m. [DOI] [PubMed] [Google Scholar]
- 56.Li J, Hoop CL, Kodali R, Sivanandam VN, Van der Wel PCA. Amyloid-like fibrils from a domain-swapping protein feature a parallel, in-register conformation without native-like interactions. J Biol Chem. 2011;286:28988–28995. doi: 10.1074/jbc.M111.261750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mishra R, Hoop CL, Kodali R, Sahoo B, van der Wel PC, Wetzel R. Serine phosphorylation suppresses huntingtin amyloid accumulation by altering protein aggregation properties. J Mol Biol. 2012;424:1–14. doi: 10.1016/j.jmb.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lewandowski JR, van der Wel PCA, Rigney M, Grigorieff N, Griffin RG. Structural complexity of a composite amyloid fibril. Journal of the American Chemical Society. 2011;133:14686–14698. doi: 10.1021/ja203736z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–2917. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 60.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nature Cell Biology. 2009;11:219–U232. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW, Maggio JE. Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry. 2000;39:6288–95. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 62.Cannon MJ, Williams AD, Wetzel R, Myszka DG. Kinetic analysis of beta-amyloid fibril elongation. Anal Biochem. 2004;328:67–75. doi: 10.1016/j.ab.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 63.Yanagi K, Sakurai K, Yoshimura Y, Konuma T, Lee YH, Sugase K, Ikegami T, Naiki H, Goto Y. The Monomer-Seed Interaction Mechanism in the Formation of the beta2-Microglobulin Amyloid Fibril Clarified by Solution NMR Techniques. J Mol Biol. 2012;422:390–402. doi: 10.1016/j.jmb.2012.05.034. [DOI] [PubMed] [Google Scholar]
- 64.Schor M, Vreede J, Bolhuis PG. Elucidating the Locking Mechanism of Peptides onto Growing Amyloid Fibrils through Transition Path Sampling. Biophysical Journal. 2012;103:1296–1304. doi: 10.1016/j.bpj.2012.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walters RH, Murphy RM. Aggregation kinetics of interrupted polyglutamine peptides. J Mol Biol. 2011;412:505–19. doi: 10.1016/j.jmb.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Walters RH, Jacobson KH, Pedersen JA, Murphy RM. Elongation kinetics of polyglutamine Peptide fibrils: a quartz crystal microbalance with dissipation study. J Mol Biol. 2012;421:329–47. doi: 10.1016/j.jmb.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doran TM, Anderson EA, Latchney SE, Opanashuk LA, Nilsson BL. Turn Nucleation Perturbs Amyloid beta Self-Assembly and Cytotoxicity. Journal of Molecular Biology. 2012;421:315–328. doi: 10.1016/j.jmb.2012.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol. 2007;17:48–57. doi: 10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 69.Toyama BH, Weissman JS. Annual Review of Biochemistry. Vol. 80. Annual Reviews; Palo Alto: 2011. Amyloid structure: conformational diversity and consequences; pp. 557–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walters RH, Murphy RM. Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J Mol Biol. 2009;393:978–92. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Digambaranath JL, Campbell TV, Chung A, McPhail MJ, Stevenson KE, Zohdy MA, Finke JM. An accurate model of polyglutamine. Proteins-Structure Function and Bioinformatics. 2011;79:1427–1440. doi: 10.1002/prot.22970. [DOI] [PubMed] [Google Scholar]
- 72.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 73.O’Nuallain B, Shivaprasad S, Kheterpal I, Wetzel R. Thermodynamics of abeta(1–40) amyloid fibril elongation. Biochemistry. 2005;44:12709–18. doi: 10.1021/bi050927h. [DOI] [PubMed] [Google Scholar]
- 74.Takegoshi K, Nakamura S, Terao T. C-13-H-1 dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem Phys Lett. 2001;344:631–637. doi: 10.1063/1.2364503. [DOI] [PubMed] [Google Scholar]
- 75.Bennett AE, Rienstra CM, Auger M, Lakshmi KV, Griffin RG. Heteronuclear decoupling in rotating solids. J Chem Phys. 1995;103:6951–6958. [Google Scholar]
- 76.Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16:546–55. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]