

Abstract
A series of N-substituted lobelane analogues was synthesized and evaluated for their [3H]dihydrotetrabenazine binding affinity at the vesicular monoamine transporter and for their inhibition of vesicular [3H]dopamine uptake. Compound 19a, which contains an N-1,2(R)-dihydroxypropyl group, had been identified as a potential clinical candidate for the treatment of methamphetamine abuse.
The rewarding and reinforcing effects of methamphetamine (METH) are believed to be through its interaction with the vesicular monoamine transporter-2 (VMAT2) and the dopamine (DA) transporter (DAT), resulting in elevated synaptic DA concentrations.1 The involvement of VMAT2 in the mechanism of METH addiction is demonstrated also by the reduced drug-seeking behavior exhibited by heterozygous VMAT2 knockout mice compared to wild-type mice in condition place preference (CPP) experiments.2 Thus, VMAT2 has been considered as a potential target for development of treatments for METH abuse.3 Over the past decade, a research focus in our laboratories has been the discovery and development of novel therapeutic agents that target VMAT2.4 The validation of VMAT2 as a therapeutic target for METH abuse was based on (−)-lobeline (1, Fig. 1), a natural alkaloid from Lobelia inflata. Lobeline inhibits METH-evoked DA release from rat striatal slices and attenuates METH-induced behavioral effects in rats.5–7 The inhibitory effects of lobeline on METH are thought to result from its interaction with VMAT2. Lobeline binds to the tetrabenazine (TBZ) binding site on VMAT2 and inhibits DA uptake at VMAT2 at a concentration that does not interact with DAT and the serotonin transporter (SERT).8 However, lobeline is also a nicotinic acetylcholine receptor (nAChR) ligand exhibiting high affinity at the α4β2* subtype.8 Thus, the involvement of nAChRs as part of the mechanism by which lobeline inhibits the behavioral effects of METH cannot be ruled out. Structural modification of the lobeline molecule identified lobelane (2, Fig. 1) as a compound of interest. Compared to lobeline, lobelane has negligible activity at nAChRs, but has increased potency and selectivity in inhibiting VMAT2 function.9,10 Importantly, lobelane also dose-dependently decreases METH self-administration (SA) in rats without altering food-maintained responding.11 Collectively, these results suggest that VMAT2 is a valid target for METH abuse.
Fig. 1.
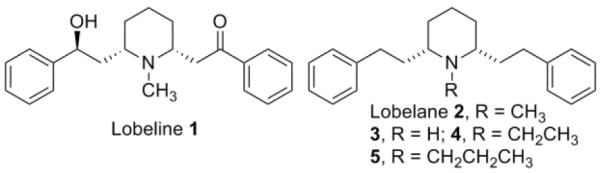
Structures of lobeline 1, lobelane 2, and lobelane analogues.
To identify analogues with higher potency and selectivity in inhibiting VMAT2 function, extensive structure-activity relationship (SAR) studies have been carried out based on the lobelane scaffold, including structural modifications of the piperidine ring, the two phenyl rings, and the ethylene linkers bridging the piperidine ring and the phenyl rings.4,9,12 These modifications have also included changing the chirality of the piperidinyl C-2 and C-6 stereo centers, altering the positions of the two phenethylsubstituents on the piperidine ring, replacing the piperidine ring with other aza-heterocyclic ring systems, adding various substituent groups on the phenyl rings, and varying the distance between the piperidine ring and the two phenyl rings.4,9,12 A number of lead compounds have been identified based on these modifications; however, similar to lobelane, most of these analogues also exhibit low water solubility, which limits in vivo evaluation of their pharmacological properties. In addition, despite their high potency in the in vitro VMAT2 assays, in vivo preliminary screens indicate that these lead compounds have limited efficacy in inhibiting METH SA in rats (unpublished results). We reasoned that poor pharmacokinetic (PK) properties of these analogues might be responsible for the low in vivo efficacy.
In the present study, we focused on structural modifications that would increase the hydrophilicity of these lobelane analogues by introducing polar functional groups into the lobelane molecule. During our previous SAR studies, we observed that the N-methyl group of lobelane is not required for VMAT2 binding and inhibition, since nor-lobelane (3, Fig. 1) has equal potency to lobelane (Table 1).10 In addition, replacement of the N-methyl group with an N-ethyl (compound 4) or an N-n-propyl group (compound 5)12a only slightly decreased the inhibitory effect of [3H]DA uptake at VMAT2 when compared to lobelane (Table 1). These preliminary results indicate that modifications to the N-methyl group can be tolerated. Herein, we further investigate the structural modification of the N-methyl group of lobelane and its analogues.
Table 1.
Analogue inhibition of [3H]DTBZ binding to rat synaptic vesicle membranes and inhibition of [3H]DA uptake into rat striatal synaptic vesicles.
| Compd. | [3H]DTBZ | VMAT2 [3H]DA Uptake |
|---|---|---|
| Ki ± SEM (μM)a | Ki ± SEM (μM)a | |
| 1 | 2.76 ± 0.64b | 0.47 ± 0.045b |
| 2 | 0.97 ± 0.00b | 0.045 ± 0.002b |
| 3 | 2.31 ± 0.21b | 0.044 ± 0.008b |
| 4 | 3.41 ± 0.67c | 0.060 ± 0.005 |
| 5 | 1.87 ± 0.25c | 0.087 ± 0.04 |
| 6 | 4.4 ± 0.76 | 0.34 ± 0.13 |
| 7 | 1.43 ± 0.136 | 0.17 ± 0.01 |
| 8 | 22.57 ± 0.23 | 4.49 ± 0.35 |
| 9 | 5.59 ± 0.94 | 0.51 ± 0.089 |
| 10 | 9.59 ± 1.47 | 0.18 ± 0.024 |
| 11 | 3.49 ± 0.63 | 0.68 ± 0.18 |
| 13 | > 100 | 0.054 ± 0.042 |
Each Ki value represents data from at least three independent experiments, evaluated in duplicate;
data taken from reference 10;
data taken from reference 12b.
Analogues 6–13, 14a, and 14b were synthesized starting from the common intermediate, nor-lobelane (3), which was prepared as previously reported (Scheme 1 and 2).12aN-Alkylation of 3 with methyl bromoacetate or chloroacetonitrile afforded analogues 6 and 8, respectively. Reduction of 6 and 8 with DIBAL-H furnished the corresponding amino alcohol analogue 7 and the diamino analogue 9. 9 was converted into analogue 10 by N-methylation under the treatment of NaBH3CN/(CH2O)n and to guanidino analogue 11 by reacting with N,N′-di-(tert-butoxycarbonyl)thiourea, followed by treatment with CF3COOH to remove the two BOC protective groups. Conversion of 3 to the corresponding hydrazino compound 12 was accomplished by a two-step procedure, i.e., N-nitrosation and a subsequent LAH reduction. Without further purification after work-up, the unstable compound 12 was converted into analogue 13 using the same N-methylation procedure as described for analogue 10 (Scheme 1). Analogues 14a and 14b, which contain an N-1,2(R)-dihydroxypropyl group and N-1,2(S)-dihydroxypropyl group, respectively, were prepared by reacting nor-lobelane with S-glycidol or R-glycidol, respectively (Scheme 2).
Scheme 1.
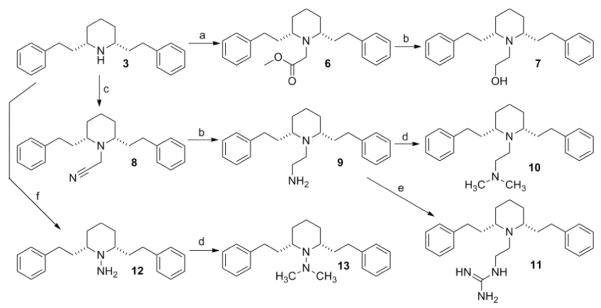
Reagents and conditions: a) BrCH2COOMe, K2CO3, CH3CN, reflux, 72 h, 77%; (b) DIBAL-H, toluene, 0 °C, 1 h, 83% for 7, 79% for 9; (c) ClCH2CN, K2CO3, NaI, CH3CN, reflux, 96 h, 66%; (d) NaCNBH3, (CH2O)n, HOAc, MeOH, rt, 4 h, 60% for 10, 35% for 13; (e) i. N,N′-di-(tertbutoxycarbonyl)thiourea, DMF, rt, 18 h, 58%; ii. CF3COOH, CH2Cl2, rt, 30 min, 74%; (f) i. NaNO2, HOAc, MeOH/H2O, 0 °C-reflux, 20 h, 82%; ii. LAH, THF, reflux, 1 h, 33%.
Scheme 2.
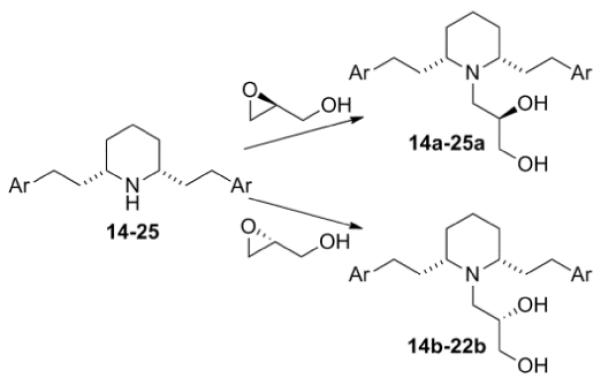
Synthesis of analogues containing N-1,2-dihydroxypropyl group.
Phenyl ring modified nor-lobelane analogues 15–25 were synthesized as previously reported.12a15a–25a and 15b–22b were synthesized by applying the same method for the synthesis of analogues 14a and 14b, from their corresponding precursors, 15–25 (Scheme 2, Table 2). Similarly, analogue 28a, a homologue of 19a, was prepared from the corresponding nor-compound 28. 28 was synthesized by initial Kumada coupling13 between 2,6-dibromopyridine (26) and 4-methoxybenzylmagnesium chloride to form compound 27, followed by catalytic hydrogenation over Adams catalyst (PtO2) (Scheme 3).
Table 2.
Analogue inhibition of [3H]DTBZ binding to rat synaptic vesicle membranes and inhibition of [3H]DA uptake into rat striatal synaptic vesicles.
| Compd. | Ar | [3H]DTBZ | VMAT2 [3H]DA Uptake |
|---|---|---|---|
| Ki ± SEM (μM)a | Ki ± SEM (μM)a | ||
| 14a |
|
0.56 ± 0.08 | 0.19 ± 0.05 |
| 14b | 1.28 ± 0.13 | 0.86 ± 0.12 | |
| 15a |
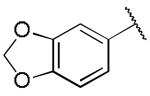
|
1.30 ± 0.05 | 0.16 ± 0.04 |
| 15b | 5.61 ± 0.62 | 0.76 ± 0.04 | |
| 16a |
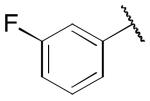
|
1.00 ± 0.16 | 0.19 ± 0.06 |
| 16b | 1.08 ± 0.38 | 1.03 ± 0.16 | |
| 17a |
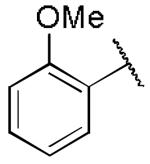
|
1.04 ± 0.73 | 0.49 ± 0.06 |
| 17b | 1.87 ± 0.69 | 0.79 ± 0.08 | |
| 18a |
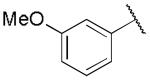
|
0.46 ± 0.22 | 0.14 ± 0.02 |
| 18b | 2.73 ± 0.68 | 0.52 ± 0.04 | |
| 19a |
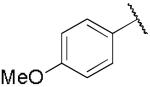
|
8.29 ± 2.79 | 0.029 ± 0.008 |
| 19b | 7.74 ± 2.34 | 0.18 ± 0.04 | |
| 20a |
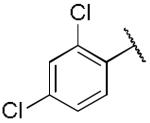
|
10.4 ± 0.65 | 0.14 ± 0.04 |
| 20b | 13.9 ± 0.38 | 0.09 ± 0.04 | |
| 21a |
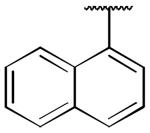
|
0.31 ± 0.07 | 0.03 ± 0.002 |
| 21b | 0.13 ± 0.07 | 0.08 ± 0.01 | |
| 22a |
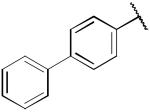
|
> 100 | 0.79 ± 0.23 |
| 22b | 90.2 ± 9.70 | 2.25 ± 1.30 | |
| 23 |
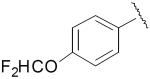
|
1.75 ± 0.36 | 0.023 ± 0.006 |
| 23a | 0.53 ± 0.053 | 0.060 ± 0.006 | |
| 24 |
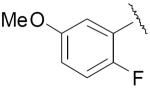
|
0.93 ± 0.22 | 0.039 ± 0.002 |
| 24a | 0.21 ± 0.02 | 0.16 ± 0.035 | |
| 25 |
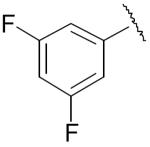
|
5.24 ± 1.81 | 0.043 ± 0.003 |
| 25a | 0.62 ± 0.10 | 0.104 ± 0.004 | |
| 28 | 13.1 ± 7.4 | 0.048 ± 0.005 | |
| 28a | - | 91 ± 40 | 0.57 ± 0.05 |
Each Ki value represents data from at least three independent experiments, evaluated in duplicate; The data of 14a–22a and 14b–22b were taken from reference 16.
Scheme 3.
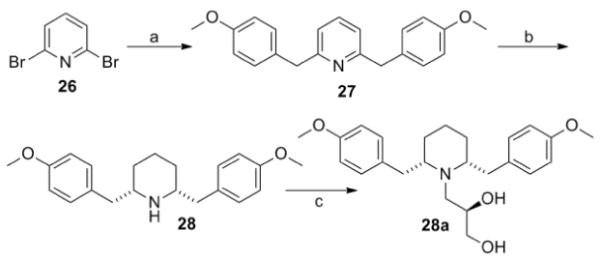
Reagents and conditions: (a) MeOPhCH2MgCl, NiCl2(dppp), THF, rt, 18 h, 97%; (b) H2, PtO2, HOAc, 50 psi, rt, 36 h, 84%; (c) (S)-glycidol, EtOH, reflux, 48 h, 76%.
The ability of the above analogues14 to interact with VMAT2 was assessed by determining their inhibition of [3H]dihydrotetrabenazine ([3H]DTBZ) binding to rat brain vesicle membranes, and their inhibition of [3H]DA uptake into rat striatal synaptic vesicles.10 The results are summarized in Table 1 and 2. Initial structural exploration of N-substitution patterns based on lobelane led to analogues 6–13 (Table 1), 14a, and 14b (Table 2). Compound 13, in which the N-methyl group is replaced by an N,N-dimethylhydrazino group, was the most potent analogue in this series, inhibiting [3H]DA uptake at VMAT2, being equipotent to lobelane. Analogue 8, in which the N-methyl group is replaced by an N-acetonitrile group, displayed 100-fold lower potency in inhibiting [3H]DA uptake at VMAT2 compared to lobelane. The rest of the analogues in this series were 4–19 fold less potent compared to lobelane in the same assay. Interestingly, the most potent inhibitor of DA uptake at VMAT2, 13, exhibited no affinity at the [3H]DTBZ binding site, indicating that DA uptake at VMAT2 is not via interaction with the DTBZ recognition binding site on VMAT2. For this series of analogues, there is no correlation between affinity for the VMAT2 binding site and inhibition of DA uptake at VMAT2, which is in consistent with our previously reported studies on other lobelane analogues.10,15
Compound 14a, which bears an N-1,2(R)-dihydroxypropyl group, was chosen for further structural optimization based on preliminary data from in vivo screens (decrease in METH SA in rats). We incorporated the chiral N-1,2-dihydroxypropyl moiety into a series of phenyl ring substituted lobelane analogues, including 15–22 and their corresponding N-methylated analogues. Compared to compound 14a, these analogues exhibited equal or improved binding affinity and potency for inhibiting [3H]DA uptake at VMAT2, as shown in our previous study.15 Accordingly, analogues 15a–22a, which contain the N-1,2(R)-dihydroxypropyl group, and 15b–22b, which contain the N-1,2(S)-dihydroxypropyl group, were synthesized and evaluated (Scheme 2 and Table 2). SAR on this series of analogues was discussed recently.16 In general, analogues containing N-1,2-dihydroxypropyl moieties analogues exhibited lower potencies for inhibiting [3H]DA uptake at VMAT2 compared to their corresponding N-methyl analogues. There are no apparent preferences in terms of [3H]DTBZ binding affinities between N-1,2(R)-dihydroxypropyl containing analogues and their corresponding N-1,2(S)-dihydroxypropyl containing enantiomers. N-1,2(R)-Dihydroxypropyl containing analogues generally had a higher potency at the [3H]DA uptake assay than their corresponding S-enantiomers, with the exception of an equipotent pair of analogues, 20a and 20b. Analogue 19a (Ki = 29 nM) had the highest potency of the series inhibiting [3H]DA uptake into synaptic vesicles, and was chosen for further neurochemical and behavioral studies.
Studies showed that 19a had the best pharmacological profile in this series,16 including over 50-fold selectivity for VMAT2 over DAT, SERT, and nAChRs; and decreasing METH-evoked DA release from vesicles and striatal slices without intrinsic activity. 19a was also shown to have great selectivity over a series of off-target receptors/proteins (unpublished data).
Our recent studies have demonstrated that 19a effectively decreases METH SA and CPP in rats without altering food-maintained responding. In addition, tolerance to these effects is not observed after repeated dosing of 19a.17 Importantly, 19a is predicted to have low abuse liability, since it is not self-administered and does not produce CPP.17 Compared to lobeline and lobelane, 19a exhibited a significantly increased duration of action to inhibit METH SA within a behavioral session. In this respect, we reasoned that 19a likely has a better PK profile than lobeline and lobelane due to the introduction of the hydrophilic N-1,2-dihydroxypropyl moiety. We are currently characterizing the PK profile of this molecule and results will be reported in due course.
We have continued the structural optimization studies based on 19a. These studies include modifying the substituents on the two phenyl rings to improve metabolic stability (analogues 23a–25a, Table 2) and modifying the two ethylene linkers bridging the phenyl rings and the central piperidine ring (analogue 28a). Further evaluation and modification of these compounds is ongoing currently.
Conclusions
In summary, a series of lobelane analogues, in which the N-methyl group of lobelane has been replaced by various hydrophilic moieties, has been synthesized and evaluated in the [3H]DTBZ binding assay and the vesicular [3H]DA uptake assay. Compound 19a, which contains a N-1,2(R)-dihydroxypropyl group, has been identified as a potential clinical candidate for the treatment of METH abuse.
Acknowledgements
This research was supported by NIH grant R01DA13519, U01DA13519, T32DA01617, and TR000117.
Notes and references
- 1.Sulzer D, Sonders MS, Poulsen NW, Galli A. Prog. Neurobiol. 2005;75:406. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, Jackson-Lewis V, Przedborski S, Uhl GR. Proc. Natl. Acad. Sci. USA. 1997;94:9938. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Zheng G, Dwoskin LP, Crooks PA. AAPS J. 2006;8:E682. doi: 10.1208/aapsj080478. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dwoskin LP, Crooks PA. Biochem. Pharmacol. 2002;63:89. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- 4.Crooks PA, Zheng G, Vartak AP, Culver JP, Zheng F, Horton DB, Dwoskin LP. Curr. Top. Med. Chem. 2011;11:1103. doi: 10.2174/156802611795371332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller DK, Crooks PA, Teng L, Witkin JM, Munzar P, Goldberg SR, Acri JB, Dwoskin LP. J. Pharmacol. Exp. Ther. 2001;296:1023. [PubMed] [Google Scholar]
- 6.Harrod SB, Dwoskin LP, Crooks PA, Klebaur JE, Bardo MT. J. Pharmacol. Exp. Ther. 2001;298:172. [PubMed] [Google Scholar]
- 7.Harrod SB, Dwoskin LP, Green TA, Gehrke BJ, Bardo MT. Psychopharmacology (Berl) 2003;165:397. doi: 10.1007/s00213-002-1289-6. [DOI] [PubMed] [Google Scholar]
- 8.(a) Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. J. Pharmacol. Exp. Ther. 1997;280:1432. [PubMed] [Google Scholar]; (b) Teng L, Crooks PA, Sonsalla PK, Dwoskin LP. J. Neurochem. 1998;71:258. doi: 10.1046/j.1471-4159.1998.71010258.x. [DOI] [PubMed] [Google Scholar]
- 9.Zheng G, Dwoskin LP, Deaciuc AG, Norrholm SD, Crooks PA. J. Med. Chem. 2005;48:5551. doi: 10.1021/jm0501228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nickell JR, Krishnamurthy S, Norrholm S, Deaciuc G, Siripurapu KB, Zheng G, Crooks PA, Dwoskin LP. J. Pharmacol. Exp. Ther. 2010;332:612. doi: 10.1124/jpet.109.160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neugebauer NM, Harrod SB, Stairs DJ, Crooks PA, Dwoskin LP, Bardo MT. Eur. J. Pharmacol. 2007;571:33. doi: 10.1016/j.ejphar.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Zheng G, Dwoskin LP, Deaciuc AG, Zhu J, Jones MD, Crooks PA. Bioorg. Med. Chem. 2005;13:3899. doi: 10.1016/j.bmc.2005.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zheng G, Dwoskin LP, Deaciuc AG, Crooks PA. Bioorg. Med. Chem. Lett. 2005;15:4463. doi: 10.1016/j.bmcl.2005.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zheng G, Dwoskin LP, Deaciuc AG, Crooks PA. Bioorg. Med. Chem. Lett. 2008;18:6509. doi: 10.1016/j.bmcl.2008.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Vartak AP, Nickell JR, Chaqkutip J, Dwoskin LP, Crooks PA. J. Med. Chem. 2009;52:7878. doi: 10.1021/jm900770h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumada M. Pure Appl. Chem. 1980;52:669. [Google Scholar]
- 14. Selected spectra data: compound 6, 1H NMR (300 MHz, CDCl3) δ 7.12–7.32 (m, 10H); 3.65 (s, 3H), 3.35 (s, 2H), 2.77 (m, 2H), 2.68 (t, J = 8.1 Hz, 4H), 1.77–1.90 (m, 3H), 1.50–1.65 (m, 4H), 1.20–1.40 (m, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 173.3, 142.6, 128.5, 128.4, 125.8, 67.8, 51.8, 46.3, 36.5, 32.6, 27.4, 24.9 ppm; EI-MS m/z 365 (M+); compound 7, 1H NMR (300 MHz, CDCl3) δ 7.14–7.34 (m, 10H); 3.50 (br s, 2H), 2.50–2.78 (m, 8H), 1.95–2.05 (m, 2H), 1.52–1.65 (m, 4H), 1.32–1.45 (m, 4H) ppm; 13C NMR (75 MHz, CDCl3) δ 141.9, 128.53, 128.50, 126.0, 62.6, 60.5, 46.9, 36.0, 33.5, 26.2, 23.1 ppm; EI-MS m/z 336 (M−1)+; compound 8, 1H NMR (300 MHz, CDCl3) δ 7.15–7.33 (m, 10H); 3.67 (s, 2H), 2.68 (t, J = 8.4 Hz, 4H), 2.52 (m, 2H), 1.62–1.92 (m, 8H), 1.30–1.52 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3) δ 142.1, 128.5, 128.3, 126.0, 115.5, 59.9, 36.3, 35.7, 31.0, 30.2, 24.3 ppm; EI-MS m/z 332 (M+); compound 9, 1H NMR (300 MHz, CDCl3) δ 7.05–7.30 (m, 10H); 2.40–2.75 (m, 10H), 1.78–1.95 (m, 2H), 1.30–1.65 (m, 8H) ppm; 13C NMR (75 MHz, CDCl3) δ 142.5, 128.4, 128.3, 125.7, 62.1, 49.0, 43.1, 36.7, 33.3, 26.6, 24.0 ppm; EI-MS m/z 335 (M−1)+; compound 10, 1H NMR (300 MHz, CDCl3) δ 7.15–7.32 (m, 10H); 2.55–2.75 (m, 8H), 2.37 (t, J = 8.1 Hz, 2H), 2.22 (s, 6H), 1.85–2.00 (m, 2H), 1.78 (m, 1H), 1.50–1.72 (m, 4H), 1.33–1.45 (m, 3H) ppm; 13C NMR (75 MHz, CDCl3) δ 142.4, 128.5, 128.4, 125.8, 62.5, 60.0, 46.1, 43.6, 36.2, 33.1, 27.0, 24.4 ppm; EI-MS m/z 364 (M+); compound 13, 1H NMR (300 MHz, CDCl3) δ 7.15–7.35 (m, 10H); 2.66–2.80 (m, 2H), 2.54 (s, 6H), 2.40–2.60 (m, 4H), 1.95–2.10 (m, 2H), 1.78–1.90 (m, 2H), 1.58–1.75 (m, 4H), 1.23–1.38 (m, 2H) ppm; 13C NMR (75 MHz, CDCl3) δ 143.3, 128.5, 128.4, 125.6, 61.1, 40.9, 36.7, 32.3, 31.9, 24.1 ppm; EI-MS m/z 336 (M+); compound 19a, 1H NMR (300 MHz, CDCl3) δ 7.07 (dd, J = 8.4, 2.1 Hz, 4H), 6.82 (d, J = 8.4 Hz, 4H), 3.78 (s, 6H), 1.22–1.63 (m, 7H), 3.70 (dd, J = 11.4, 3.3 Hz, 1H), 3.45–3.54 (m, 1H), 3.37 (dd, J = 11.4, 3.9 Hz, 1H), 1.72–1.92 (m, 3H), 2.42–2.74 (m, 8H) ppm; 13C NMR (75 MHz, CDCl3) δ 157.8, 134.2, 134.1, 129.4, 114.0, 68.2, 64.9, 63.1, 61.6, 55.5, 46.5, 36.5, 36.2, 32.8, 32.7, 26.6, 25.9, 23.1 ppm; LC-ESI-MS m/z 428 (M+1)+. All compounds are estimated >95% pure based on GC-MS or LC-MS data.
- 15.Nickell JR, Zheng G, Deaciuc G, Crooks PA, Dwoskin LP. J. Pharmacol. Exp. Ther. 2011;336:724. doi: 10.1124/jpet.110.172882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horton DB, Siripurapu KB, Zheng G, Crooks PA, Dwoskin LP. J. Pharmacol. Exp. Ther. 2011;339:286. doi: 10.1124/jpet.111.184770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beckmann JS, Denehy ED, Zheng G, Crooks PA, Dwoskin LP, Bardo MT. Psychopharmacology (Berl) 2012;220:395. doi: 10.1007/s00213-011-2488-9. [DOI] [PMC free article] [PubMed] [Google Scholar]