

Abstract
Elucidating the biogeography of bacterial communities on the human body is critical for establishing healthy baselines from which to detect differences associated with diseases. To obtain an integrated view of the spatial and temporal distribution of the human microbiota, we surveyed bacteria from up to 27 sites in 7–9 healthy adults on four occasions. We found that community composition was determined primarily by body habitat. Within habitats, interpersonal variability was high, while individuals exhibited minimal temporal variability. Several skin locations harbored more diverse communities than the gut and mouth, and skin locations differed in their community assembly patterns. These results indicate that our microbiota, although personalized, varies systematically across body habitats and time: such trends may ultimately reveal how microbiome changes cause or prevent disease.
The human body hosts complex microbial communities whose combined membership outnumbers our own cells by at least a factor of ten (1, 2). Together, our ~100 trillion microbial symbionts (the human microbiota) endow us with crucial traits; for example, we rely on them to aid in nutrition, resist pathogens, and educate our immune system (1, 3). To understand the full range of human genetic and metabolic diversity, it is necessary to characterize the factors influencing the diversity and distribution of the human microbiota (4, 5).
Determining our microbiota’s role in disease predisposition and pathogenesis will depend critically upon first defining “normal” states (5). Prior studies of healthy individuals have focused on particular body habitats including the gut (6, 7), skin (8–10), and oral cavity (11, 12), and have revealed microbial communities that were highly variable both within and between people. However, our microbial habitats are not isolated from one another; instead, each person comprises a complex yet interconnected landscape, consisting of many body habitats harboring distinctive microbiotas (1). We currently lack an integrated “whole-body” view of the microbial communities associated with healthy people over time.
Here, we address three general questions regarding the biogeography of the human microbiota in healthy adults: How is bacterial diversity partitioned across body habitats, people, and time? How does diversity at a variety of skin locales compare to that found in other body habitats? Do skin communities assemble differently at different sites? We performed an intensive survey of human-associated bacterial communities using a multiplexed barcoded pyrosequencing approach. Microbiota samples were donated on June 17th and 18th and September 17th and 18th, 2008. Volunteers were unrelated individuals of both sexes (13) and the following body habitats were sampled: gut (stool), oral cavity, external auditory canal [EAC; including earwax (cerumen) if present], inside the nostrils (nares), hair on the head, and skin surfaces (fig. S1). Up to 18 skin locations were sampled on each day and we subsequently performed a skin community assembly experiment. For each sample, variable region 2 (V2) of the bacterial 16S rRNA gene was PCR-amplified using a primer-set with a unique error-correcting barcode (14). Using this approach, we generated a data set consisting of >1,070,000 high-quality, classifiable 16S rRNA gene sequences with an average of 1,315 ± 420 (SD) sequences per sample (n = 815; Table S1).
The sequences collected for this study provide an overview of the healthy human microbiota. Across all body habitats we detected members of 22 bacterial phyla, but most sequences (92.3%) were related to four phyla: Actinobacteria (36.6%), Firmicutes (34.3%), Proteobacteria (11.9%), and Bacteroidetes (9.5%). Each habitat harbored a characteristic microbiota (figs. S2 to S4) and a relatively stable set of abundant taxa across people and over time (fig. S4) (13).
We assessed differences in overall bacterial community composition using a phylogeny-based metric, UniFrac (15). A relatively small UniFrac distance implies that two communities are similar, consisting of lineages sharing a common evolutionary history. UniFrac-based principal coordinates analysis (PCoA) revealed strong primary clustering by body habitat, rather than by host sex, individual, or day (Fig. 1 and fig. S5). Moreover, hierarchical clustering of UniFrac- and phylotype-based distances (phylotypes defined at ≥97% sequence identity; fig. S6) revealed a nested structure, with communities grouping first by body habitat, then by host individual, and finally by month. Accordingly, we found that composition varied significantly less within habitats than between habitats. Within habitats, variation was significantly less within individuals sampled over time than between individuals on a given day. Finally, after accounting for habitat and host individual, variation was significantly less over 24 hours than over 3 months (P < 0.01 for each comparison, one-tailed t tests; Fig. 1E and fig. S7). Hierarchical clustering of UniFrac distances among people’s daily composite “whole-body” communities (as defined with respect to our study) revealed perfect grouping by host individual and month (fig. S8), further emphasizing that our seemingly personalized microbiota remains relatively stable over time.
Fig. 1.

16S rRNA gene surveys reveal hierarchical partitioning of human-associated bacterial diversity. (A to D) Communities clustered using PCoA of the unweighted UniFrac distance matrix. Each point corresponds to a sample colored by (A) body habitat, (B) host sex, (C) host individual, or (D) collection date. The same plot is shown in each panel. F, female; M, male. (E and F) Mean (± SEM) unweighted UniFrac distance between communities. In (E) habitats are weighted equally and in (F) skin comparisons are within sites. (G) UPGMA clustering of composite communities from the indicated locales. Leaves are colored according to body habitat as in (A). R, right; L, left.
Despite the strong inter- and intrapersonal structuring of bacterial diversity, a high degree of spatial and temporal variability was also evident. We estimated community overlap by examining the fraction of shared phylotypes and evolutionary history (i.e., branch length) within a phylogenetic tree. Study-wide, ~12% of phylotypes (20% of branch lengths) appeared on all dates, while 3% of phylotypes (9% of branch lengths) appeared in all individuals, and only 0.1% of phylotypes (1% of branch lengths) appeared in all body habitats (fig. S9). No dominant phylotype was distributed among all of the body habitats of any person on any given day at our level of survey effort.
Body habitats differed in the degree to which their bacterial communities exhibited compositional variation. While intrapersonal differences (over time) were smaller than interpersonal differences (on each day) within all habitats examined (Fig. 1F and fig. S10), oral cavity communities were significantly less variable in terms of membership alone, both within and between people, than all other habitats (P < 0.01 for each comparison, one-tailed t tests; Fig. 1F and fig. S10A). Gut community structure was highly variable among people, but exhibited minimal variability within people over time (Fig. 1F and fig. S10). Skin (within sites), hair, nostril, and EAC communities had the highest levels of intrapersonal variability in membership over time, and were roughly on par with the gut in terms of interpersonal variability (Fig. 1F and fig. S10A). These results indicate that the size of the community “core” (the set of phylotypes shared among all individuals) will depend on the body habitat examined, and is likely to be larger in the oral cavity than in other habitats such as the gut or skin.
Compositional variation in skin bacterial communities was also attributable to differences among sites within hosts: the average site-to-site UniFrac distance within people was higher than the inter- and intrapersonal variability observed within sites. To gain insight into the shared community structure of skin sites in relation to one another and other body habitats, we performed hierarchical clustering of weighted UniFrac distances, which account for relative abundances as well as membership (16) (Fig. 1G and fig. S11). We found that right and left sides of the body grouped together with the exception of index fingers, which clustered with their respective palms. Clustering revealed a “head” group, including the forehead, external nose, external ears (pinnae), and hair, dominated by Propionibacterineae (60–80%; fig. S12) and an “arm” group, including volar aspect of forearms, palms, and index fingers, where Propionibacterineae were less abundant (20–40%; fig. S12). Sites on the trunk and legs clustered separately and were dominated by Staphylococcus spp. [armpits (axillae) and soles of feet] or Corynebacterium spp. [navel (umbilicus) and backs of knees (popliteal fossae)] (fig. S12). It is proposed that site-to-site clustering of skin bacterial communities is driven by differences in skin environmental characteristics (10). Although the nostrils (nares) and EACs clustered with skin, they also harbored upper-respiratory commensals (e.g., Branhamella spp.) and taxa likely derived from earwax, respectively (fig. S4). The labia minora was divergent, as Lactobacillus spp., a common inhabitant of the female urogenital system, dominated this skin site (fig. S12). Finally, the oral cavity (mouth rinse samples) and dorsal tongue, which clustered together, along with the gut were most divergent from skin and other communities. These patterns were also evident when we mapped the relative abundances of core (i.e., shared by all people in our study) and peripheral taxa found within the 27 communities onto the human body (fig. S13).
Skin sites vary dramatically in their level of bacterial diversity (10) (fig. S14). Moreover, we found that high-diversity skin locations harbored as many or more phylotypes (fig. S15) and significantly more phylogenetic diversity (i.e., branch length; Fig. 2A) than the gut or oral cavity given our survey effort. Indeed, most people on most days had at least one, and oftentimes many skin sites harboring diversity as high or higher than their gut. On average, high-diversity skin sites included the forearm, palm, index finger, back of the knee, and sole of the foot. Other sites (e.g., the forehead) had lower diversity (fig. S14). Skin sites were also compositionally distinct (fig. S16), as highlighted by PCoA of UniFrac distances among forehead (low-diversity) and forearm (high-diversity) communities (Fig. 2B). Importantly, site-to-site differences in skin diversity were inter- and intrapersonally robust: forehead diversity was lower than palm diversity in each person on each day (Fig. 2C) and this was also true for forehead versus forearm communities (fig. S17).
Fig. 2.

Site-to-site variation on skin surfaces. (A) Rarefaction curves for communities sampled from skin and other habitats. Phylogenetic diversity is in units of branch length. Mean ± 95% confidence interval shown. (B) PCoA plot as in Fig. 1 showing only dorsal tongue, forehead, and volar forearm samples. (C) Individual rarefaction curves for forehead and palm communities.
These and others’ results (10) indicate that skin bacterial communities exhibit predictable biogeographic patterns. However, it is unclear whether these patterns arise due to differences in current environmental factors (e.g., local chemistry, nutrient availability), historical exposures (i.e., microbes available to colonize), or both (17, 18). To address this question, and to gain insight into the community assembly patterns of skin bacterial communities, we carried out an experiment in which plots on the foreheads and left volar forearms of volunteers were disinfected, inoculated with foreign microbiotas (i.e., defined historical exposures), and tracked over time (13) (fig. S18).
Skin bacterial community assembly proceeded differently on the forehead than on the volar forearm. At 2, 4, and 8 hours post-transplant, forearm plots (n = 16) inoculated with tongue bacteria were more similar to tongue communities than to native forearm communities in composition, diversity, and the relative abundance of Propionibacterium spp. (Fig. 3 and fig. S19). Conversely, forehead plots (n = 16) inoculated with tongue bacteria grew more similar to native forehead communities over time, as seen in overall structure and the relative abundance of Propionibacterium spp. (Fig. 3). Thus on the forehead, factors additional to the history of exposure to tongue bacteria shaped community assembly. Forearm and forehead plots (n = 16 each) inoculated with each other’s microbiota appeared to assemble communities that were more similar to their initial native microbiota than to the transplants (Fig. 3). Intrapersonal and same- and opposite-sex interpersonal transplants performed similarly (figs. S20 and S21). While acknowledging that our conclusions might change given a longer observation period, we suggest that environmental characteristics play a stronger role in shaping skin bacterial communities at sebaceous sites such as the forehead than at dry sites such as the forearm, either by selecting for the native microbiota, against the foreign microbiota, or by supporting more rapid growth and/or recolonization from sites protected from disturbance.
Fig. 3.
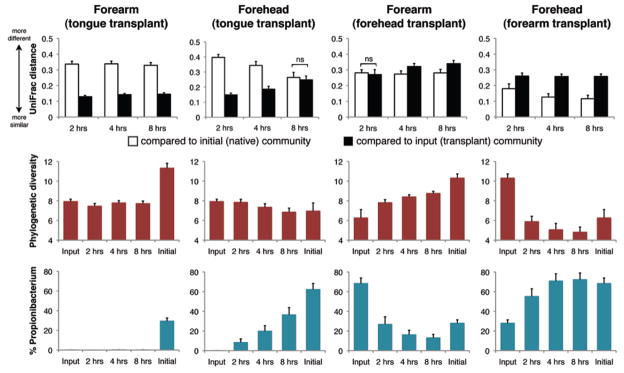
Community assembly on forehead versus volar forearm skin surfaces. (Upper) Mean (± SEM) weighted UniFrac distance between communities. At each time point, P < 0.01 unless indicated; two-tailed t tests. ns, not significant. (Middle) Mean (± SEM) phylogenetic diversity controlled for sampling effort. (Bottom) Mean (± SEM) relative abundance of Propionibacterium spp.
These findings have a variety of implications for the practice of medicine, both from the perspective of prevention and therapeutics. For example, they emphasize the need to (i) specify body habitat when conducting in-patient microbial surveillance studies designed to examine the flow of normal and pathogenic organisms into and out of different body sites in patients and their health care providers, (ii) determine the local biotic and abiotic conditions of sub-sites of a given body habitat such as the skin in order to understand why some sub-sites are more or less resistant to invasion, and (iii) designate those sites that are amenable to transplantation of microbial communities with natural or engineered metabolic capacities that would be beneficial to a host.
Our work also ties together two emerging themes from studies of human-associated microbial communities: high levels of variability among individuals in every body habitat studied to date, including the gut (6, 7), skin (8–10), and oral cavity (11, 12), and relative stability within individuals (7, 10). These patterns suggest that the search for microbial factors associated with disease, although difficult to ascertain due to the high intrinsic levels of variability among healthy individuals, may be achievable using broad profiling techniques such as those employed here.
Supplementary Material
Acknowledgments
We thank the volunteers for their participation; J. Manchester for pyrosequencing; D. McDonald and J. Kuczynski for bioinformatics support; K. Ramirez and R. Gesumaria for sample collection and processing; L. Kyro for graphical design; and M. Blaser, P. Hugenholtz, G. Huttley, N. Pace, and members of the Knight lab for valuable feedback. This work was supported in part by HHMI, and by grants from the NIH (DK78669, DK64540), the Bill and Melinda Gates Foundation, and the Crohns and Colitis Foundation of America. Sequences were deposited in the European Read Archive (accession number ERA000159).
Footnotes
References and Notes
- 1.Wilson M. Bacteriology of humans: an ecological perspective. Blackwell Publishing; Malden, MA: 2008. [Google Scholar]
- 2.Savage DC. Annu Rev Microbiol. 1977;31:107. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 3.Dethlefsen L, McFall-Ngai M, Relman DA. Nature. 2007;449:811. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lederberg J. Science. 2000;288:287. doi: 10.1126/science.288.5464.287. [DOI] [PubMed] [Google Scholar]
- 5.Turnbaugh PJ, et al. Nature. 2007;449:804. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eckburg PB, et al. Science. 2005;308:1635. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ, et al. Nature. 2009;457:480. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Z, Tseng CH, Pei ZH, Blaser MJ. Proc Natl Acad Sci USA. 2007;104:2927. doi: 10.1073/pnas.0607077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fierer N, Hamady M, Lauber CL, Knight R. Proc Natl Acad Sci USA. 2008;105:17994. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grice EA, et al. Science. 2009;324:1190. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aas JA, Paster BJ, Stokes LN, Olsen I, Dewhirst FE. J Clin Microbiol. 2005;43:5721. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nasidze I, Li J, Quinque D, Tang K, Stoneking M. Genome Research. 2009;19:636. doi: 10.1101/gr.084616.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.See supporting material on Science Online
- 14.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Nature Methods. 2008;5:235. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lozupone C, Knight R. Appl Environ Microbiol. 2005;71:8228. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lozupone CA, Hamady M, Kelley ST, Knight R. Appl Environ Microbiol. 2007;73:1576. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martiny JBH, et al. Nature Rev Microbiol. 2006;4:102. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 18.Rawls JF, Mahowald MA, Ley RE, Gordon JI. Cell. 2006;127:423. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.