

Abstract
The aim of this investigation was to test the hypothesis that extracellular guanosine regulates extracellular adenosine levels. Rat preglomerular vascular smooth muscle cells were incubated with adenosine, guanosine, or both. Guanosine (30 μmol/l) per se had little effect on extracellular adenosine levels. Extracellular adenosine levels 1 h after addition of adenosine (3 μmol/l) were 0.125 ± 0.020 μmol/l, indicating rapid disposition of extracellular adenosine. Extracellular adenosine levels 1 h after addition of adenosine (3 μmol/l) plus guanosine (30 μmol/l) were 1.173 ± 0.061 μmol/l, indicating slow disposition of extracellular adenosine. Cell injury increased extracellular levels of endogenous adenosine and guanosine, and the effects of cell injury on endogenous extracellular adenosine were modulated by altering the levels of endogenous extracellular guanosine with exogenous purine nucleoside phosphorylase (converts guanosine to guanine) or 8-aminoguanosine (inhibits purine nucleoside phosphorylase). Extracellular guanosine also slowed the disposition of extracellular adenosine in rat preglomerular vascular endothelial cells, mesangial cells, cardiac fibroblasts, and kidney epithelial cells and in human aortic and coronary artery vascular smooth muscle cells and coronary artery endothelial cells. The effects of guanosine on adenosine levels were not mimicked or attenuated by 5-iodotubericidin (adenosine kinase inhibitor), erythro-9-(2-hydroxy-3-nonyl)-adenine (adenosine deaminase inhibitor), 5-aminoimidazole-4-carboxamide (guanine deaminase inhibitor), aristeromycin (S-adenosylhomocysteine hydrolase inhibitor), low sodium (inhibits concentrative nucleoside transporters), S-(4-nitrobenzyl)−6-thioinosine [inhibits equilibrative nucleoside transporter (ENT) type 1], zidovudine (inhibits ENT type 2), or acadesine (known modulator of adenosine levels). Guanosine also increases extracellular inosine, uridine, thymidine, and cytidine, yet decreases extracellular uric acid. In conclusion, extracellular guanosine regulates extracellular adenosine levels.
Keywords: guanosine, guanosine 5′-monophosphate, adenosine, adenosine 5′-monophosphate, purine nucleoside phosphorylase
adenosine is formed in the extracellular compartment, activates multiple cell surface receptors, and thereby regulates cellular function (31). Because adenosine and guanosine are structurally very similar, it seems likely that evolution also would utilize guanosine as a signaling molecule. Indeed, many of the same stimuli that increase extracellular adenosine also increase extracellular guanosine (53). However, there is only limited evidence for cell surface receptors for guanosine (85) and it remains unclear as to whether such receptors exist (84). We hypothesize, therefore, that guanosine functions as a significant extracellular signaling molecule without the need for guanosine receptors. Specifically, we hypothesized that extracellular guanosine is a critically important physiological regulator of extracellular adenosine levels and that extracellular guanosine regulates extracellular adenosine by interfering with the disposition of extracellular adenosine. The purpose of the experiments described in this report was to address this hypothesis.1
METHODS
Chemicals.
Adenosine, guanosine, guanosine 5′-monophosphate (GMP), guanosine 3′,5′-cyclic monophosphate (cGMP), α,β-methylene-adenosine-5′-diphosphate (AMPCP), iodoacetate, 5-iodotubericidin, erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA), acadesine, 5-aminoimidazole-4-carboxamide (AIC), aristeromycin, S-(4-nitrobenzyl)-6-thioinosine (NBMPR), zidovudine (AZT), uridine, thymidine, cytosine, and purine nucleoside phosphorylase (PNP) were from Sigma-Aldrich (St. Louis, MO). 8-Aminoguanosine and pentostatin were from Toronto Research Chemicals (North York, Ontario, Canada).
Cell culture.
Rat preglomerular vascular smooth muscle cells (64), rat preglomerular vascular endothelial cells (8, 26), rat glomerular mesangial cells (40), and rat cardiac fibroblasts (15) were cultured from kidneys and hearts harvested from male Wistar-Kyoto rats (Taconic Farms, Germantown, NY) as previously described in detail (see references). The University of Pittsburgh Institutional Animal Care and Use Committee approved all procedures. Rat mixed renal epithelial cells were purchased from Cell Biologics (Chicago, IL). Human aortic vascular smooth muscle cells, human coronary artery smooth muscle cells, and human coronary artery endothelial cells were obtained from Lonza (Allendale, NJ).
Real-time PCR for equilibrative (ENTs) and concentrative (CNTs) nucleoside transporters.
RNA was isolated (TRIzol reagent; Life Technologies), and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad). Rat ENT1 primers were as follows: forward, 5′- ggcttcttcacctctgttgc-3′; reverse, 5′- ttgagctgcaggtaatgtcg-3′; 167 bp amplification product. Rat ENT2 primers were as follows: forward, 5′-ctgctcttcaacgtcatgga-3′; reverse, 5′-atcctgccagaagatgatgg-3′; 177 bp amplification product. Rat CNT1 primers were as follows: forward, 5′-acgctcagaacctcttggaa-3′; reverse, 5′-ggacgtaggagcagatgagc-3′; 186 bp amplification product. Rat CNT2 primers were as follows: forward, 5′-ggtgcctgtgtttccttcat-3′; reverse, 5′-ctgaggctggactccttgtc-3′; 218 bp amplification product. Rat CNT3 primers were as follows: forward, 5′-gattgcagcctgtgcactaa-3′; reverse, 5′-gaatgaccgccaagatcagt-3′; 209 bp amplification product. β-actin primers were as follows: forward, 5′-actcttccagccttccttc-3′; reverse, 5′-atctccttctgcatcctgtc-3′; 171 bp amplification product. Real-time polymerase chain reaction (PCR) analysis was performed using SYBR Green PCR Master Mix (Applied Biosystems; Foster City, CA) in the AB 7300 Real-Time PCR System (Applied Biosystems). Threshold cycle (Ct) for β-actin was subtracted from Ct for target to calculate 2ΔCt.
General protocol.
Experiments were performed in confluent cells (passage number between 2 and 5) in 24-well plates under standard cell culture conditions. On the day of study, cells were washed twice with 1 ml of phosphate-buffered saline and then incubated for 1 h with 0.5 ml of phosphate-buffered saline containing the indicated treatments. After the 1-h incubation, the medium was collected and frozen (−80°C) for later analysis of adenosine or guanosine as described below.
Analytical method for purines.
Samples were injected onto an Agilent Zorbax eclipse XDB-C-18 column (3.5 μm beads; 2.1 × 100 mm) and quantified using a triple quadrupole mass spectrometer (TSQ Quantum-Ultra, ThermoFisher Scientific, San Jose, CA) operating in the selected reaction monitoring mode with a heated electrospray ionization source. The mobile phase, which was delivered by an ultra pressure liquid chromatographic system (Accela, ThermoFisher Scientific, San Jose, CA) at 300 μl/min, was a linear gradient of buffer A (0.1% formic acid in water) and buffer B (0.1% formic acid in methanol). The gradient (A/B) was as follows: 0 to 2 min, 98.5%/1.5%; 2 to 4 min, to 98%/2%; 5 to 6 min, to 92%/8%; 7 to 8 min, to 85%/15%; 9 to 11.5 min, to 98.5%/1.5%. Sample tray temperature was set at 4°C, and the column temperature was kept at 20°C. Instrument parameters were as follows: ion spray voltage, 3.8 kV; ion transfer tube temperature, 270°C; source vaporization temperature, 220°C; Q2 CID gas, argon at 1.5 mTorr; sheath gas, nitrogen at 50 psi; auxiliary gas, nitrogen at 40 psi; Q1/Q3 width, 0.7/0.7 units full-width half-maximum; source CID, off; scan width, 0.5 units; scan time, 0.05 s; tube lens offset, 123 V. Mass transitions were: adenosine, 268→136; 13C10-adenosine (internal standard), 278→141; inosine, 269→137; 3′,5′-cAMP, 330→136; guanosine, 284→152; uridine, 245→113; cytidine, 244→112; thymidine, 243→127; hypoxanthine, 137→69; xanthine, 153→136; uric acid, 169→141.
Statistical analyses.
Statistical analysis was performed with Student's unpaired t-test or by 1-factor or 2-factor analysis of variance (ANOVA) with post hoc comparisons using a Fisher's least significant difference (LSD) test. The criterion of significance was P < 0.05. All values in text and figures are means ± SE.
RESULTS
Extracellular guanosine inhibits adenosine disposition in rat preglomerular vascular smooth muscle cells.
Rat preglomerular vascular smooth muscle cells were incubated for 1 h with no treatments, with exogenous adenosine only (0.3, 1 or 3 μmol/l), with exogenous guanosine only (10 or 30 μmol/l), or with combinations of adenosine plus guanosine. As shown in Fig. 1A, in the absence of added adenosine, guanosine at 10 and 30 μmol/l had little, if any, effect on extracellular levels of adenosine, indicating little, if any, direct effect of guanosine on adenosine production or release. However, when exogenous adenosine was added to the medium, guanosine now caused a concentration-related increase in extracellular adenosine levels. In this regard, the ability of a given concentration of guanosine to enhance extracellular adenosine levels was proportional to the concentration of added adenosine, thus giving rise to a highly statistically significant (P < 0.000001) interaction between guanosine and adenosine. In cells in which extracellular adenosine was not added to the medium, extracellular adenosine levels were near or below the detection limit (∼0.0002 μmol/l) of our assay. One hour after addition of 3 μmol/l of exogenous adenosine, adenosine levels were low (0.125 ± 0.020 μmol/l), indicating rapid and near complete (96%) disposition of extracellular adenosine by a monolayer of cells in culture. In the presence of 30 μmol/l of guanosine per se, extracellular adenosine levels remained very low (0.077 ± 0.008 μmol/l). However, extracellular adenosine levels 1 h after addition of adenosine (3 μmol/l) plus guanosine (30 μmol/l) were 1.173 ± 0.061 μmol/l, indicating slow and incomplete (61%) disposition of extracellular adenosine. Thus these experiments confirm that guanosine significantly inhibits the disposition of extracellular adenosine.
Fig. 1.

A: rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations of adenosine (0, 0.3, 1, or 3 μmol/l) plus various concentrations of guanosine (0, 10, or 30 μmol/l). B: rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence (control) or presence of guanosine (30 μmol/l), guanosine 5′-monophosphate (GMP; 30 μmol/l), or GMP plus α,β-methylene-adenosine-5′-diphosphate (AMPCP; 100 μmol/l). For A and B, the medium was assayed for adenosine by mass spectrometry. Values represent means ± SE. <DL, less than detection limit.
Extracellular GMP inhibits extracellular adenosine disposition in rat preglomerular vascular smooth muscle cells via conversion to guanosine.
Because extracellular GMP can be metabolized to guanosine via ecto-5′-nucleotidase (72), extracellular GMP should mimic the effects of guanosine. To test this, rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l), GMP (30 μmol/l), or GMP plus AMPCP [100 μmol/l; inhibitor of ecto-5′-nucleotidase (99)]. As illustrated in Fig. 1B, GMP, like guanosine, increased extracellular adenosine levels. This effect was likely mediated by conversion of GMP to guanosine because GMP was less effective than guanosine and inhibition of ecto-5′-nucleotidase abolished the ability of GMP to elevate extracellular adenosine.
Extracellular guanine does not affect extracellular adenosine disposition in rat preglomerular vascular smooth muscle cells.
It is conceivable that extracellular guanosine inhibits extracellular adenosine disposition via conversion to extracellular guanine by an ecto-purine nucleoside phosphorylase (71). To test this, rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l) and either without or with 8-aminoguanosine [30 μmol/l; purine nucleoside phosphorylase inhibitor (82)]. As depicted in Fig. 2A, 8-aminoguanosine did not inhibit the ability of added guanosine to elevate extracellular adenosine levels. Moreover, added guanine (30 μmol/l) did not elevate extracellular adenosine levels when incubated with 3 μmol/l of added adenosine (Fig. 2B). Thus, it can be concluded that guanosine, not guanine, regulates extracellular adenosine.
Fig. 2.

A: rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l) and either without or with 8-aminoguanosine (8-AG; 30 μmol/l; purine nucleoside phosphorylase inhibitor). B: rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanine (30 μmol/l). For A and B, the medium was assayed for adenosine by mass spectrometry. Values represent means ± SE.
Fig. 8.

Rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l) and either without or with the following: 5-iodotubericidin (IDO; 0.1 μmol/l; adenosine kinase inhibitor; A); erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA; 10 μmol/l; adenosine deaminase inhibitor; B); IDO plus EHNA (C); or S-(4-nitrobenzyl)-6-thioinosine (NBMPR; 10 μmol/l; equilibrative nucleoside transporter type 1 inhibitor; D). The medium was assayed for adenosine by mass spectrometry. Values represent means ± SE. aSignificantly different from corresponding no-guanosine group; bsignificantly different from PBS-guanosine group.
Extracellular guanosine, GMP, and cGMP inhibit the disposition of AMP-derived extracellular adenosine in rat preglomerular vascular smooth muscle cells.
Extracellular nucleotides are an important source of extracellular adenosine (31). Thus, extracellular guanosine and GMP should inhibit the disposition of extracellular adenosine that is derived from the metabolism of extracellular AMP to extracellular adenosine. Because ecto-phosphodiesterase can metabolize extracellular cyclic nucleotides (42), cGMP may also affect extracellular adenosine levels by its conversion to GMP and hence to guanosine. To test these hypotheses, rat preglomerular vascular smooth muscle cells were incubated without or with AMP (10 μmol/l) for 1 h in the absence or presence of guanosine, GMP, or cGMP (10 and 30 μmol/l). Exogenous guanosine, GMP, and cGMP per se had little, if any, effect on extracellular adenosine levels (Fig. 3, A, B, and C, respectively). In contrast, in the presence of added AMP, exogenous guanosine, GMP, and cGMP increased extracellular adenosine levels (Fig. 3, A, B, and C, respectively), as evidenced by a highly significant statistical interaction between guanosine and AMP (P < 0.000001), GMP and AMP (P < 0.000001), and cGMP and AMP (P < 0.000001). Consistent with cGMP → GMP → guanosine → inhibition of extracellular adenosine disposition, the rank order potency was guanosine > GMP > cGMP. Consistent with AMP → adenosine, when extracellular adenosine levels in the absence of guanosine were approximately the same in the presence of exogenous AMP (10 μmol/l) and adenosine (3 μmol/l), the increases in extracellular adenosine by exogenous guanosine were nearly identical (Fig. 4A). Also consistent with AMP → adenosine and GMP → guanosine, in the presence of AMPCP (100 μmol/l) to block ecto-5′-nucleotidase, GMP (10 and 30 μmol/l) did not affect extracellular adenosine when incubated with exogenous AMP (10 μmol/l) (Fig. 4B).
Fig. 3.

A: rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations of guanosine (0, 10, or 30 μmol/l) without and with adenosine 5′-monophosphate (AMP; 10 μmol/l). B: rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations of GMP (0, 10, or 30 μmol/l) without and with AMP (10 μmol/l). C: rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations of guanosine 3′,5′-cyclic monophosphate (cGMP; 0, 10, or 30 μmol/l) without and with AMP (10 μmol/l). For A–C, the medium was assayed for adenosine by mass spectrometry. Values represent means ± SE.
Fig. 4.

A: rat preglomerular vascular smooth muscle cells were incubated for 1 h with either AMP (10 μmol/l) or adenosine (3 μmol/l) without and with guanosine (30 μmol/l). B: rat preglomerular vascular smooth muscle cells were incubated with AMP (10 μmol/l) plus AMPCP (100 μmol/l) for 1 h in the presence of various concentrations of GMP (0, 10, or 30 μmol/l). For A and B, the medium was assayed for adenosine by mass spectrometry. Values represent means ± SE.
Endogenous extracellular guanosine inhibits the disposition of endogenous extracellular adenosine in rat preglomerular vascular smooth muscle cells.
To test the concept that the endogenous extracellular guanosine regulates the disposition of endogenous extracellular adenosine, rat preglomerular vascular smooth muscle cells were incubated for 1 h in the absence and presence of a iodoacetate [50 μmol/l; metabolic toxin (62)] to induce cellular injury, and without or with either 8-aminoguanosine (30 μmol/l) or purine nucleoside phosphorylase (1 U/ml) to block or accelerate, respectively, the metabolism of guanosine. Iodoacetate increased both extracellular levels of guanosine (Fig. 5A) and adenosine (Fig. 5B). 8-Aminoguanosine (blocks guanosine metabolism) enhanced the ability of iodoacetate to elevate both extracellular guanosine (Fig. 5C) and adenosine (Fig. 5D). Addition of PNP [metabolizes guanosine but not adenosine (28, 78)] abolished iodoacetate-induced extracellular guanosine (Fig. 5E) and nearly abolished iodoacetate-induced extracellular adenosine (Fig. 5F). Moreover, as shown in Fig. 6, cell injury with iodoacetate significantly increased extracellular guanosine (Fig. 6A), had only a small effect on extracellular uridine (Fig. 6B), and had no effect on extracellular cytidine (Fig. 6C) or thymidine (Fig. 6D). These results imply that cell injury releases primarily guanosine, but releases less uridine, cytidine, and thymidine and that endogenously released guanosine regulates extracellular adenosine levels.
Fig. 5.

Rat preglomerular vascular smooth muscle cells were incubated for 1 h in the absence and presence of iodoacetate (50 μmol/l) and without (A and B) or with either 8-aminoguanosine (30 μmol/l; C and D) or purine nucleoside phosphorylase (1 U/ml; E and F). Medium was assayed for guanosine and adenosine by mass spectrometry. Values represent means ± SE. <DL, less than detection limit.
Fig. 6.

Rat preglomerular vascular smooth muscle cells were incubated for 1 h in the absence (basal) and presence of iodoacetate (50 μmol/l), and medium was assayed for guanosine (A), uridine (B), cytidine (C), and thymidine (D) by mass spectrometry. Values represent means ± SE. <DL, less than detection limit.
Extracellular guanosine inhibits adenosine disposition in many cell types.
To determine whether extracellular guanosine regulates extracellular adenosine disposition in many cell types, we examined the ability of a wide range of concentrations of exogenous guanosine (0.3 to 300 μmol/l) to increase extracellular adenosine levels when incubated with added adenosine (3 μmol/l). Guanosine concentration-dependently increased extracellular adenosine levels in rat preglomerular vascular smooth muscle cells (Fig. 7A), rat preglomerular vascular endothelial cells (Fig. 7B), rat glomerular mesangial cells (Fig. 7C), rat cardiac fibroblasts (Fig. 7D), rat renal epithelial cells (Fig. 7E), human aortic vascular smooth muscle cells (Fig. 7F), human coronary artery smooth muscle cells (Fig. 7G), and human coronary artery endothelial cells (Fig. 7H).
Fig. 7.

Rat preglomerular vascular smooth muscle cells (A), rat preglomerular vascular endothelial cells (B), rat mesangial cells (C), rat cardiac fibroblasts (D), rat kidney epithelial cells (E), human aortic vascular smooth muscle cells (F), human coronary artery vascular smooth muscle cells (G), and human coronary artery endothelial cells (H) were incubated for 1 h with various concentrations of guanosine [0 (basal), 0.3, 1, 3, 10, 30, 100, or 300 μmol/l] plus adenosine (3 μmol/l). The medium was assayed for adenosine by mass spectrometry. Values represent means ± SE. aSignificantly different from basal.
Mechanism by which extracellular guanosine inhibits adenosine disposition.
Two major enzymes involved in adenosine disposition are adenosine kinase and adenosine deaminase (16). Therefore we examined in rat preglomerular vascular smooth muscle cells the role of these enzymes in mediating the effects of guanosine on adenosine levels. Neither administration of 5-iodotubericidin [adenosine kinase inhibitor (80, 87, 88); 0.1 μmol/l; Fig. 8A] nor administration of erythro-9-(2-hydroxy-3-nonyl)-adenine [adenosine deaminase inhibitor (2, 52, 56–58); 10 μmol/l; Fig. 8B] nor simultaneous administration of 5-iodotubericidin and erythro-9-(2-hydroxy-3-nonyl)-adenine (Fig. 8C) mimicked the efficacy of guanosine with respect to increasing adenosine levels in the presence of added adenosine. Moreover, neither 5-iodotubericidin (Fig. 8A) nor erythro-9-(2-hydroxy-3-nonyl)-adenine (Fig. 8B) nor 5-iodotubericidin plus erythro-9-(2-hydroxy-3-nonyl)-adenine (Fig. 8C) attenuated the ability of guanosine to increase adenosine levels in the presence of added adenosine.
The equilibrative nucleoside transporters types 1 and 2 (ENT1 and ENT2) and concentrative nucleoside transporters types 1, 2, and 3 (CNT1, CNT2, and CNT3) mediate in part the transport of adenosine across cell membranes (29, 94), and ENT1 in particularly is highly expressed in many cell types and importantly mediates transport of adenosine across cell membranes (30). Therefore we examined in rat preglomerular vascular smooth muscle cells the relative expression of mRNAs for ENT1, ENT2, CNT1, CNT2, and CNT3 using quantitative real-time PCR. CNT3 mRNA was not detectable, and ENT1 was the most highly expressed. The expression of ENT2, CNT1, and CNT2 expressed as a percentage of ENT1 expression was as follows: 60.9 ± 7.5%, 3.8 ± 0.1%, and 6.1 ± 0.6%. Because the expression of ENT1 was greater than the expression of the other nucleoside transporters, we examined in rat preglomerular vascular smooth muscle cells the role of ENT1 in mediating the effects of guanosine on adenosine levels. S-(4-nitrobenzyl)-6-thioinosine [potent inhibitor of rat ENT1 (94); 10 μmol/l; Fig. 8D] neither mimicked the efficacy of guanosine nor attenuated the efficacy of guanosine with regard to increasing adenosine levels in the presence of added adenosine. Also, simultaneous inhibition of adenosine kinase, adenosine deaminase, and ENT1 not only did not mimic the efficacy of guanosine, this combination actually enhanced the efficacy of guanosine to increase adenosine levels in the presence of added adenosine (Fig. 9A).
Fig. 9.
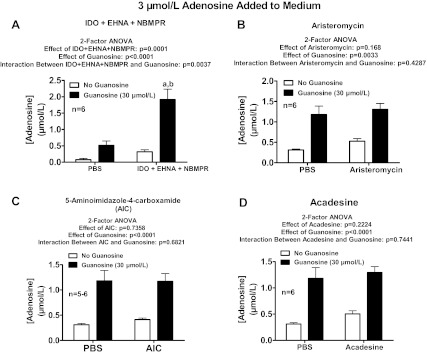
Rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l) and either without or with the following: 5-iodotubericidin (IDO; 0.1 μmol/l; adenosine kinase inhibitor) plus erythro-9-(2-hydroxy-3-nonyl)-adenine (EHNA; 10 μmol/l; adenosine deaminase inhibitor) plus S-(4-nitrobenzyl)-6-thioinosine (NBMPR; 10 μmol/l; equilibrative nucleoside transporter type 1 inhibitor; A); aristeromycin (10 μmol/l; S-adenosylhomocysteine hydrolase inhibitor; B); 5-aminoimidazole-4-carboxamide (100 μmol/l; guanine deaminase inhibitor; C); or 5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (acadesine; 100 μmol/l; D). The medium was assayed for adenosine by mass spectrometry. Values represent means ± SE. aSignificantly different from corresponding no-guanosine group; bsignificantly different from PBS-guanosine group.
The enzyme S-adenosylhomocysteine hydrolase can also metabolize adenosine (12, 13, 49, 73). Therefore we examined in rat preglomerular vascular smooth muscle cells the role of this enzyme in mediating the effects of guanosine on adenosine levels. However, aristeromycin [inhibitor of S-adenosylhomocysteine hydrolase (1, 9, 11, 36, 41); 10 μmol/l; Fig. 9B] neither mimicked the efficacy of guanosine nor attenuated the efficacy of guanosine with regard to increasing adenosine levels in the presence of added adenosine.
Next, we entertained the possibility that adenosine and guanosine could interact at the level of guanine deaminase (48). Therefore we examined in rat preglomerular vascular smooth muscle cells the role of this enzyme in mediating the effects of guanosine on adenosine levels. 5-Aminoimidazole-4-carboxamide [inhibitor of guanine deaminase (35); 100 μmol/l; Fig. 9C] did not mimic nor attenuate the efficacy of guanosine with regard to increasing adenosine levels in the presence of added adenosine.
Acadesine is well known to modulate adenosine levels by multiple mechanisms (20, 65). Therefore we examined in rat preglomerular vascular smooth muscle cells whether acadesine alters the effects of guanosine on adenosine levels. Acadesine (100 μmol/l; Fig. 9D) did not mimic nor attenuate the efficacy of guanosine with regard to increasing adenosine levels in the presence of added adenosine.
In the course of our mechanistic studies, we also examined the effects of pentostatin (2′-deoxycoformycin; 300 μmol/l), which, like erythro-9-(2-hydroxy-3-nonyl)-adenine, is an adenosine deaminase inhibitor (60, 79, 83), on the efficacy of guanosine to increase adenosine levels. Pentostatin not only mimicked the effects of guanosine, but guanosine was unable to further increase adenosine levels in the presence of pentostatin (Fig. 10A). Although this supported a role for adenosine deaminase, this conclusion could not be reconciled with the lack of effects of erythro-9-(2-hydroxy-3-nonyl)-adenine. We hypothesized therefore that pentostatin, because it has a structure similar to guanosine, might have inhibited adenosine transport by one or more concentrative nucleoside transporters (CNTs) and that inhibition by CNTs by guanosine and pentostatin was the commonality shared by these compounds. However, replacement of sodium chloride in the phosphate-buffered saline [CNTs are sodium-dependent transporters (29)] with N-methyl-d-glucamine neither mimicked the efficacy of guanosine nor attenuated the efficacy of guanosine with regard to increasing adenosine levels in the presence of added adenosine (Fig. 10B). Moreover, guanosine increased inosine levels in both normal and low-sodium buffer (Fig. 10C), a finding that ruled out any involvement of inhibition of the transformation of adenosine to inosine (i.e., adenosine deaminase activity) in the ability of guanosine to elevate adenosine levels.
Fig. 10.
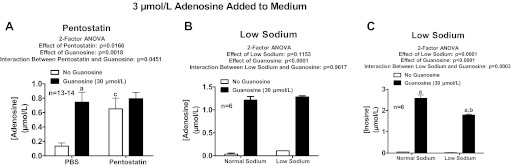
Rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l) and either without or with the following: pentostatin (300 μmol/l; adenosine deaminase inhibitor; A) or low sodium in medium (B and C). The medium was assayed for adenosine or adenosine and inosine by mass spectrometry. Values represent means ± SE. aSignificantly different from corresponding no-guanosine group; bsignificantly different from normal sodium-guanosine group; csignificantly different from PBS-no-guanosine group.
The experiments described above ruled out all known enzymes that metabolize adenosine and seemed to rule out CNTs and ENT1. Although equilibrative nucleoside transporter type 3 (ENT3) exists, ENT3 is an intracellular protein and most likely not involved in the transport of adenosine across cell membranes (3). There are reports of yet another equilibrative nucleoside transporter called ENT4 (also called PMAT); however, this transporter has limited ability to transport nucleosides (19) and functions only at acidic pHs (4) (our experiments were performed at pH 7.4). So the only remaining candidate mechanism by which guanosine might regulate adenosine disposition appeared to be inhibition of ENT2. Although rat ENT2 is known to be insensitive to S-(4-nitrobenzyl)-6-thioinosine (94), rat ENT2 does transport AZT, even at levels in the low μmol/l range (95). Therefore we examined the ability of AZT at an extremely high concentration (2 mmol/l; higher concentrations were toxic and caused cells to detach) to mimic and inhibit the effects of guanosine. AZT did not mimic the efficacy of guanosine nor did AZT attenuate the efficacy of guanosine with regard to increasing adenosine levels in the presence of added adenosine (Fig. 11A).
Fig. 11.
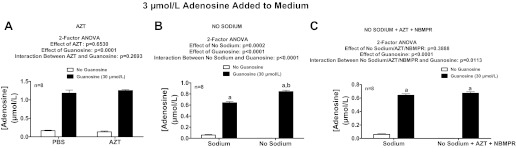
Rat preglomerular vascular smooth muscle cells were incubated with adenosine (3 μmol/l) for 1 h in the absence or presence of guanosine (30 μmol/l) and either without or with the following: zidovudine (AZT; 2 mmol/l; ENT2 inhibitor; A) or no-sodium buffer (B) or AZT plus NBMPR in a no-sodium buffer (C). The medium was assayed for adenosine by mass spectrometry. Values represent means ± SE. aSignificantly different from corresponding no-guanosine group; bsignificantly different from normal sodium-guanosine group.
Having ruled out inhibition of ENTs as the mechanism of the guanosine-adenosine interaction, we reconsidered a possible role of CNTs because our initial experiments (Fig. 10) were performed in low-sodium buffer, i.e., N-methyl-d-glucamine replaced NaCl. However, the buffer still contained a low concentration of sodium in the form of Na2HPO4. Therefore, we examined the effects of guanosine on adenosine levels in buffer in which NaCl was replaced with N-methyl-d-glucamine and Na2HPO4 was replaced with Trizma phosphate dibasic. In this nominally “no-sodium” buffer, guanosine still increased adenosine levels (Fig. 11B). Also, simultaneous inhibition of CNTs (with no-sodium buffer), ENT1 (with NBMPR), and ENT2 (with AZT) did not mimic or inhibit the effects of guanosine on adenosine levels (Fig. 11C), thus ruling out compensation by ENTs when CNTs were blocked and vice versa. These experiments suggest that guanosine inhibits a mechanism of adenosine disposition that is important yet unrecognized.
Comparison of the effects of extracellular guanosine, uridine, thymidine, and cytosine on extracellular adenosine levels.
To determine whether other endogenous nucleosides might also regulate extracellular levels of adenosine, we compared in head-to-head experiments the ability of guanosine, uridine, thymidine, and cytosine to increase extracellular levels of adenosine, inosine, and 3′,5′-cAMP in preglomerular vascular smooth muscle cells treated with exogenous adenosine. As shown in Fig. 12A, guanosine was more potent than either uridine or cytosine with regard to increasing extracellular adenosine levels. Although guanosine and thymidine were equipotent and efficacious in their ability to increase extracellular adenosine, guanosine was unique compared with thymidine, uridine, or cytosine in causing large increases in extracellular inosine (Fig. 12B) and increases in the signaling molecule 3′,5′-cAMP (Fig. 12C). To confirm the effect of guanosine on inosine levels, in a separate experiment we incubated cells with inosine (3 μmol/l) in the absence and presence of guanosine, uridine, thymidine, and cytidine (3, 10, and 100 μmol/l) and measured inosine, hypoxanthine, xanthine, and uric acid. Although all nucleosides increased inosine, guanosine (Fig. 13A) was 10-fold more potent than uridine (Fig. 13B) and thymidine (Fig. 13C) and 30-fold more potent than cytidine (Fig. 13D). None of the nucleosides affected hypoxanthine (Fig. 13, E–H). As expected, guanosine increased xanthine levels because guanosine is metabolized to guanine and guanine deaminase converts guanine to xanthine (22) (Fig. 14A); however, uridine, thymidine, and cytidine did not affect xanthine levels (Fig. 14, B–D). Despite causing a large increase in xanthine (precursor of uric acid), guanosine actually suppressed uric acid levels below the detection limit of our assay (Fig. 14E). In contrast, uridine, thymidine, and cytidine increased uric acid levels (Fig. 14, F–H). Taken together the data suggest that guanosine is unique compared with the other nucleosides in that it potently increases extracellular levels of both adenosine and inosine and yet suppresses levels of extracellular uric acid.
Fig. 12.

Rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations of guanosine, uridine, thymidine, or cytosine plus adenosine (3 μmol/l). The medium was assayed for adenosine (A), inosine (B), and 3′,5′-cAMP (C) by mass spectrometry. Values represent means ± SE. aSignificantly different from guanosine at the corresponding concentration.
Fig. 13.

Rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations [0 (basal), 3, 10, 100 μmol/l] of guanosine (A and E), uridine (B and F), thymidine (C and G), or cytidine (D and H) and in the presence of inosine (3 μmol/l). The medium was assayed for inosine (A–D) and hypoxanthine (E–H) by mass spectrometry. Values represent means ± SE. aSignificantly different from basal in corresponding panel; bsignificantly different from corresponding effects of uridine, thymidine, and cytidine.
Fig. 14.

Rat preglomerular vascular smooth muscle cells were incubated for 1 h with various concentrations [0 (basal), 3, 10, 100 μmol/l] of guanosine (A and E), uridine (B and F), thymidine (C and G), or cytidine (D and H) and in the presence of inosine (3 μmol/l). The medium was assayed for xanthine (A–D) and uric acid (E–H) by mass spectrometry. Values represent means ± SE. aSignificantly different from basal in corresponding panel; bsignificantly different from corresponding effects of uridine, thymidine, and cytidine.
Comparison of the effects of extracellular guanosine on extracellular levels of adenosine, uridine, thymidine, and cytosine.
Because guanosine elevates extracellular inosine levels, we considered that guanosine may also increase extracellular levels of other nucleosides. To test this, rat preglomerular vascular smooth muscle cells were incubated for 1 h without or with guanosine (30 μmol/l) and without or with adenosine, uridine, thymidine, or cytidine (each at 3 μmol/l). In the absence of added nucleosides, guanosine had little, if any, effect on levels of adenosine, uridine, thymidine, or cytidine; however, in the presence of nucleosides, guanosine markedly increased extracellular levels of adenosine, uridine, thymidine, and cytidine (Fig. 15, A–D). These findings indicate that guanosine likely inhibits the disposition of not only adenosine and inosine, but also uridine, thymidine, and cytidine.
Fig. 15.
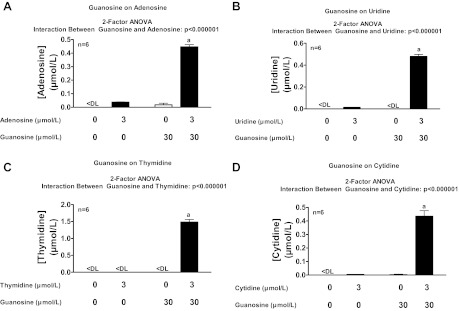
Rat preglomerular vascular smooth muscle cells were incubated for 1 h without or with guanosine (30 μmol/l) and without or with adenosine (A), uridine (B), thymidine (C), or cytidine (D) (each at 3 μmol/l). Medium was assayed for adenosine, uridine, thymidine, and cytidine by mass spectrometry. Values represent means ± SE. <DL, less than detection limit. aSignificantly different from all other groups in panel.
DISCUSSION
Adenosine is a ubiquitous, potent, and critically important endogenous autocrine/paracrine factor that regulates the kidneys (90), heart (66), liver (70), brain (77), lungs (63, 92), bladder (97, 98), skeletal muscle (39, 61), adipose tissue (25), autonomic nervous system (69, 93) and immune system (5, 51), protects against ischemia-reperfusion injury (16) and modulates fibrosis (7) and accelerates wound healing (21, 91). These broad-ranging actions of adenosine are mediated by A1, A2A, A2B, and A3 adenosine receptors (24, 45, 86), and all four adenosine receptor subtypes are members of the 7-transmembrane spanning superfamily of G protein-coupled receptors.
Although adenosine is produced both within and outside cells, there is increasing evidence that biosynthesis of adenosine in the extracellular compartment via ecto-enzymes is the most important source of adenosine that engages cell surface receptors and thus regulates cell function (16–18, 30–33). In this regard, dephosphorylation of extracellular adenosine nucleotides (both linear and cyclic) to adenosine monophosphates [AMPs; in particular 5′-AMP (16–18, 30–33), but also perhaps 2′-AMP and 3′-AMP (43, 44)] followed by dephosphorylation of AMPs to adenosine is critical to the formation rate of extracellular adenosine. However, the extracellular levels of adenosine at steady state would depend not only on the production rate of adenosine in the extracellular compartment, but also on the disposition rate of adenosine from the extracellular compartment (30). The balance of these two processes would then determine the steady-state level of adenosine in the interstitial space and therefore the degree of activation of adenosine cell-surface receptors.
Regarding the disposition of extracellular adenosine, adenosine has an ultrashort half-life (seconds to minutes) in the extracellular compartment (16), indicating very efficient disposition mechanisms. Consequently, endogenous inhibitors of the disposition of adenosine could be a critically important mechanism controlling extracellular levels of adenosine (16).
The results of the present study indicate that guanosine indeed is an endogenous inhibitor of adenosine disposition from the extracellular compartment. In the presence of added adenosine, guanosine profoundly increases extracellular adenosine levels and in proportion to the concentration of added adenosine. As reported previously by others (14, 76), we too find that in the absence of added adenosine, guanosine has little effect on extracellular adenosine levels, indicating that extracellular guanosine does not drive the extracellular formation of adenosine or increase release of adenosine from cells. The only explanation for our results is that guanosine regulates the disposition of extracellular adenosine. This regulation is not mediated by guanine because guanine does not mimic the effects of guanosine, and blocking the enzyme that converts guanosine to guanine does not alter the ability of guanosine to regulate adenosine disposition.
Linear guanine nucleotides are released from tissues by injurious stimuli and are metabolized by ecto-enzymes to guanosine (84). Moreover, cGMP is transported into the extracellular compartment by members of the multidrug resistance protein (MRP) family, including MRP4, MRP5, and MRP8 (74), and studies support the concept that extracellular cyclic nucleotides can be metabolized by ecto-phosphodiesterases to linear nucleotides (42). Therefore, if guanosine regulates the disposition of extracellular adenosine, then so should linear and cyclic guanine nucleotides. Indeed, the present study shows that extracellular GMP and cGMP also inhibit adenosine disposition, although not as potently as guanosine, which is consistent with the concept that guanosine is the final mediator of this effect. Also consistent with this concept is that blockade of the enzyme that converts GMP to guanosine abolishes the ability of GMP to inhibit adenosine disposition.
In theory, extracellular guanosine could alter the disposition of adenosine from the extracellular compartment by blocking one or more processes involved in the removal of extracellular adenosine. One possibility in this regard is that guanosine could directly inhibit enzymes that metabolize extracellular adenosine. For example ecto-adenosine deaminase metabolizes extracellular adenosine to inosine (10, 23, 37, 54), and it is conceivable that guanosine could block this enzyme. Another possibility is that guanosine could interfere with the transport of adenosine from the extracellular compartment to the intracellular compartment. Although adenosine is not per se permeable to cell membranes, adenosine is escorted through the cell membrane by CNTs (6, 29, 68) and ENTs (3, 50, 96), and it is conceivable that guanosine could interfere with transport of adenosine by CNTs or ENTs. A third possibility is that guanosine could block intracellular metabolism of adenosine. For example, adenosine is metabolized within cells to inosine by adenosine deaminase (16), 5′-AMP by adenosine kinase (16) and S-adenosylhomocysteine (SAH) by SAH hydrolase (13, 49, 73). Even guanine deaminase may have activity to metabolize adenosine to inosine (35, 48). Guanosine in theory could block any of these enzymes.
The results of the present study rule out the involvement of adenosine kinase, SAH hydrolase, guanine deaminase, ENT1, ENT2, and CNTs in the interaction between extracellular guanosine and extracellular adenosine. Although pentostatin, which is considered a potent adenosine deaminase inhibitor (60), mimics and blocks the effects of extracellular guanosine on extracellular adenosine levels, it is unlikely that this is related to adenosine deaminase because erythro-9-(2-hydroxy-3-nonyl)-adenine, also an inhibitor of adenosine deaminase, does not alter the interaction between extracellular guanosine and extracellular adenosine. Moreover, extracellular guanosine increases both extracellular adenosine and inosine levels, which rules out any involvement of adenosine deaminase in the interaction. The fact that extracellular guanosine also blocks the disposition of extracellular uridine, thymidine, and cytidine argues against a mechanism of action involving inhibition of metabolism because guanosine would have to inhibit simultaneously the metabolism of inosine, uridine, thymidine, and cytidine, which seems unlikely.
Because our exhaustive experiments appear to rule out all known likely possibilities by which extracellular guanosine could reduce the disposition of extracellular adenosine, we hypothesize that extracellular guanosine decreases the disposition of extracellular adenosine by inhibiting a transporter or transporters not previously recognized as importantly regulating extracellular adenosine, inosine, uridine, thymidine, and cytidine. Inasmuch as ∼2,000 genes in the human genome code for transporters or transporter-related proteins this hypothesis is not unreasonable.
An important issue is whether endogenous extracellular guanosine reaches significant concentrations to inhibit the disposition of extracellular adenosine. Cardiac microdialysis studies in pigs show that myocardial ischemia increases interstitial levels of guanosine to ∼10 μmol/l (89). This is clearly within the range necessary for guanosine to modulate adenosine disposition from the extracellular compartment. Our current findings further support the conclusion that endogenous extracellular guanosine regulates extracellular adenosine disposition. In this regard, the metabolic poison, iodoacetate, increases both extracellular guanosine and adenosine, and the ability of iodoacetate to increase extracellular adenosine is augmented when guanosine metabolism is inhibited and attenuated when guanosine metabolism is increased.
Another important issue is whether endogenous extracellular guanosine inhibits the disposition of extracellular adenosine in a wide range of cells, or only in selected cell types. The studies presented here indicate that extracellular guanosine inhibits extracellular adenosine disposition in eight different cell types, including both rat and human cells. Thus, although the concentration of extracellular guanosine required to modulate extracellular adenosine disposition varies according to the cell type, the mechanism seems to exist generally. An exception is mouse cortical neurons, which we observed removed extracellular adenosine only slowly and by a process not inhibited by guanosine (data not shown).
It is conceivable that extracellular uridine, thymidine, and cytosine, like extracellular guanosine, also regulate the disposition of extracellular adenosine. Our head-to-head comparison of the four nucleosides indicates that guanosine is considerably more potent than cytidine, somewhat more potent than uridine, and equipotent with thymidine with regard to attenuating the disposition of extracellular adenosine. From a physiological perspective, however, whether uridine and thymidine are as important as guanosine with regard to regulating extracellular levels of adenosine depends on whether cell injury increases extracellular levels of uridine or thymidine as much as guanosine. Our experiments show that cell injury with iodoacetate increases extracellular guanosine levels approximately twofold more than extracellular adenosine, indicating robust increases in extracellular guanosine with injury. Moreover, experiments with iodoacetate show that removing extracellular guanosine from the medium of injured cells nearly abolishes injury-induced increases in extracellular adenosine. This would imply that the other nucleosides play little, if any, role, either because they are not released sufficiently by cell injury or have insufficient efficacy or some combination of these factors. Indeed, we observe that in rat preglomerular vascular smooth muscle cells in culture, cell injury releases predominantly guanosine. However, it is likely that under different conditions (cell types, modes of injury) other nucleosides are released and may contribute to the modulation of extracellular adenosine levels. Another striking difference between guanosine versus uridine, cytidine, and thymidine is that guanosine causes a remarkable increase in extracellular inosine, whereas uridine, cytidine, and thymidine do not.
Because guanosine is metabolized to guanine and guanine is converted to xanthine by guanine deaminase (22), it is not surprising that our experiments show that extracellular guanosine increases extracellular xanthine. What is surprising is that despite the large increase in xanthine levels, extracellular guanosine suppresses levels of uric acid. In contrast, uridine, thymidine, and cytidine increase extracellular uric acid levels. This suggests that guanosine inhibits xanthine oxidase, but additional experiments are required to corroborate this hypothesis.
In conclusion, taken together, our results suggest a unique physiological role for extracellular guanosine. Cell injury increases extracellular guanosine, and extracellular guanosine elevates adenosine and inosine, while decreasing uric acid. Importantly, adenosine is antiinflammatory (27, 55), as is inosine (59), which also binds to and stimulates adenosine receptors (34, 38, 67, 81). Moreover, uric acid is proinflammatory (46, 75). By increasing adenosine and inosine and reducing uric acid, guanosine may protect tissues/organs against injury. This hypothesis is consistent with the fact that exogenous guanosine protects kidneys from ischemia-reperfusion injury (47). Our results suggest that the protective effects of guanosine should be examined in a wide range of animal models of inflammation and tissue/organ injury and ultimately in humans.
GRANTS
This work was supported by National Institutes of Health Grants HL-109002, DK-091190, HL-069846, DK-068575, and DK-079307.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.K.J., T.C.J., J.D.V., and D.G.G. conception and design of the research; E.K.J., D.C., T.C.J., J.D.V., and D.G.G. analyzed the data; E.K.J., D.C., T.C.J., and J.D.V. interpreted the results of the experiments; E.K.J. prepared the figures; E.K.J. drafted the manuscript; E.K.J., D.C., T.C.J., J.D.V., and D.G.G. edited and revised the manuscript; E.K.J., D.C., T.C.J., J.D.V., and D.G.G. approved the final version of the manuscript; D.C., T.C.J., J.D.V., and D.G.G. performed the experiments.
Footnotes
This article is the topic of an Editorial Focus by George R. Dubyak (15a).
REFERENCES
- 1. Ault-Riche DB, Lee Y, Yuan CS, Hasobe M, Wolfe MS, Borcherding DR, Borchardt RT. Effects of 4′-modified analogs of aristeromycin on the metabolism of S-adenosyl-l-homocysteine in murine L929 cells. Mol Pharmacol 43: 989–997, 1993 [PubMed] [Google Scholar]
- 2. Baker DC, Hanvey JC, Hawkins LD, Murphy J. Identification of the bioactive enantiomer of erythro-3-(adenin-9-yl)-2-nonanol (EHNA), a semi-tight binding inhibitor of adenosine deaminase. Biochem Pharmacol 30: 1159–1161, 1981 [DOI] [PubMed] [Google Scholar]
- 3. Baldwin SA, Beal PR, Yao SYM, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflügers Arch 447: 735–743, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Barnes K, Dobrzynski H, Foppolo S, Beal PR, Ismat F, Scullion ER, Sun L, Tellez J, Ritzel MWL, Claycomb WC, Cass CE, Young JD, Billeter-Clark R, Boyett MR, Baldwin SA. Distribution and functional characterization of equilibrative nucleoside transporter-4, a novel cardiac adenosine transporter activated at acidic pH. Circ Res 99: 510–519, 2006 [DOI] [PubMed] [Google Scholar]
- 5. Bours MJL, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 112: 358–404, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Casado FJ, Lostao MP, Aymerich I, Larrayoz IM, Duflot S, Rodriguez-Mulero S, Pastor-Anglada M. Nucleoside transporters in absorptive epithelia. J Physiol Biochem 58: 207–216, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Chan ESL, Cronstein BN. Adenosine in fibrosis. Mod Rheumatol 20: 114–122, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng D, Ren J, Gillespie DG, Mi Z, Jackson EK. Regulation of 3′,5′-cAMP in preglomerular smooth muscle and endothelial cells from genetically-hypertensive rats. Hypertension 56: 1096–1101, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiang PK, Guranowski A, Segall JE. Irreversible inhibition of S-adenosylhomocysteine hydrolase by nucleoside analogs. Arch Biochem Biophys 207: 175–184, 1981 [DOI] [PubMed] [Google Scholar]
- 10. Ciruela F, Saura C, Canela EI, Mallol J, Lluis C, Franco R. Adenosine deaminase affects ligand-induced signalling by interacting with cell surface adenosine receptors. FEBS Lett 380: 219–223, 1996 [DOI] [PubMed] [Google Scholar]
- 11. De Clercq E. S-adenosylhomocysteine hydrolase inhibitors as broad-spectrum antiviral agents. Biochem Pharmacol 36: 2567–2575, 1987 [DOI] [PubMed] [Google Scholar]
- 12. Deussen A, Lloyd HG, Schrader J. Contribution of S-adenosylhomocysteine to cardiac adenosine formation. J Mol Cell Cardiol 21: 773–782, 1989 [DOI] [PubMed] [Google Scholar]
- 13. Deussen A, Pexa A, Loncar R, Stehr SN. Effects of homocysteine on vascular and tissue adenosine: a stake in homocysteine pathogenicity? Clin Chem Lab Med 43: 1007–1010, 2005 [DOI] [PubMed] [Google Scholar]
- 14. Di Iorio P, Kleywegt S, Ciccarelli R, Traversa U, Andrew CM, Crocker CE, Werstiuk ES, Rathbone MP. Mechanisms of apoptosis induced by purine nucleosides in astrocytes. Glia 38: 179–190, 2002 [DOI] [PubMed] [Google Scholar]
- 15. Dubey RK, Gillespie DG, Mi Z, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation 96: 2656–2666, 1997 [DOI] [PubMed] [Google Scholar]
- 15a. Dubyak GR. Dueling nucleosides: regulation of extracellular adenosine by guanosine. Focus on “Extracellular guanosine regulates extracellular adenosine levels.” Am J Physiol Cell Physiol (January 16, 2013). doi:10.1152/ajpcell.00012.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology 111: 904–915, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schonfeld C, Loffler M, Reyes G, Duszenko M, Karhausen J, Robinson A, Westerman KA, Coe IR, Colgan SP. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med 202: 1493–1505, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eltzschig HK, Weissmuller T, Mager A, Eckle T. Nucleotide metabolism and cell-cell interactions. Methods Mol Biol 341: 73–87, 2006 [DOI] [PubMed] [Google Scholar]
- 19. Engel K, Zhou M, Wang J. Identification and characterization of a novel monoamine transporter in the human brain. J Biol Chem 279: 50042–50049, 2004 [DOI] [PubMed] [Google Scholar]
- 20. Engler RL. Harnessing nature's own cardiac defense mechanism with acadesine, an adenosine regulating agent: importance of the endothelium. J Card Surg 9: 482–492, 1994 [DOI] [PubMed] [Google Scholar]
- 21. Feoktistov I, Biaggioni I, Cronstein BN. Adenosine receptors in wound healing, fibrosis and angiogenesis. Handb Exp Pharm: 383–397, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fernandez JR, Sweet ES, Welsh WJ, Firestein BL. Identification of small molecule compounds with higher binding affinity to guanine deaminase (cypin) than guanine. Bioorg Med Chem 18: 6748–6755, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franco R, Valenzuela A, Lluis C, Blanco J. Enzymatic and extraenzymatic role of ecto-adenosine deaminase in lymphocytes. Immunol Rev 161: 27–42, 1998 [DOI] [PubMed] [Google Scholar]
- 24. Fredholm BB. Adenosine receptors as drug targets. Exp Cell Res 316: 1284–1288, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fredholm BB, Johansson S, Wang YQ. Adenosine and the regulation of metabolism and body temperature. Adv Pharmacol 61: 77–94, 2011 [DOI] [PubMed] [Google Scholar]
- 26. Frye CA, Patrick CW., Jr Isolation and culture of rat microvascular endothelial cells. In Vitro Cell Dev Biol Anim 38: 208–212, 2002 [DOI] [PubMed] [Google Scholar]
- 27. Gessi S, Merighi S, Fazzi D, Stefanelli A, Varani K, Borea PA. Adenosine receptor targeting in health and disease. Expert Opin Investig Drugs 20: 1591–1609, 2011 [DOI] [PubMed] [Google Scholar]
- 28. Giblett ER. ADA and PNP deficiencies: how it all began. Ann NY Acad Sci 451: 1–8, 1985 [DOI] [PubMed] [Google Scholar]
- 29. Gray JH, Owen RP, Giacomini KM. The concentrative nucleoside transporter family, SLC28. Pflügers Arch 447: 728–734, 2004 [DOI] [PubMed] [Google Scholar]
- 30. Grenz A, Bauerle JD, Dalton JH, Ridyard D, Badulak A, Tak E, McNamee EN, Clambey E, Moldovan R, Reyes G, Klawitter J, Ambler K, Magee K, Christians U, Brodsky KS, Ravid K, Choi DS, Wen J, Lukashev D, Blackburn MR, Osswald H, Coe IR, Nürnberg B, Haase VH, Xia Y, Sitkovsky M, Eltzschig HK. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J Clin Invest 122: 693–710, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Grenz A, Homann D, Eltzschig HK. Extracellular adenosine: a safety signal that dampens hypoxia-induced inflammation during ischemia. Antioxid Redox Signal 15: 2221–2234, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grenz A, Zhang H, Eckle T, Mittelbronn M, Wehrmann M, Kohle C, Kloor D, Thompson LF, Osswald H, Eltzschig HK. Protective role of ecto-5′-nucleotidase (CD73) in renal ischemia. J Am Soc Nephrol 18: 833–845, 2007 [DOI] [PubMed] [Google Scholar]
- 33. Grenz A, Zhang H, Hermes M, Eckle T, Klingel K, Huang DY, Muller CE, Robson SC, Osswald H, Eltzschig HK. Contribution of E-NTPDase1 (CD39) to renal protection from ischemia-reperfusion injury. FASEB J 21: 2863–2873, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Guinzberg R, Cortes D, Diaz-Cruz A, Riveros-Rosas H, Villalobos-Molina R, Pina E. Inosine released after hypoxia activates hepatic glucose liberation through A3 adenosine receptors. Am J Physiol Endocrinol Metab 290: E940–E951, 2006 [DOI] [PubMed] [Google Scholar]
- 35. Gupta NK, Glantz MD. Isolation and characterization of human liver guanine deaminase. Arch Biochem Biophys 236: 266–276, 1985 [DOI] [PubMed] [Google Scholar]
- 36. Guranowski A, Montgomery JA, Cantoni GL, Chiang PK. Adenosine analogues as substrates and inhibitors of S-adenosylhomocysteine hydrolase. Biochemistry 20: 110–115, 1981 [DOI] [PubMed] [Google Scholar]
- 37. Hashikawa T, Hooker SW, Maj JG, Knott-Craig CJ, Takedachi M, Murakami S, Thompson LF. Regulation of adenosine receptor engagement by ecto-adenosine deaminase. FASEB J 18: 131–133, 2004 [DOI] [PubMed] [Google Scholar]
- 38. Hasko G, Kuhel DG, Nemeth ZH, Mabley JG, Stachlewitz RF, Virag L, Lohinai Z, Southan GJ, Salzman AL, Szabo C. Inosine inhibits inflammatory cytokine production by a posttranscriptional mechanism and protects against endotoxin-induced shock. J Immunol 164: 1013–1019, 2000 [DOI] [PubMed] [Google Scholar]
- 39. Hespel P, Richter EA. Role of adenosine in regulation of carbohydrate metabolism in contracting muscle. Adv Exp Med Biol 441: 97–106, 1998 [DOI] [PubMed] [Google Scholar]
- 40. Inoue T, Mi Z, Gillespie DG, Jackson EK. Cyclooxygenase inhibition reveals synergistic action of vasoconstrictors on mesangial cell growth. Eur J Pharmacol 361: 285–291, 1998 [DOI] [PubMed] [Google Scholar]
- 41. Ishikura T, Nakamura H, Sugawara T, Itoh T, Nomura A, Mizuno Y. Inhibition of S-adenosylhomocysteine hydrolase by purine nucleoside analogues. Nucleic Acids Symp Ser: 119–122, 1983 [PubMed] [Google Scholar]
- 42. Jackson EK, Dubey RK. Role of the extracellular cAMP-adenosine pathway in renal physiology. Am J Physiol Renal Physiol 281: F597–F612, 2001 [DOI] [PubMed] [Google Scholar]
- 43. Jackson EK, Ren J, Cheng D, Mi Z. Extracellular cAMP-adenosine pathways in the mouse kidney. Am J Physiol Renal Physiol 301: F565–F573, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jackson EK, Ren J, Mi Z. Extracellular 2′,3′-cAMP is a source of adenosine. J Biol Chem 284: 33097–33106, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jacobson KA. Introduction to adenosine receptors as therapeutic targets. Handb Exp Pharmacol: 1–24, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang H, Yang XF. Uric acid, hyperuricemia and vascular diseases. Front Biosci 17: 656–669, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kelly KJ, Plotkin Z, Dagher PC. Guanosine supplementation reduces apoptosis and protects renal function in the setting of ischemic injury. J Clin Invest 108: 1291–1298, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim J, Park SI, Ahn C, Kim H, Yim J. Guanine deaminase functions as dihydropterin deaminase in the biosynthesis of aurodrosopterin, a minor red eye pigment of Drosophila. J Biol Chem 284: 23426–23435, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kloor D, Osswald H. S-Adenosylhomocysteine hydrolase as a target for intracellular adenosine action. Trends Pharmacol Sci 25: 294–297, 2004 [DOI] [PubMed] [Google Scholar]
- 50. Kong W, Engel K, Wang J. Mammalian nucleoside transporters. Curr Drug Metab 5: 63–84, 2004 [DOI] [PubMed] [Google Scholar]
- 51. Kumar V, Sharma A. Adenosine: an endogenous modulator of innate immune system with therapeutic potential. Eur J Pharmacol 616: 7–15, 2009 [DOI] [PubMed] [Google Scholar]
- 52. Lambe CU, Nelson DJ. Pharmacokinetics of inhibition of adenosine deaminase by erythro-9-(2-hydroxy-3-nonyl)adenine in CBA mice. Biochem Pharmacol 31: 535–539, 1982 [DOI] [PubMed] [Google Scholar]
- 53. Lazarowski ER, Sesma JI, Seminario-Vidal L, Kreda SM. Molecular mechanisms of purine and pyrimidine nucleotide release. Adv Pharmacol 61: 221–261, 2011 [DOI] [PubMed] [Google Scholar]
- 54. Lecka J, Bloch-Boguslawska E, Molski S, Komoszynski M. Extracellular purine metabolism in blood vessels (Part II): Activity of ecto-enzymes in blood vessels of patients with abdominal aortic aneurysm. Clin Appl Thromb Hemost 16: 650–657, 2010 [DOI] [PubMed] [Google Scholar]
- 55. Linden J. Regulation of leukocyte function by adenosine receptors. Adv Pharmacol 61: 95–114, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lum CT, Schmidtke JR, Sutherland DE, Najarian JS. Inhibition of human T-cell rosette formation by the adenosine deaminase inhibitor erythro-9-(2-hydroxy-3-nonyl) adenine hydrochloride (EHNA). Clin Immunol Immunopathol 10: 258–261, 1978 [DOI] [PubMed] [Google Scholar]
- 57. Lum CT, Sutherland DE, Najarian JS. Inhibition of PHA and NaIO4 mitogenesis by the adenosine deaminase inhibitors erythro-9-(2-hydroxy-3-nonyl) adenine (EHNA) and 2-deoxycoformycin (2-dCF). Clin Immunol Immunopathol 12: 453–459, 1979 [DOI] [PubMed] [Google Scholar]
- 58. Lupidi G, Cristalli G, Marmocchi F, Riva F, Grifantini M. Inhibition of adenosine deaminase from several sources by deaza derivatives of adenosine and EHNA. J Enzym Inhib 1: 67–75, 1985 [DOI] [PubMed] [Google Scholar]
- 59. Mabley JG, Pacher P, Murthy KGK, Williams W, Southan GJ, Salzman AL, Szabo C. The novel inosine analogue INO-2002 exerts an anti-inflammatory effect in a murine model of acute lung injury. Shock 32: 258–262, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Major PP, Agarwal RP, Kufe DW. Clinical pharmacology of deoxycoformycin. Blood 58: 91–96, 1981 [PubMed] [Google Scholar]
- 61. Marshall JM. Roles of adenosine in skeletal muscle during systemic hypoxia. Clin Exp Pharmacol Physiol 29: 843–849, 2002 [DOI] [PubMed] [Google Scholar]
- 62. McKee RW, Parks ME, Dickey A. Influence of iodoacetate on glycolytic intermediates and on respiration in Ehrlich-Lettre ascites carcinoma cells. Arch Biochem Biophys 124: 450–455, 1968 [DOI] [PubMed] [Google Scholar]
- 63. Mohsenin A, Blackburn MR. Adenosine signaling in asthma and chronic obstructive pulmonary disease. Curr Opin Pulm Med 12: 54–59, 2006 [DOI] [PubMed] [Google Scholar]
- 64. Mokkapatti R, Vyas SJ, Romero GG, Mi Z, Inoue T, Dubey RK, Gillespie DG, Stout AK, Jackson EK. Modulation by angiotensin II of isoproterenol-induced cAMP production in preglomerular microvascular smooth muscle cells from normotensive and genetically hypertensive rats. J Pharmacol Exp Ther 287: 223–231, 1998 [PubMed] [Google Scholar]
- 65. Mullane K. Acadesine: the prototype adenosine regulating agent for reducing myocardial ischaemic injury. Cardiovasc Res 27: 43–47, 1993 [DOI] [PubMed] [Google Scholar]
- 66. Mustafa SJ, Morrison RR, Teng B, Pelleg A. Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharm: 161–188, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Naydenova Z, Rose JB, Coe IR. Inosine and equilibrative nucleoside transporter 2 contribute to hypoxic preconditioning in the murine cardiomyocyte HL-1 cell line. Am J Physiol Heart Circ Physiol 294: H2687–H2692, 2008 [DOI] [PubMed] [Google Scholar]
- 68. Pastor-Anglada M, Errasti-Murugarren E, Aymerich I, Casado FJ. Concentrative nucleoside transporters (CNTs) in epithelia: from absorption to cell signaling. J Physiol Biochem 63: 97–110, 2007 [DOI] [PubMed] [Google Scholar]
- 69. Pelleg A, Katchanov G, Xu J. Autonomic neural control of cardiac function: modulation by adenosine and adenosine 5′-triphosphate. Am J Cardiol 79: 11–14, 1997 [DOI] [PubMed] [Google Scholar]
- 70. Peng Z, Fernandez P, Wilder T, Yee H, Chiriboga L, Chan ESL, Cronstein BN. Ecto-5′-nucleotidase (CD73)-mediated extracellular adenosine production plays a critical role in hepatic fibrosis. Nucleosides Nucleotides Nucleic Acids 27: 821–824, 2008 [DOI] [PubMed] [Google Scholar]
- 71. Rathbone M, Pilutti L, Caciagli F, Jiang S. Neurotrophic effects of extracellular guanosine. Nucleosides Nucleotides Nucleic Acids 27: 666–672, 2008 [DOI] [PubMed] [Google Scholar]
- 72. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev 161: 95–109, 1998 [DOI] [PubMed] [Google Scholar]
- 73. Riksen NP, Rongen GA, Blom HJ, Russel FGM, Boers GHJ, Smits P. Potential role for adenosine in the pathogenesis of the vascular complications of hyperhomocysteinemia. Cardiovasc Res 59: 271–276, 2003 [DOI] [PubMed] [Google Scholar]
- 74. Sager G. Cyclic GMP transporters. Neurochem Int 45: 865–873, 2004 [DOI] [PubMed] [Google Scholar]
- 75. Sanchez-Lozada LG, Nakagawa T, Kang DH, Feig DI, Franco M, Johnson RJ, Herrera-Acosta J. Hormonal and cytokine effects of uric acid. Curr Opin Nephrol Hypertens 15: 30–33, 2006 [DOI] [PubMed] [Google Scholar]
- 76. Schmidt AP, Bohmer AE, Schallenberger C, Antunes C, Tavares RG, Wofchuk ST, Elisabetsky E, Souza DO. Mechanisms involved in the antinociception induced by systemic administration of guanosine in mice. Br J Pharmacol 159: 1247–1263, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sebastiao AM, Ribeiro JA. Adenosine receptors and the central nervous system. Handb Exp Pharm: 471–534, 2009 [DOI] [PubMed] [Google Scholar]
- 78. Seegmiller JE. Overview of possible relation of defects in purine metabolism to immune deficiency. Ann NY Acad Sci 451: 9–19, 1985 [DOI] [PubMed] [Google Scholar]
- 79. Siaw MF, Mitchell BS, Koller CA, Coleman MS, Hutton JJ. ATP depletion as a consequence of adenosine deaminase inhibition in man. Proc Natl Acad Sci USA 77: 6157–6161, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sinclair CJ, Powell AE, Xiong W, LaRiviere CG, Baldwin SA, Cass CE, Young JD, Parkinson FE. Nucleoside transporter subtype expression: effects on potency of adenosine kinase inhibitors. Br J Pharmacol 134: 1037–1044, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Souza LF, Horn AP, Gelain DP, Jardim FR, Lenz G, Bernard EA. Extracellular inosine modulates ERK 1/2 and p38 phosphorylation in cultured Sertoli cells: possible participation in TNF-alpha modulation of ERK 1/2. Life Sci 77: 3117–3126, 2005 [DOI] [PubMed] [Google Scholar]
- 82. Stoeckler JD, Cambor C, Kuhns V, Chu SH, Parks RE., Jr Inhibitors of purine nucleoside phosphorylase, C(8) and C(5′) substitutions. Biochem Pharmacol 31: 163–171, 1982 [DOI] [PubMed] [Google Scholar]
- 83. Tedde A, Balis ME, Ikehara S, Pahwa R, Good RA, Trotta PP. Animal model for immune dysfunction associated with adenosine deaminase deficiency. Proc Natl Acad Sci USA 77: 4899–4903, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Thauerer B, zur Nedden S, Baier-Bitterlich G. Purine nucleosides: endogenous neuroprotectants in hypoxic brain. J Neurochem 121: 329–342, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Traversa U, Bombi G, Camaioni E, Macchiarulo A, Costantino G, Palmieri C, Caciagli F, Pellicciari R. Rat brain guanosine binding site. Biological studies and pseudo-receptor construction. Bioorg Med Chem 11: 5417–5425, 2003 [DOI] [PubMed] [Google Scholar]
- 86. Trincavelli ML, Daniele S, Martini C. Adenosine receptors: what we know and what we are learning. Curr Top Med Chem 10: 860–877, 2010 [DOI] [PubMed] [Google Scholar]
- 87. Ugarkar BG, Castellino AJ, DaRe JS, Ramirez-Weinhouse M, Kopcho JJ, Rosengren S, Erion MD. Adenosine kinase inhibitors. 3. Synthesis, SAR, and antiinflammatory activity of a series of l-lyxofuranosyl nucleosides. J Med Chem 46: 4750–4760, 2003 [DOI] [PubMed] [Google Scholar]
- 88. Ugarkar BG, DaRe JM, Kopcho JJ, Browne CE, 3rd, Schanzer JM, Wiesner JB, Erion MD. Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J Med Chem 43: 2883–2893, 2000 [DOI] [PubMed] [Google Scholar]
- 89. Valen G, Owall A, Takeshima S, Goiny M, Ungerstedt U, Vaage J. Metabolic changes induced by ischemia and cardioplegia: a study employing cardiac microdialysis in pigs. Eur J Cardiothorac Surg 25: 69–75, 2004 [DOI] [PubMed] [Google Scholar]
- 90. Vallon V, Muhlbauer B, Osswald H. Adenosine and kidney function. Physiol Rev 86: 901–940, 2006 [DOI] [PubMed] [Google Scholar]
- 91. Valls MD, Cronstein BN, Montesinos MC. Adenosine receptor agonists for promotion of dermal wound healing. Biochem Pharmacol 77: 1117–1124, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Vass G, Horvath I. Adenosine and adenosine receptors in the pathomechanism and treatment of respiratory diseases. Curr Med Chem 15: 917–922, 2008 [DOI] [PubMed] [Google Scholar]
- 93. Westfall DP, Shinozuka K, Forsyth KM, Bjur RA. Presynaptic purine receptors. Ann NY Acad Sci 604: 130–135, 1990 [DOI] [PubMed] [Google Scholar]
- 94. Yao SY, Ng AM, Muzyka WR, Griffiths M, Cass CE, Baldwin SA, Young JD. Molecular cloning and functional characterization of nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei) equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat tissues. J Biol Chem 272: 28423–28430, 1997 [DOI] [PubMed] [Google Scholar]
- 95. Yao SY, Ng AM, Sundaram M, Cass CE, Baldwin SA, Young JD. Transport of antiviral 3′-deoxy-nucleoside drugs by recombinant human and rat equilibrative, nitrobenzylthioinosine (NBMPR)-insensitive (ENT2) nucleoside transporter proteins produced in Xenopus oocytes. Mol Membr Biol 18: 161–167, 2001 [DOI] [PubMed] [Google Scholar]
- 96. Young JD, Yao SYM, Sun L, Cass CE, Baldwin SA. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica 38: 995–1021, 2008 [DOI] [PubMed] [Google Scholar]
- 97. Yu W, Robson SC, Hill WG. Expression and distribution of ectonucleotidases in mouse urinary bladder. PLos One 6: e18704, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Yu W, Zacharia LC, Jackson EK, Apodaca G. Adenosine receptor expression and function in bladder uroepithelium. Am J Physiol Cell Physiol 291: C254–C265, 2006 [DOI] [PubMed] [Google Scholar]
- 99. Zimmermann H. 5′-Nucleotidase: molecular structure and functional aspects. Biochem J 285: 345–365, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]