

Abstract
We have shown in vitro a hypoxia-induced time-dependent increase in facilitative glucose transporter isoform 3 (GLUT3) expression in N2A murine neuroblasts. This increase in GLUT3 expression is partially reliant on a transcriptional increase noted in actinomycin D and cycloheximide pretreatment experiments. Transient transfection assays in N2A neuroblasts using murine glut3-luciferase reporter constructs mapped the hypoxia-induced enhancer activities to −857- to −573-bp and −203- to −177-bp regions. Hypoxia-exposed N2A nuclear extracts demonstrated an increase in HIF-1α and p-Creb binding to HRE (−828 to −824 bp) and AP-1 (−187 to −180 bp) cis-elements, respectively, in electromobility shift and supershift assays, which was confirmed by chromatin immunoprecipitation assays. In addition, the interaction of CBP with Creb and HIF-1α and CREST with CBP in hypoxia was detected by coimmunoprecipitation. Furthermore, small interference (si)RNA targeting Creb in these cells decreased endogenous Creb concentrations that reduced by twofold hypoxia-induced glut3 gene transcription. Thus, in N2A neuroblasts, phosphorylated HIF-1α and Creb mediated the hypoxia-induced increase in glut3 transcription. Coactivation by the Ca++-dependent CREST and CBP proteins may enhance cross-talk between p-Creb-AP-1 and HIF-1α/HRE of the glut3 gene. Collectively, these processes can facilitate an adaptive response to hypoxic energy depletion targeted at enhancing glucose transport and minimizing injury while fueling the proliferative potential of neuroblasts.
Keywords: hypoxia-inducible factor-1α, glucose transport, neuroblastic proliferation, coactivator complex, Creb-binding protein, CREST
glucose, an essential substrate for oxidative metabolism of the brain, is transported across the blood-brain barrier and into neurons and glia by a family of structurally related membrane-spanning glycoproteins called the facilitative glucose transporters (4, 11, 22). Of the major isoforms cloned to date (4, 8, 11, 22, 34, 41, 45, 57, 58), at least three of the isoforms are expressed in mammalian neurons (GLUT3, GLUT4, GLUT8) (3, 10, 16, 48). Of these, GLUT3 is the predominant neuronal isoform that fuels the process of neurotransmission (19, 29, 38).
More recently, we have observed that murine brain glut3 gene transcription peaks at embryonic day 12, when progenitor cells are engaged in the process of neurogenesis (54). The normal process of progenitor-neuroblastic cell proliferation encountered in the periventricular region and necessary for the formation of the ultimate complement of neurons by transitioning into neuronal differentiation (35, 52) is reliant on the synthesis of GLUT3. The mechanism(s) that mediates transcription of neuroblastic glut3 gene during embryonic development is unknown.
We have also observed that murine brain glut3 transcription and expression peak again during postnatal development, coinciding with synaptogenesis (54). Previous in vivo investigations have demonstrated that hypoxia (Hx) and hypoxic-ischemia increase postnatal murine brain GLUT3 protein concentrations in a time-dependent manner, thereby protecting neuronal glucose uptake in the face of oxygen deficiency (64). The mechanism for this Hx-induced adaptive increase in postnatal neuronal GLUT3 concentrations was recently determined to be transcriptional when examined in vivo (54), although the molecular mechanism remains to be deciphered.
In addition, various cancerous transformation of cells, including gliomas (27) and neuroblastomas (18), has exhibited increased glucose uptake mediated by increased expression of glucose transporters, which includes GLUT3 (32). This enhanced GLUT3 expression is adaptive in response to the cellular Hx encountered, as the cellular proliferative rate outstrips the supply of oxygen, creating local Hx (12). In this situation as well, the molecular mechanisms responsible for increased GLUT3 expression require unraveling.
Previously, we have demonstrated that various transactivators, including Sp3/Sp1 and cAMP response element-binding protein (Creb), regulate GLUT3 expression in murine neuroblasts maintained under normoxic conditions (42, 43). Additionally, in the case of embryonic stem cells lacking Hx-inducible factor-1α (HIF-1α), lower levels of basal GLUT3 that failed to demonstrate Hx-induced increase in GLUT3 mRNA were expressed (21). This suggests a possible role for HIF-1α in glut3 transcription. On the basis of these collective investigations, we hypothesized that Hx induces an adaptive increase in neuroblast GLUT3 expression via transcriptional molecular mechanisms to maintain glucose transport. To test this hypothesis, we undertook the present study in murine N2A neuroblasts that are a prototype of cancerous cells while also serving as a surrogate for progenitor cells that give rise to neurons. We observed that the CREST/Creb-binding protein (CBP)/phosphorylated Creb (p-Creb) and the CREST-CBP-HIF-1α complexes play pivotal roles in Hx-induced glut3 transcriptional control. This transcriptional control enhances GLUT3-mediated neuroblastic glucose transport under oxygen deprivation whether encountered as neuronal progenitors (neuroblasts) during development or following cancerous transformation into neuroblastomas.
MATERIALS AND METHODS
Animals.
Balb/c mice were purchased from The Jackson Laboratories (Bar Harbor, ME). The protocol for the care and use of animals was approved by the Animal Research Committee of the University of California Los Angeles (UCLA) in accordance with the guidelines set by the National Institutes of Health. Mice were allowed access to laboratory chow and water ad libitum and were maintained on 12:12-h light-dark cycles.
Hx studies.
Twenty-one-day-old mice were subjected to normoxia (Nx; FiO2 = 0.21, flow rate = 1.2 l/min) or Hx (FiO2 = 0.08, flow rate = 1.2 l/min), as described previously (54). The mice recovered in their cages for 4 h of reoxygenation, after which they were euthanized (pentobarbital sodium at 100 mg/kg ip) so that their brains could be harvested. Whole brains were removed from the crania and employed for the subsequent preparation of nuclear extracts.
Cells.
N2A and murine neuroblasts (American Tissue Culture Collection, Rockville, MD) were grown in culture dishes in airtight chambers at 37°C with 20% oxygen, 5% CO2, and 75% nitrogen and maintained in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine and 10% fetal bovine serum. These culture conditions were considered to be the normoxic controls. Hx was induced by bleeding in nitrogen with 3% oxygen and 5% CO2 into airtight chambers in which the culture dishes were maintained.
Cell viability.
N2A cell survival rate under Nx and Hx conditions for 24 and 48 h was assessed by the XTT method, following the manufacturer's instructions (Sigma Chemical, St. Louis, MO). The absorbance was measured at 450 nm using a spectrophotometer and correlated directly with cell viability.
Cytotoxicity assay.
Lactate dehydrogenase (LDH) was measured in the N2A supernatant. Following the addition of LDH substrate, the enzymatic reaction occurred at room temperature for 30 min in the dark. After cessation of the reaction, the color reaction was assessed at 490 nm using the ELISA plate reader (Molecular Devices, Sunnyvale, CA). In addition, cell death was measured as percent cytotoxicity calculated as LDH released into the supernatant divided by that in the cell lysate X 100 (Promega, Madison, WI).
Cell proliferation.
Cells were grown in 96-well plates, and cell proliferation assay was conducted at 24 and 48 h according to the manufacturer's instructions (Promega). After a 20-min incubation, absorbance was recorded at 490 nm using an ELISA plate reader (Molecular Devices).
2-Deoxyglucose uptake.
Glucose uptake experiments were undertaken as described previously (29). Briefly, N2A cells grown on 24-well plates (Becton-Dickinson) were washed with 1 ml of d-glucose-free extracellular fluid (ECF) buffer consisting of 122 mM NaCl, 25 mM NaHCO3, 3 mM KCl, 1.4 mM CaCl2, 1.2 mM MgSO4, 0.4 mM K2HPO4, and 10 mM HEPES (pH 7.4) at 37°C and preincubated for 10 min with ECF buffer in the presence or absence of 20 μM cytochalasin B, a nonspecific inhibitor of facilitative glucose transporters (GLUT). Glucose uptake was initiated by adding 200 μl of d-glucose-free ECF buffer containing 0.5 μCi of [1-14C]2-deoxy-d-glucose ([14C]2-DG; Sigma Chemical) in the presence or absence of 20 μM cytochalasin B. After 10 s, uptake was terminated by immediately removing the radioactive solution, and the cells were washed with ice-cold d-glucose-free ECF buffer. The cells were then solubilized in 1 N NaOH and subsequently neutralized. Radioactivity was measured using a liquid scintillation counter, and the cellular protein content was determined using a protein assay kit (Bio-Rad Laboratories, Hercules, CA), with bovine serum albumin as a standard. The uptake rate of [14C]2-DG was calculated by subtracting the uptake of [14C]2-DG in the presence of cytochalasin B from that in the absence of cytochalasin B to evaluate the GLUT-mediated cellular uptake of [14C]2-DG. 2-DG uptake was expressed per milligram of protein and proliferative index to overcome any change in uptake that was due to increased cellular proliferation.
Immunolocalization of GLUT3.
N2A cells grown in a monolayer on coverslips were fixed in 4% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4, for 20 min at room temperature and washed with phosphate-buffered saline (PBS), as described previously (48). The N2A cells were incubated for 1 h with the rabbit anti-mouse GLUT3 antibody (1:500 dilution) (48), followed by a wash with PBS. The cells were subsequently incubated with a mixture of Texas red-labeled donkey anti-rabbit IgG (Jackson Immunoresearch, West Grove, PA) and 4′,6-diamino-2-phenylindole dihydrochloride (Sigma Chemical) for 45 min. N2A cells were then washed with PBS, mounted using an anti-bleaching mounting medium (48), and examined under a Nikon E-600 (Nikon, Melville, NY) equipped with a cooled charge-coupled device camera (CoolSnap HQ Monochrome; Roper Scientific, Tucson, AZ). Negative controls consisted of normal serum or primary antibody in the presence of the GLUT3 peptide used to generate the antibody, which abolished the immunoreactivity entirely (48).
Subcellular fractionation studies.
Differential centrifugation was performed as described previously (48). Briefly, after N2A cells were maintained in Nx and Hx conditions for 24 h, they were subjected to differential centrifugation and layered onto a sucrose cushion (20 mM HEPES, 1 mM EDTA, 1.12 M sucrose). The plasma membrane (PM) was layered on top of this cushion while the low-density microsome (LDM) pellet was collected. These two membrane fractions were resuspended in 1× Laemmli buffer (50–100 μl) and stored at −70°C until assayed.
GLUT3, other GLUT isoforms, and Creb protein studies.
Fifteen micrograms of either total N2A cellular protein determined by the Bio-Rad dye-binding assay (6) or subfractions of PM and LDM assessed by bicinchoninic acid protein assay (Pierce, Rockford, IL) were solubulized in 50 mM Tris, pH 6.8, containing 2% SDS. Western blot analysis was carried out as described previously (43). Proteins separated by SDS-PAGE were transferred onto nitrocellulose membranes. The membranes were incubated with primary antibodies against GLUT3 (1:1,000) (48), GLUT1 (1:1,000) (48), GLUT4 (1:1,000) (53), GLUT8 (1:500) (48), and Creb (1:1,000) (Upstate Biotechnology, Lake Placid, NY). To standardize the interlane loading, membranes were reincubated with an antibody raised against vinculin (1:6,000 dilution; Sigma Chemical) (24). Detection of protein bands was carried out by subjecting immunoblots to the enhanced chemiluminescence system (Amersham Biosciences, Picataway, NJ) using horseradish peroxidase-labeled anti-mouse or anti-rabbit IgG (Sigma Chemical). The visualized protein bands were quantified in Image Quant 5.2 software (GE Healthcare Bioscience).
RNA studies.
Total RNA was extracted from confluent cultured N2A cells according to the manufacturer's instructions using the RNeasy lipid tissue mini kit (Qiagen, Valencia, CA). The extracted RNA was subjected to Northern blot and reverse transcription with real-time PCR analyses, as described previously (42, 43).
Reverse transcription and quantitative real-time polymerase chain reaction was accomplished using total RNA isolated from N2A cells under Nx and Hx conditions. Real-time PCR primers and probes were designed (Table 1) using the Primer Express computer software (Applied Biosystems, Foster City, CA), as described previously (53). Taqman PCR was carried out in triplicate using a StepOnePlus real-time PCR system (Applied Biosystems), and quantification of the amplified product was done against the amplification of 18S as the internal control. The cycling consisted of 12 min at 95°C, followed by 40 cycles of 95°C for 30 s, 59°C for 30 s, and 72°C for 30 s. Relative quantification of PCR amplification products was based on differences between the target and 18S control using the comparative critical threshold method, as described previously (53).
Table 1.
Primers used to make the 5′ deletional constructs
| DNA Construct, bp | Sequence Information | Nucleotide Orientation |
|---|---|---|
| −1,553 | 5′-gcc tac gtg tat gtc tg | Sense |
| −857 | 5′-cct agg cct cag tgt cac | Sense |
| −573 | 5′-cct aga gcc gcg gca agt g | Sense |
| −203 | 5′-caa cta aaa gca gca ctg a | Sense |
| −29 | 5′-cac gag gag gat gtg gta aaa ag | Sense |
| +237 | 5′-gtt cct cgg gtc cta cag | Antisense |
Northern blot analysis.
A 32P-labeled 650-bp fragment of the murine glut3 cDNA extending from exon 2 to exon 5 served as the probe (38). This probe was obtained by amplification of a mouse glut3 clone using a sense primer (5′-catctctggtgttcgccgtg-3′) and an antisense primer (5′-tctgtagcttggtcttcctc-3′). Interlane loading variability was standardized by rehybridization of the stripped filters with a 32P-labeled rat S2 rRNA probe (9).
Actinomycin D and cycloheximide studies.
Cells were maintained for 24 h under Nx or Hx conditions, following which actinomycin D (7 μg/ml) or cycloheximide (20 μg/ml) was added and the cells were maintained in the assigned conditions (Nx or Hx) for timed intervals spanning 0, 1.5, 3, and 6 h. In addition, cells at ∼60% confluence were pretreated with actinomycin D (7 μg/ml) or cycloheximide (20 μg/ml) prior to them being subjected for 0, 7, or 24 h to either Nx or Hx conditions. The cells were quickly washed once in ice-cold PBS, and total RNA was extracted for detecting GLUT3 mRNA by Northern blot analysis.
Transient transfection assays.
A 1.8-kb fragment of the mouse glut3 DNA spanning the −1,553- to +237-bp region was amplified by PCR and cloned into a vector containing firefly luciferase reporter gene (pGL2-basic; Promega, Madison, WI). Subsequently, serial 5′-deletional mouse glut3-Luc fusion gene constructs were created by using a PCR-based strategy, employing primers listed in Table 1. The sequence and orientation of the individual clones were confirmed by sequencing. Transient transfection of cultured cells was carried out using the Superfect transfection reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. pRL-Tk plasmid DNA (thymidine kinase promoter-driven Renilla luciferase; Promega) was cotransfected with each individual transfectant to standardize the results for transfection efficiency.
Luciferase enzyme activity assay.
The luciferase reporter activity was assessed by the dual-luciferase assay (Promega). Briefly, 36–48 h posttransfection the cells were quickly washed with ice-cold PBS and lysed in lysis buffer (Promega). The supernatant on centrifugation at 10,000 rpm for 10 min was stored at −70°C until analysis. Twenty microliters of this cellular extract was mixed with 100 μl of the luciferase assay buffer, and the firefly luciferase activity was measured as light output (10 s) in a Zylux luminometer (Fisher Scientific, Pittsburgh, PA) (42, 43). Subsequently, the Renilla luciferase activity was used to standardize the glut3 promoter-driven firefly luciferase activity for transfection efficiency. The corrected glut3 promoter-driven luciferase activity was expressed as a fold increase over pGL2-basic promoterless luciferase activity. The SV40 promoter-driven luciferase activity served as the positive control in every transfection experiment.
siRNA transfection experiments.
siRNA was constructed to target the mouse Creb (Dharmacom, Dallas, TX) sequence between the +810- to +830-bp coding region by using the following oligonucleotides: sense 5′-gagagagguccgucuaauguu-3′ and antisense 5′-cauuagacggaccucugucuu-3′ (42, 59). Complementary oligonucleotides were converted to a 2′-hydroxyl annealed and desalted duplex strand with a nine-base spacer, thereby creating a short-hairpin RNA that was driven by the RNA polymerase III promoter followed by a (T)5 RNA polymerase III transcriptional stop signal. Cotransfections into N2A cells exposed to Nx or Hx were performed by using the −203-bp glut3-luciferase DNA construct (2 μg) and the constructed siRNA targeted at Creb (100 nM) in six-well plates using Trans-It-LT1 and Trans-IT-TKO (Mirus, Madison, WI), respectively, as transfection reagents, (7, 59). Luciferase activity was measured in N2A cellular extract at 48 h posttransfection by the dual-luciferase assay system (Promega) (42) using the same controls. In addition, 15 μg of protein from siRNA-transfected N2A cell lysates was subjected to Western blot analysis for detection of endogenous Creb amounts.
Electromobility shift assay.
Nuclear extracts from the N2A cells and whole brains from 21-day-old old mice maintained in Hx and Nx were prepared using nuclear extract kits (Active Motif, Carlsbad, CA). Five micrograms of Nx and Hx nuclear extracts were incubated for 30 min at room temperature with 0.2 pmol of 32P-labeled DNA oligoprobe. Synthesized double-stranded oligonucleotides using the primers listed in Table 2 were end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. The DNA-protein complexes were separated from the unbound DNA by electrophoresis through a 5% nondenaturing polyacrylamide gel in 90 mM Tris borate and 2 mM EDTA buffer (42, 43). The gels were dried and subjected to autoradiography in the presence of intensifying screens (Perkin-Elmer Life Sciences) at −80°C. Competition experiments included the addition of five- to 500-fold excess of unlabeled DNA oligonucleotides, whereas supershift experiments included the subsequent incubation with antibodies against HIF-1α, Sp1, Creb, and p-Creb (Active Motif) for 30 min at room temperature prior to gel electrophoresis (42, 43).
Table 2.
Primers used for gel shift analysis
| Site | Primer |
|---|---|
| CRE/AP-1 sites | |
| 5′-agcagcactgactctactctgcg-sense | |
| 5′-cgcagagtagagtcagtgctgct-antisense | |
| AP-1 site mutation | 5′-agcagcactgaaaaaactcgcg-sense |
| 5′-cgcgagttttttcagtgctgct-antsense | |
| CRE site mutation | 5′-agcagcaaaaaatctactctgcg-3′-sense |
| 5′-cgcagagtagattttttgctgct-3′-antisense | |
| CRE and AP-1 site mutation | 5′-agcagcaaaaaaaaaactctgcg-3′-sense |
| 5′-cgcagagttttttttttgctgct-3′-antisense | |
| HRE site | 5′-ttatcacgtgctcagctg-sense |
| 5′-cagctgagcacgtgataa-antisense | |
| HRE site mutation | 5′-ttatcaaaagctcagctg-sense |
| 5′-cagctgagcttttgataa-antisense |
CRE, cAMP response element; AP-1, activator protein-1; HRE, hypoxia-responsive element. In the primer sets, letters in boldface indicate when there is a mutation, and underlined letters indicate when nucleotides that are part of the site are demonstrated.
ELISA quantifying protein-DNA binding.
Ten micrograms of nuclear extracts obtained from N2A cells maintained in Nx or Hx for 48 h were used. ELISA was undertaken to quantify the DNA binding ability of specific nuclear factors, namely HIF-1α, Creb, and p-Creb, that are present in N2A nuclear extracts using protein-binding consensus DNA sequences [hypoxia-responsive element (HRE) and cAMP response element (CRE)] and antibodies targeted at these three nuclear factors. Specificity of the DNA binding by these nuclear proteins was determined by abolition of DNA binding when a mutant DNA consensus sequence was employed. The Trans AM transcription assay kits (Active Motif) were used according to manufacturer's instructions.
Chromatin immunoprecipitation assay.
The chromatin immunoprecipitation (ChIP) assay was performed as described previously (5, 42). Chromatin was extracted from fixed N2A murine neuroblasts (∼2 × 107 in a 150-mm culture dish) exposed to Nx or Hx, of which 10% was set aside as the input chromatin. One hundred microliters of the staphylococcus A cell-precleared and -sheared chromatin lysate obtained from 107 N2A cells was incubated in the absence or presence of 1 μg of the rabbit anti-HIF-1α (H-206, sc-10790; Upstate Biotechnology), anti-Creb (Upstate Biotechnology), anti-p-Creb (Upstate Biotechnology), or anti-Sp3 (D-20, sc-644; Santa Cruz Biotechnology, Santa Cruz, CA) polyclonal antibodies and shaken on a rotator at 4°C. Then, 1 μg of an anti-rabbit secondary antibody (Sigma Chemical) was added to the sample and incubated at room temperature for 15 min. In certain experiments, incubation with rabbit anti-CBP, anti-p300 polyclonal antibodies (Santa Cruz Biotechnology), or anti-CREST (a gift from Dr. Anirvan Ghosh, University of California San Diego, La Jolla, CA) polyclonal antibody was undertaken. Following ChIP, reversal of the formaldehyde cross-link, and precipitation and purification of DNA (∼4 ng) that was complexed with the immunoprecipitated protein, this DNA was used as a template in each PCR (5, 42). The primers used for PCR were designed and synthesized by Retrogen (San Diego, CA). The sequences of all primers used and the PCR conditions are shown in Table 3.
Table 3.
Primers and PCR conditions used in ChIP assays
| DNA Binding Site (Location, PCR Product Size) | Sequence (5′ to 3′) |
|---|---|
| PCR conditions: 95°C for 2 min, 30 cycles of 95°C for 30 s (denaturation), 58°C for 30 s (annealing), and 72°C for 120 s (extension) | |
| HIF-1α/HRE (size = ∼175 bp) | |
| Forward (−989 bp) | gcttcagttcagtccatcag |
| Reverse (−816 bp) | gtcgactcgtgcactatttg |
| Internal control (∼700 bp downstream; size = 354 bp) | |
| Forward (−94 bp) | aggctgtcggctcttg |
| Reverse (+260 bp) | gtatccagccaatgttctcg |
| PCR conditions: 95°C for 2 min, 30 cycles at 95°C for 30 s (denaturation), 55°C for 30 s (annealing), and 72°C for 90 s (extension) | |
| Creb/p-Creb(+Sp3)-CRE/AP-1 (size = ∼185 bp) | |
| Forward (−192 bp) | agcagcactgactctactctgcg |
| Reverse (−7 bp) | ttactacatcctcctcctcgtgg |
| Internal control (∼600 bp upstream; size = ∼665 bp) | |
| Forward (−1,481 bp) | caagttcgagtatagccagg |
| Reverse (−816 bp) | gtcgactcgtgcactatt |
| Creb/p-Creb(+Sp3)-HRE [internal control (∼250 bp upstream; size = ∼250 bp)] | |
| Forward (−1,481 bp) | caagttcgagtatagccagg |
| Reverse (−1,231 bp) | agaatccaagccacttggtg |
ChIP, chromatin immunoprecipitation; HIF-1α, hypoxia-inducible factor-1α; HRE, hypoxia-responsive element; Creb, cAMP response element-binding protein; p-Creb, phosphorylated Creb.
Nuclear protein concentrations.
N2A cells were grown to 60–70% confluency in either Nx or Hx conditions in 150-mm culture dishes (HeLa cells served as the positive control), and the cells were cross-linked with 10 mM dimethyl adipimidate in ice-cold 1× PBS along with 0.25% DMSO in it for 45 min at room temperature. The cells were scraped and washed thoroughly in 1× PBS with 100 mM PMSF at 4°C. Nuclear fraction was isolated from the cells using a nuclear extraction kit (Active Motif), and the nuclear protein was estimated using the Bradford reagent (Pierce Biotechnology, Rockford, IL). Nuclear concentrations of HIF-1α, Creb, p-Creb, Sp3, CBP, p300, and CREST were assessed by 7.5% SDS-PAGE and Western blot analysis, with vinculin (24) as the internal control (42, 43).
Coimmunoprecipitation of nuclear proteins.
Coimmunoprecipitation (co-IP) of Creb, HIF-1α, or CBP was accomplished by immunoprecipitating chromatin from N2A with 1 μg of the anti-HIF-1α, CBP, or p-Creb primary antibodies. The immunoprecipitated antibody-antigen-DNA complex was subjected to a reversal of the cross-link, followed by Western blot analysis as described above using either the polyclonal anti-pCreb (dilution 1:1,000), monoclonal anti-HIF-1α (1:1,000), or polyclonal anti-CBP (1:1,000) as primary antibodies (Santa Cruz Biotechnology), with the HeLa nuclear extract serving as the antigen-positive control (42).
Data analysis.
All data are depicted as means ± SE. The difference between two groups was validated by Student's t-test; differences between more than two groups were determined by the one-way analysis of variance, and intergroup differences were validated by the Fisher's protected least significant difference test. Significance was defined as a P < 0.05.
RESULTS
N2A neuroblast cellular health.
Figure 1A demonstrates the time-dependent survival of N2A cells in response to Hx exposure. Whereas time-dependent changes in cell survival are observed with maximal values seen at 48 h related to cell proliferation, at 24 and 48 h there was no difference in cell survival between the Nx and Hx conditions. At 72 h, a slight but significant Hx-induced decrease in cell survival was observed. Hence, most of the subsequent experiments were undertaken between 24 and 48 h. Figure 1B demonstrates the effect of Hx on cellular LDH and release (supernatant), percent cytotoxicity, and cell proliferation at 24 and 48 h. Hx led to an increase in cellular LDH at both time points, with no change in the release. There was no Hx-induced cytotoxicity to the N2A cells that was detectable. Hx led to increased cell proliferation at 24 and 48 h.
Fig. 1.

N2A cell viability, state, glucose transport, and glucose transporter proteins. Cell viability assessed by XTT analysis (n = 6/time point/condition; A), cell state by cellular (cell) and released [supermatant (sup)] lactate dehydrogenase, cellular %cytotoxicity (Cx; n = 4 per time point per condition), and proliferation index (n = 8–9/time point/condition; B) when exposed to normoxia (Nx) and hypoxia (Hx) for 24, 48, and 72 h. *P < 0.05 and **P < 0.01 vs. time-matched Nx group. Cytochalasin B inhibitable [1-14C]2-deoxy-d-glucose ([14C]2-DG) uptake (transport)/mg protein (C) or cell proliferation index (D) in N2A cells when exposed to Nx and Hx (n = 9–10/time point/condition) for 18, 24, and 48 h. **P < 0.01 and ***P < 0.01 vs. time-matched Nx group. Western blot analyses demonstrate total GLUT1 (E) and GLUT3 (F) protein concentrations when exposed to Nx and Hx for 18, 24, and 48 h (n = 6/time point/condition). Representative Western blots are shown at E and F, top, with vinculin serving as the internal control. *P < 0.02 and **P < 0.01 vs. the time-matched Nx group.
Neuroblast cell glucose uptake and glucose transporter proteins.
Figure 1C demonstrates the time-dependent effect of Hx and Nx on N2A cellular 2-deoxy-[14C]glucose transport (cytochalasin B inhibitable glucose uptake). A significant increase in cellular glucose transport was noted in response to Hx compared with Nx at 24 and 48 h when expressed per milligram of cellular protein. Figure 1D demonstrates Hx-induced increase in glucose transport beginning at 24 and 48 h, with no change earlier at 18 h, even when the cell proliferation index was held constant.
To decipher the mechanism by which glucose transport was increased, we examined the effect of Hx on GLUT (GLUT3, GLUT4, and GLUT8) isoforms expressed by neurons. First, however, we studied GLUT1, normally not expressed by neurons in situ (10) but induced in in vitro cultures, and noted that Hx-induced increase was seen as a trend that was not significant at 18 h, which further increased at 24 h, remaining the same at 48 h with no further increase (Fig. 1E). In contrast, Hx-induced increase in total N2A GLUT3 protein occurred only at 24 h, increasing further to peak at 48 h but not earlier at 18 h, compared with time-matched Nx exposed cells (Fig. 1F). This pattern seen with GLUT3 (and not GLUT1) paralleled that seen with N2A glucose uptake under Hx conditions compared with time-matched Nx (Fig. 2D). Furthermore, no change in N2A total GLUT4 or GLUT8 protein concentrations due to Hx was observed (data not shown). Therefore, hypoxia increased only GLUT1 and GLUT3 in N2A cells.
Fig. 2.

Subcellular localization of GLUT3 protein. Immunolocalization experiments demonstrate GLUT3 immunoreactivity (bright) at 4 (A and B), 24 (C and D), and 48 h (E and F) in N2A cells exposed to Nx (A, C, and E) and Hx (B, D, and F), as seen in representative photomicrographs. At the 24- and 48-h time points, an increase in plasma membrane (PM)-associated GLUT3 immunoreactivity (arrows) was observed in Hx vs. the predominantly cytoplasmic distribution seen in Nx. Scale bar, 50 μm. Subfractionation experiments, shown as a representative Western blot, demonstrate the presence of GLUT3 entirely in the N2A cellular PM in Nx after 24 h. Hx for 24 h increased the PM-associated GLUT3 concentrations with the appearance of low-density microsome (LDM)-associated GLUT3 (G, top). Semiquantification demonstrates the effect of Hx and Nx (n = 3/subfraction/condition) on PM- and LDM-associated GLUT3 concentrations in the bar graph (G, bottom). *P < 0.05 vs. respective Nx group.
Since previous reports had established that Hx increases GLUT1 in cultured neural cells by posttranscriptional mechanisms (56), we pursued understanding the mechanisms behind Hx-induced increase in N2A GLUT3 concentrations. Additionally, in vivo, whereas GLUT1 is neither expressed by neurons (10) nor regulated by hypoxic exposure (64), GLUT3 is expressed primarily in neurons (16) and enhanced by hypoxic exposure (54, 64). Thus our subsequent experiments focused on GLUT3 alone.
GLUT3 protein subcellular distribution.
We next examined the effect of Hx on GLUT3 subcellular distribution in N2A cells. In timed experiments that included both immunolocalization (Fig. 2, A–F) and subcellular fractionation followed by Western blot analysis (Fig. 2G), an increase in Hx-induced PM-associated GLUT3 protein was observed compared with the Nx group, particularly at 24 and 48 h (Fig. 2, A–G). Corresponding Western blots demonstrate an increase in PM-associated GLUT3 concentrations at 24 h in Hx-exposed cells compared with the Nx-exposed cells (Fig. 2G).
GLUT3 mRNA and transcription studies.
Paralleling these protein observations is an increase in neuronal GLUT3 mRNA concentrations (Fig. 3A) seen at 24 and 48 h with Hx. This increase in Hx-induced GLUT3 mRNA at 24 and 48 h is dependent on the presence of extracellular glucose. In the absence of glucose, the Hx-induced effect on GLUT3 mRNA is attenuated. Hence, all of the experiments were conducted in the presence of 5 mM glucose. Figure 3B demonstrates the confirmed changes in GLUT3 mRNA in response to Hx assessed by reverse transcription and quantitative real-time PCR. Again at 24 and 48 h, we observed an increase in GLUT3 mRNA in the N2A cells. Figure 3C shows that actinomycin D added to cells after exposure to Hx for 24 h partially inhibits transcription. A time-dependent decrease in the Hx-related enhancement of N2A GLUT3 mRNA concentrations is observed. Similarly, cycloheximide treatment of cells following exposure to Hx for 24 h caused a time-dependent decline in the Hx-induced augmentation of GLUT3 mRNA concentrations, supporting the need for protein synthesis in transcription of GLUT3 (Fig. 3D). Actinomycin D and cycloheximide (Fig. 3E) addition prior to Hx exposure led to a complete suppression or a drastic reduction of GLUT3 mRNA, respectively, with both Hx and Nx by 24 h, supporting a major role for transcription of GLUT3 under both conditions.
Fig. 3.

GLUT3 mRNA expression studies. Representative Northern blots in the presence (+) and absence (−) of glucose (5 mM; n = 2/time point/condition) with ribosomal S2 mRNA as the internal control (A, bottom panel) and reverse transcription-quantitative PCR (n = 5–6/time point/condition; B) demonstrate GLUT3 mRNA (A, top) in N2A cells exposed to Nx and Hx in a time-dependent manner. *P < 0.01 vs. 24-h Nx; †P < 0.001 vs. 48-h Nx; #P < 0.01 vs. 24-h Hx. Actinomycin D (Act D; 7 μg/ml; C and E) or cycloheximide (Cyx; 20 μg/ml; D and E) treatment over 0, 1.5, 3, and 6 h following 24 h of Nx or Hx (C and D) or pretreatment of N2A cells before exposure to Nx or Hx for 0 [control (con)], 7, and 24 h (E). Blots in C, D, and E, top, demonstrate the corresponding representative Northern blots (top blots), with ribosomal S2 mRNA (bottom blots) serving as the internal control. The results are depicted as a percent of the 0-h Nx (or con) value (C and D: n = 4/time point/condition/treatment, *P < 0.05 vs. the 0 time value in the same condition; E: n = 2/time point/condition/treatment, *P < 0.002 vs. the cycloheximide Nx counterpart and +P < 0.005 vs. the respective con for Nx or Hx).
Transient transfection and glut3-luciferase reporter studies.
Figure 4 demonstrates that the luciferase activity assay results in transient transfection experiments, using various 5′-deletional glut3-luciferase (Luc) DNA constructs (Table 1). The −857-bp glut3-Luc and −203-bp glut3-Luc activity is increased with Hx over that seen with Nx by 100% each compared with the other regions of the glut3 DNA. Previously, the −203-bp to the +1-bp region of the glut3 gene has been characterized extensively and noted to have a Sp binding site that binds Sp1/Sp3 and a CRE/AP-1 site that binds Creb (42, 43) and activates GLUT3 expression in Nx. A computer analysis of the −857-bp to the −573-bp region of the glut3 gene demonstrated the presence of one of the potential four hypoxia regulatory elements (HRE) seen in the 5′-flanking region of the gene. The HRE core consensus sequence is (a/g)cgt(g/c)c (46, 50), with the HRE sequence in the VEGF gene being tacgtg (14) and in the mouse glut3 gene being acgtg (−824 to −828 bp), tcgtg (−1,362 to −1,358 bp), acgtg (−1,548 to −1,544 bp), and tcgtg (−2,224 to −2,220 bp).
Fig. 4.
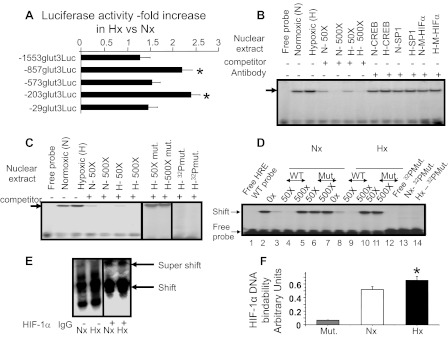
Glut3-luciferase reporter assays, gel shift assays targeting hypoxia-responsive element (HRE), super-gel shift assays, and ELISA detecting hypoxia-inducible factor-1α (HIF-1α)/HRE DNA binding ability. A: luciferase (Luc) activity of transiently transfected 5′ deletional constructs consisting of −1,553, −857, −573, −203, and −29 bp of the glut3 DNA fused to the Luc reporter DNA in N2A cells exposed to Nx or Hx for 24–48 h. The fold increase in values of Hx vs. Nx is depicted (n = 7/DNA construct/condition). *P < 0.05 vs. luciferase activity emitted by the N2A cells containing the −29-bp glut3-Luc construct. Gel shifts in N2A cells detect the glut3-HRE site. B: representative gel using a 32P end-labeled DNA probe that spans the glut3 gene containing the HRE site (free probe; lane 1) and nuclear extracts obtained from N2A cells exposed to normoxia (N; lane 2) or hypoxia (H; lane 3) revealed a gel shift (arrow) that was competed by increasing concentrations (50×, lanes 4 and 6; 500×, lanes 5 and 7) of the unlabeled wild-type (WT) HRE-containing oligoprobe (WT-HRE; lanes 4–7). Gel shifts with the WT-HRE oligoprobe did not change in the presence of anti-cAMP response element-binding protein (Creb; lanes 8 and 9) or anti-SP1 (lanes 10 and 11) antibodies, nor were any supershifts observed (lanes 8-11). An unlabeled mutant (M) HRE oligoprobe failed to displace the gel shift observed with the WT-HRE oligoprobe, and no supershift was observed in the presence of an anti-HIFα antibody in N or H (lanes 12 and 13). Gel shifts reveal glut3-HRE nuclear protein-binding ability in N2A cells. C: Hx (H) and Nx (N) demonstrate a comparable dose-related displacement of the gel shift (lanes 4–7) obtained with N2A nuclear extracts (arrow) using the WT-HRE unlabeled oligoprobe (lanes 2 and 3; free probe seen in lane 1). In contrast, increasing concentrations of the unlabeled mutated oligoprobe (HRE mut) failed to displace the gel shift obtained with the 32P-labeled WT-HRE oligoprobe (lanes 8 and 9). When a 32P-labeled oligoprobe containing the HRE mutation (HRE mut) was used instead of the WT-HRE oligoprobem, no comparable gel shift was observed in Hx or Nx (lanes 10 and 11). Vertical lines indicate approximation of noncontiguous lanes. Gel shifts reveal glut3-HRE nuclear protein binding ability in 21-day-old mice brains. D: representative gel using a 32P-end labeled DNA probe that spans the glut3 gene containing the HRE site (WT; lane 1) and nuclear extracts obtained from whole brain of 21-day-old mice maintained in Nx (lane 2) or Hx (lane 7) revealed a gel shift that was competed by increasing concentrations (50×, lanes 3 and 8; 500×, lanes 4 and 9) of the unlabeled WT-HRE oligoprobe, but not with increasing concentrations (50×, lanes 5 and 10; 500×, lanes 6 and 11) of the unlabeled oligoprobe containing the mutated HRE sequence (Mut). 32P-labeled oligoprobe containing the HRE mutation (32P-Mut; lane 12) failed to bind nuclear proteins, resulting in no gel shift with nuclear extracts obtained from brains subjected to Nx (lane 13) and Hx (lane 14). E: super-gel shifts detect HIF-1α/HRE binding. Representative polyacrylamide gel demonstrates a supershift only in the presence (+; lanes 3 and 4) and not in the absence (−; lanes 1 and 2) of the HIF-1α IgG, confirming binding to the 32P-labeled DNA oligoprobe containing the HRE site by HIF-1α within nuclear extracts obtained from N2A cells exposed to Nx (lane 3) and Hx (lane 4). Vertical lines indicate approximation of noncontiguous lanes. F: ELISA detected DNA binding ability of HIF-1α. Nuclear extracts from N2A cells exposed to Nx or Hx were treated with DNA containing the consensus HRE sequence and an anti-HIF-1α antibody. DNA binding was noted in both Nx and Hx but was absent when DNA containing a mutant consensus HRE sequence (Mut) was used as a negative control (n = 7/condition). An increase in HIF-1α/DNA binding ability is observed with Hx (n = 7) vs. Nx (n = 7). *P < 0.04 vs. Nx.
Gel shift, super-gel shift, and ELISA analyses: HRE and HIF-1α.
An oligoprobe was made, consisting of the HRE-like region (−824 to −828 bp) of the glut3 gene (Table 2), and electrophoretic mobility gel shift assay was undertaken with nuclear extracts obtained from N2A cells. A gel shift was observed, which competed with unlabeled HRE sequences of the glut3 gene (including the −824- to −828-bp region) in a dose-dependent manner (Fig. 4, B and C). The unlabeled mutant HRE sequence containing oligoprobe (Table 2) failed to compete with the gel shift band, and the 32P-labeled mutant HRE sequence containing oligoprobe (table 2) failed to demonstrate a gel shift with N2A nuclear extract proteins (Fig. 4, B and C). A similar pattern of DNA protein binding and gel shifts was observed with nuclear extracts obtained from 21-day-old whole mouse brains (Fig. 4D), supporting the use of N2A cells as a surrogate for in vivo neuroblasts/neuronal progenitors/neurons. Super-gel shift assays confirmed the presence of a HIF-1α-containing protein complex that bound the HRE oligoprobe (Fig. 4E) in Hx and Nx. No significant visible difference in the supershift band density was observed between the Nx and Hx conditions. No such supershift was observed in the presence of anti-Sp1 or anti-Creb antibodies (Fig. 4B), assigning specificity to the observed HIF-1α supershift (Fig. 4E). To quantify the DNA binding of HIF-1α to HRE consensus sequences, ELISA was undertaken with nuclear extracts obtained from N2A cells maintained in Hx or Nx that revealed a statistically significant increase in Hx over that in Nx. However, when the HRE sequences were mutated, there was a near-complete loss of HIF-1α/DNA binding, confirming the specificity of this protein/DNA binding (Fig. 4F) ability.
Gel shift and super-gel shift analyses: CRE/AP-1 and creb/p-creb.
Two gel shifts were observed in nuclear extracts obtained from N2A cells maintained under Nx and Hx, with a radiolabeled oligoprobe consisting of the CRE/AP-1 site that was previously reported to bind Creb (42). These gel shifts noted in Nx and Hx were no different in band density and were similarly displaced with the unlabeled oligoprobe containing the CRE/AP-1 site of the glut3 gene in a dose-dependent manner (Fig. 5, A and B). A similar pattern was reported previously in nuclear extracts obtained from whole mouse brains (42). The consensus CRE sequence is ctgact, which is present in the glut3 gene (−187 to −184 bp), and the consensus AP-1 site sequence is tgagtca, which is present in the glut3 gene as tgactcta (−187 to −180 bp) and overlaps with the CRE region (−187 to −184 bp). Unlike the unlabeled wild-type CRE/AP-1 site containing oligoprobe, the mutant AP-1 site alone with an intact CRE site containing unlabeled oligoprobe failed to efficiently displace the DNA/nuclear protein binding compared with the wild-type oligoprobe (Fig. 5, C and D). However, this displacement study revealed that the Creb/glut3 DNA binding of the AP-1 binding site expressed less fidelity compared with the HIF-1α/glut3 DNA binding ability (HRE site; Fig. 4, B and C). In contrast to the AP-1 site mutation, when the CRE region of the glut3 gene was mutated with an intact AP-1 site, the unlabeled probe displaced the DNA protein binding as efficiently as the wild-type oligoprobe. However, when both the CRE and AP-1 sites were mutated, the unlabeled probe behaved much as the AP-1-mutated probe in displacing the DNA protein binding (Fig. 5, A and B). Furthermore, in the presence of anti-Creb (Fig. 5C) and anti-p-Creb (Fig. 5D) antibodies with the wild-type oligoprobe, supershift bands were detected. The band density of p-Creb but not Creb supershifts was visibly greater in the Hx- than the Nx-exposed cells (Fig. 5, C and D).
Fig. 5.
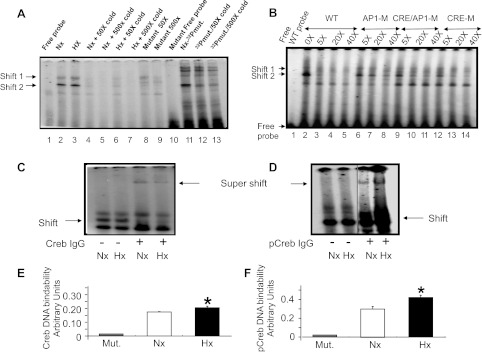
Gel shift assays targeting the cAMP response element (CRE)/activator protein (AP-1) site and super-gel shift assays and ELISA detecting Creb/phosphorylated (p)-Creb/CRE/AP-1 binding ability. Gel shifts in N2A cells detect the glut3-CRE/AP-1 site. A: representative gel demonstrates increasing concentrations (50×, lanes 4 and 6; 500×, lanes 5 and 7) of unlabeled CRE/AP-1 site containing oligoprobe, displacing the 32P-labeled probe that bound to protein in N2A nuclear extracts obtained from Nx- (lane 2) and Hx-exposed cells (lane 3). Lane 1 demonstrates the free oligoprobe without nuclear extract. When unlabeled mutated CRE/AP-1 site containing oligoprobe was added in increasing concentrations (5×, 50×, 500×), the gel shift obtained with the 32P-labeled CRE/AP-1 WT probe was displaced inefficiently (lanes 8–10) compared with that accomplished by the unlabeled WT CRE/AP1 oligoprobe. 32P-labeled mutated CRE/AP-1 oligoprobe (mut) in turn demonstrated the persistent gel shift band 2 but not gel shift band 1 (lane 11), which was further inefficiently displaced with excess unlabeled WT oligoprobe (lanes 12 and 13). Gel shifts in N2A cells differentiate between glut3 CRE and AP-1 sites. B: representative gel demonstrates increasing concentrations (5×, lanes 3, 6, 9, and 12; 20×, lanes 4, 7, 10, and 13; 40×, lanes 5, 8, 11, and 14) of unlabeled CRE/AP-1 site containing oligoprobe (lanes 3-5) that displaced the 32P-labeled WT probe that bound to protein in N2A nuclear extracts obtained from Nx-exposed cells (lane 2). In contrast, unlabeled oligoprobe containing mutation of the CRE site (CRE-M) only displaced the gel-shifted band like the WT oligoprobe (lanes 12-14), whereas mutation of the AP-1 site (AP1-M) only failed to displace the gel-shifted band (lanes 6–8), and mutation of the CRE and AP-1 sites (CRE/AP1-M; lanes 9–11) behaved as the oligoprobe containing only the AP-1 site mutation. Lane 1, free WT oligoprobe. Super-gel shift assay detected Creb and p-Creb binding to the glut3/AP-1 site in N2A cells. C and D: representative gels demonstrate a supershift confirming the binding of Creb (C) and p-Creb (D) to the 32P-labeled DNA oligoprobe containing the AP-1 site in nuclear extracts obtained from N2A cells exposed to Nx (lanes 1 and 3) and Hx (lanes 2 and 4). The supershift is observed in the presence (+; lanes 3 and 4) but not in the absence (−; lanes 1 and 2) of the anti-Creb (C) or anti-p-Creb (D) IgGs. Vertical lines in D indicate approximation of noncontiguous lanes. E and F: ELISA detected Creb/CRE and p-Creb/CRE DNA binding ability in N2A cells. Nuclear extracts from N2A cells exposed to Nx or Hx were treated with DNA containing the consensus CRE sequence and an anti-Creb (E) or anti-p-Creb (F) antibody. DNA binding was noted in both Nx and Hx but was absent when DNA containing a mutant consensus CRE sequence (Mut) was used as a negative control (Creb, n = 7–9/condition; p-Creb, n = 5/condition). An increase in Creb and p-Creb DNA binding ability is observed with Hx compared with Nx (Creb, n = 7–9/condition; p-Creb, n = 5/condition). *P < 0.02 vs. Nx.
ELISA and siRNA approaches targeting Creb/p-Creb.
Quantification of Creb and p-Creb DNA binding ability in N2A nuclear extracts to a consensus CRE sequence by ELISA revealed an increase in the cells exposed to Hx compared with Nx, although the DNA binding ability overall and the hypoxia-inducing ability were higher with p-Creb than with Creb (Fig. 5, E and F). This DNA binding ability was completely lost upon mutation of the consensus CRE region, confirming the specificity of the protein/DNA binding (Fig. 5, E and F). In addition, transient transfection assays employing siRNA targeted at Creb with the −203-bp glut3-Luc DNA constructs in N2A cells revealed a suppression of luciferase activity in cells exposed to Hx and Nx (Fig. 6A). There was a twofold suppression of luciferase activity observed under Hx [−siRNA − +siRNA = 0.526 ± 0.064 relative light units (RLU)/pg] compared with Nx (0.247 ± 0.038 RLU/pg, P < 0.01), which led to exogenous reporter gene activity that was equal to that present in Nx control cells that lack the siRNA (Fig. 6A). In addition, a siRNA-induced decline in endogenous Creb protein concentrations in nuclear extracts obtained from N2A cells maintained under Nx and Hx was observed on Western blot analysis (Fig. 6A, inset).
Fig. 6.

siRNA targeting Creb and chromatin immunoprecipitation (ChIP) assays targeting the glut3-HRE region. A: transient transfection assays using the −203-bp glut3-Luc DNA construct in N2A cells exposed to Nx and Hx for 48 h demonstrate a suppression of luciferase activity that is greater in Hx vs. Nx in the presence of siRNA (+) targeting Creb (n = 6/condition/DNA construct). *P < 0.01 vs. the Nx −siRNA-203G3Luc, #P < 0.01 vs. the Hx −siRNA-203G3Luc. No statistical difference is noted between the Hx +siRNA-203G3Luc and Nx −siRNA-203G3Luc. Top shows representative Western blots demonstrating the endogenous Creb protein (top blot) and vinculin and the internal control (bottom blot) in the siRNA-treated (+) and untreated (−) N2A nuclear extracts from cells subjected to Hx and Nx. B–F: ChIP assays detecting the PCR amplification product containing the glut3-HRE region (∼175 bp; B–F) and the ∼354-bp downstream (B) or ∼250-bp upstream internal control regions (C–F) in chromatin from Nx- or Hx-exposed N2A cells by itself (input) or when immunoprecipitated with anti-HIF-1α (B), anti-Creb (C), anti-p-Creb (D), anti-CBP (E), and anti-CREST (F) IgGs. Blots in B–F, top, demonstrate representative 2% agarose gels with internal control (top blots) and glut3-HRE (bottom blots) amplification products. M, DNA markers; IgG, nonspecific antibody [negative control, presence (+) and absence(−)]. Separate gels have been shown in different boxes, with the IgG negative control gel being the same for C–F. The bar graph depicts the ratio between densities of the ∼175-bp glut3-HRE and the ∼354-bp/∼250-bp internal control PCR products standardized to input values; n = 3/antibody/condition. *P < 0.05, **P < 0.02, and ***P < 0.001 vs. the respective Nx group.
ChIP assays targeting glut3-HRE sequences.
To confirm these findings in vivo with intact chromatin and extend them to other key members of transactivating nuclear complexes, proteins that bound the HRE and AP-1 sites of the glut3 gene were detected by the ChIP assay. Figure 6B demonstrates that HIF-1α binds the HRE-containing DNA sequences of the glut3 gene within intact chromatin under both Hx and Nx. Although no difference was evident with supershifts (Fig. 4E), and although a significant difference emerged with ELISA (Fig. 4F), we employed the more sensitive ChIP assay as validation and noted a significant increase in HIF-1α/HRE DNA binding under Hx compared with Nx conditions (Fig. 6B). To further define the protein complex that includes the HIF-1α/HRE sequences of the glut3 gene, we investigated the effect of Hx on Creb and p-Creb binding to the HRE-containing sequences of the glut3 gene. This binding can either occur directly or indirectly by interacting with the HIF-1α protein that binds HRE or proteins that interact with the HIF-1α protein itself. Under conditions of Nx and Hx, Creb interacted with the HRE sequences; however, no difference between Nx and Hx was noted in the Creb and HIF-1α-HRE complex (Fig. 6C). p-Creb interacted with the HIF-1α-HRE complex under Nx and Hx, with a significant increase under Hx (Fig. 6D). To determine the link between p-Creb, HIF-1α, and HRE, CBP interaction with the p-Creb-HIF-1α-HRE complex was examined and noted to increase with Hx compared with Nx (Fig. 6E), whereas there was no comparable increase in p300 (data not shown). Similarly, although CREST failed to demonstrate much interaction with the CBP-pCreb-HIF-1α-HRE complex under Nx, a definitely enhanced interaction was noted under Hx (Fig. 6F).
ChIP assays targeting glut3/CRE/AP-1 sequences.
Figure 7 demonstrates that Creb and p-Creb bind the CRE/AP-1 site of the glut3 gene under Hx and Nx. Whereas Creb binding to the CRE/AP-1 site was observed when cells were exposed to Nx and Hx with no difference, the p-Creb binding to the glut3 gene increased more than twofold under conditions of Hx (Fig. 7A). Paralleling the findings of p-Creb, there was no detectable interaction between CBP and Creb-p-Creb-CRE-AP-1 complex under Nx; however, CBP begins to interact with this complex under Hx (Fig. 7B). In contrast, although p300 interacts with this complex, there is no Hx-induced change (data not shown). Again HIF-1α interacted with the AP-1 site-containing complex of the glut3 gene in Nx and Hx, with a perceptible increase under Hx (Fig. 7C) possibly due to the role of CBP. Although CREST interaction with the CBP-Creb-p-Creb-CRE-AP-1 complex was enhanced with Hx (Fig. 7D), this increase was not as robust as that seen with the HRE site (Fig. 6F). Since Sp3 binds the Sp-binding site, which is in close proximity to the CRE/AP-1 site of the glut3 gene and is included when the CRE/AP-1 site of the glut3 gene is amplified by PCR as part of the ChIP assay (43), we investigated the effect of Hx on Sp3 binding to the glut3 gene as well. No significant change in the Sp3 protein/DNA binding with Hx over that seen in Nx was evident (data not shown).
Fig. 7.

ChIP assays targeting the glut3-AP-1 region and nuclear proteins. A–D: ChIP assays detecting the PCR amplification products containing the glut3-AP1 (∼185 bp) and the ∼665-bp upstream internal control regions in chromatin from Nx- or Hx-exposed N2A cells by itself (input) or when immunoprecipitated with anti-Creb and anti-p-Creb (A), anti-CBP (B), anti-HIF-1α (C), and anti-CREST (D) IgGs. Blots in A–D, top, demonstrate representative 2% agarose gels with the internal control (top blots) and the glut3-AP1 (bottom blots) containing amplification products. Separate gels are shown in different boxes. IgG negative control is the same for A–D. The bar graphs depict the ratios between densities of the glut3-HRE and the internal control PCR products standardized to input values; n = 3/antibody/condition. ***P < 0.01 and ****P < 0.001 vs. the respective Nx group. E and F: Western blot analyses demonstrate nuclear proteins from N2A cells exposed to Nx and Hx, with Hela cells serving as the positive antigen control (Con) when incubated with anti-p-Creb (E, top), anti-Creb (E, middle), and anti-HIF-1α (F, top) antibodies. E and F, bottom: TATA-binding protein (TBP) served as the internal control. The bar graphs depict quantification of the ratio between p-Creb/Creb (E) and HIF-1α (F), with both standardized to TBP; n = 3/antibody/condition. *P < 0.05 and **P < 0.03 vs. the respective Nx group. Separate gels are shown in different boxes.
Nuclear HIF-1α and creb/p-creb proteins.
To complement these experiments and determine whether there were Hx-induced changes in the absolute concentrations of these proteins in the nucleus, Western blot analysis of nuclear extracts obtained from N2A cells exposed to Hx and Nx were undertaken. There was an increase in HIF-1α protein concentrations (Fig. 7F), no change in Creb (Fig. 7E), and an increase in p-Creb protein concentrations (Fig. 7E) in Hx compared with Nx. We also assessed the nuclear CBP, p300, and CREST protein concentrations. No Hx-induced changes in these three protein concentrations within the nucleus were observed (data not shown).
Co-IP experiments to detect nuclear protein-protein interactions.
We next described nuclear protein-protein interactions. Co-IP experiments confirmed that there was an interaction between CBP and p-Creb that increased by ∼80% under Hx compared with Nx (Fig. 8, A and B) while either of the two antibodies for co-IP and Western blot analysis was used. Similarly, the interaction of CBP and HIF-1α that increased by ∼60–80% under conditions of Hx vs. Nx was proven (Fig. 8, C and D) by using both the antibodies for co-IP and Western blot analysis. However, the interaction of p-CREB with HIF-1α was visible under conditions of Hx only (Fig. 8E), in keeping with ChIP results (Fig. 6D), increasing twofold compared with the Nx value that was standardized at 100%. In contrast, the direct interaction between Sp3, particularly the 60-kDa species, and Creb was evident only under conditions of Nx but not under Hx (data not shown).
Fig. 8.

Coimmunoprecipitation (co-IP) experiments. interaction of CBP, p-Creb, and HIF-1α nuclear proteins. Representative Western blots detecting anti-p-Creb immunoprecipitation (IP) with anti-CBP (A), anti-CBP IP with anti-pCreb (B), anti-CBP IP with anti-HIF-1α (C), anti-HIF-1α IP with anti-CBP (D), and anti-p-Creb IP with anti-HIF-1α (E) antibodies are shown at top. Input = chromatin with no antibody and IgG = chromatin IP with a nonspecific antibody (negative control). The bar graphs shown at bottom depict quantification of the respective co-IPs from N2A cells exposed to Nx and Hx, presented as a percent of the Nx value, which is depicted as 100% after the respective input value is subtracted; n = 3/co-IP/condition. *P < 0.05 and **P < 0.01 vs. the respective Nx group.
DISCUSSION
Neuroblast glucose transport and glucose transporters.
We have demonstrated hypoxia-induced adaptive preservation and increase in neuroblastic glucose transport. Investigation of the mechanism behind this increased glucose transport revealed that both GLUT1 (not normally expressed by neurons in situ but induced by culturing conditions) and GLUT3 (neuronal isoform) increased upon exposure to hypoxia. Unlike this change, both GLUT4 and GLUT8 (found to a lesser extent in neurons) were unchanged in response to hypoxia. Prior investigations in cultured neural cells revealed that a hypoxia-induced adaptive increase in GLUT1 was related to posttranscriptional processes (56). In contrast, the molecular process by which hypoxia in vivo enhances postnatal murine neuronal GLUT3 (54) was unknown. Therefore, we focused on GLUT3 in the present in vitro study and, similarly to our prior in vivo investigations (54), uncovered a transcriptional increase contributing toward the hypoxia-induced increase in GLUT3 mRNA and protein. Although there exist other investigations demonstrating the impact of hypoxia on GLUT3 expression in various cell types (36, 61), including adult neural stem cells (33), the exact molecular mechanisms remain to be deciphered.
Hypoxia-induced transcription and transactivation of the glut3 gene.
Our present studies have uncovered molecular players that are unlikely partners involved in a coactivator complex that is necessary for transactivating neuroblastic glut3 gene transcription in response to hypoxia. Various studies have incriminated HIF-1α as the nuclear factor that induces hypoxia-induced gene expression related to glycolysis and angiogenesis aimed at preserving cellular metabolism (15). The presence of hypoxia and HIF-1α in certain cancers spells poor prognosis (32). Others have demonstrated a role for Creb signaling in maintaining normoxic well being of neural cells (62), mutation of which results in cell death (51). Furthermore, Creb has been observed to transcriptionally regulate Bcl2, a mitochondrial gene that prevents apoptosis (17, 62). However, synergy between p-Creb and HIF-1α in mediating hypoxia-induced neuroblastic GLUT3 expression has been discerned for the first time, contributing toward the novelty of our present study.
Examination of this discovered transcriptional process revealed that hypoxia enhanced extracellular glucose-dependent transcription of the glut3 gene. Furthermore, glut3 gene transcription requires protein synthesis, which contributes toward the overall expression product. This protein synthesis may include some key nuclear proteins that transactivate hypoxia-induced glut3 transcription. Our in vitro studies are in keeping with in vivo studies in a diabetic condition where hyperglycemia and hypoxic ischemia regulate neural glucose transporter expression (63). The cis-elements responsible for this process of glut3 gene transactivation consist of at least the HRE and AP-1 sequences present in the 5′ flanking region of the gene. The nuclear proteins involved in DNA-protein interactions that are necessary for enhancing the promoter activity of the glut3 gene include HIF-1α and p-Creb/Creb, which bind HRE and AP-1 sequences, respectively.
HIF-1α transactivates neuroblastic glut3 gene.
HIF-1α, a nuclear protein, is essential for embryonic development, the deficiency of which, or its partner HIF-1β, produces developmental defects, including neural tube defects and defective vascularization of the embryonic cephalad region with neuronal loss (21, 31). Thus, in addition to bestowing oxygen-sensing ability, HIF-1α plays a major role in neuronal proliferation and differentiation by altering the expression of specific target genes, which includes neural glut3 (21). Transactivation of genes, including the glut3 gene by HIF-1α, requires phosphorylation of the nuclear protein to facilitate nuclear translocation and DNA binding. This process is mediated by a CaM kinase that is activated by hypoxia-induced Ca++ influx into neurons (46).
CBP and HIF-1α association.
In addition to HIF-1α/HIF-1β complex activating transcription of target gene expression after binding to cognate HRE, HIF-1α-mediated activation of transcription requires the recruitment of coactivators such as CBP/p300 (60). CBP/p300 regulates chromatin structure through histone acetylation and interaction with other histone acetyl transferases (60) that acetylate specific amino acid residues involved in transcription (49). The function of both the NH2-terminal and COOH-terminal transactivation domains of HIF-1α can be enhanced by CBP, since the COOH-terminal interacts directly with the CH1 domain of CBP (20, 30). In neuroblasts, we observed that CBP interacted with HIF-1α in Nx and Hx, and the binding of CBP to the HIF-1α-HRE complex increased in response to Hx.
CREST and CBP-HIF-1α-glut3-HRE association.
However, what played a key role in driving the CBP-HIF-1α-HRE-induced glut3 transcription was the Hx-induced CREST binding to CBP as a molecular player of this protein-DNA complex. CREST has been described as a protein that is a critically important partner for CBP in regulating transcription of Creb-related target genes (2). CREST is a coactivator that demonstrates calcium dependence in mediating gene transcription. Calcium influx through voltage-sensitive calcium channels and N-methyl-d-aspartate receptors induces CREST-mediated transcription (2, 13). Hypoxia is also known to increase neuronal calcium influx (46) via similar mechanisms. While the NH2-terminal domain of CREST suppresses transactivation in the basal state, the COOH-terminal domain is involved in calcium-induced transactivation. This occurs because of CREST-related protein Synovial sarcoma translocation's interaction with CBP (2). Interaction with the NH2 terminus of CBP is via the COOH terminus of CREST; deletion of the latter reduces calcium-induced transactivation. This supports interaction, with CBP being necessary for CREST-mediated gene transcription. CREST expression is notable during brain development (prenatal and postnatal), mediating calcium signaling-regulated dendritic development. The role CREST plays in dendritic development contributes to the size of the cortex and cerebellum and helps couple synaptic activity to postnatal brain growth control (2). CREST also interacts with chromatin remodeling proteins BAF250 and BRG-1 (2). These interactions and developmental functions suggest that the CREST complex may regulate transcription by the calcium-dependent modification of chromatin structure. Because CBP is known to interact with several DNA binding proteins, it is possible that CREST may be recruited to specific gene promoters by CBP (2), as seen with the glut3 gene in our present study.
CREST binding and participation in neuroblasts was nearly absent in Nx but appeared and became evident in Hx in the present investigation. This participation induced by Hx in turn increased the contribution by CBP and HIF-1α as evident in intact chromatin (ChIP). However, unlike the role of HIF-1α in driving transcription of other key target genes such as erythropoietin and VEGF (46), the role of HIF-1α in driving neuroblastic glut3 transcription is rather limited (∼100%) compared with the more than twofold increase evident in Hx-induced glut3 transcription. Thus, this Hx-induced adaptive increase in glut3 transcription is likely regulated by other coactivators and positive cooperativity between various other DNA binding nuclear factors. Since CBP/CREST plays a major role in the HIF-1α/HRE mediated glut3 transcription, it was natural to assess the role of Creb, which binds the glut3 gene and is coactivated by CBP as well. Besides, our studies demonstrate interaction of p-Creb either directly or indirectly with the glut3-HRE region (Fig. 6D).
Creb/p-Creb and glut3 gene transactivation.
In our present investigation, besides HRE sequences the AP-1 site (−187 to 180bp) also participated in the hypoxia-induced adaptive increase in glut3 gene transcription. Previously, we observed binding of Creb/p-Creb to the AP-1 region of the glut3 gene in N2A cells and murine brain during Nx conditions (42). Activation of glut3 transcription under normoxia has also previously been reported to rely on the interaction between Sp3 and Creb nuclear proteins (43). The role of Sp3 in mediating the hypoxia-induced glut3 transcription was also investigated here. Nuclear concentrations of Sp3 (60 kDa only) and Sp3 binding to the glut3 gene did not significantly change with Hx vs. Nx. Further interaction of Sp3 with Creb occurred only under normoxia and not with hypoxia. This suggests that Sp3-directed glut3 transcription may be important for neuroblastic cell proliferation and differentiation under Nx. Under Hx, it fails to interact with Creb/p-Creb, which directs the adaptive increase in glut3 transcription.
In addition, it has been demonstrated previously that Creb function is necessary for calcium-induced dendritic growth (44). This is intriguing since GLUT3 has been shown to be localized in synapses mainly on neuronal dendrites and axons (16, 25). There is evidence for GLUT3's role in mediating neurotransmission related to calcium influx. This is accomplished by increasing glucose transport during neuronal depolarization, a process that increases neuronal calcium influx that in turn enhances synaptic activity (29, 37). This supports a role for GLUT3 in neuroblastic calcium signaling-mediated activity that preserves glucose transport and assures optimal energy supply to these cells.
Since hypoxia also enhances neuronal Ca++ influx (46), we explored the role of p-Creb in glut3 gene transcription under hypoxic conditions. We observed that p-Creb/AP-1 binding increased significantly with hypoxia both in vitro (super-gel shift assays) and within intact chromatin (ChIP) of neuroblasts. This increase in DNA binding corresponded with the hypoxia-induced increase in nuclear p-Creb concentrations. In contrast to p-Creb, Creb concentrations did not change in hypoxia, and there was no difference in Creb binding of the AP-1 site in the glut3 gene. This is key and specific to the glut3 gene, although some Hx-related increased binding to the CRE consensus sequence (ELISA) lower than that of p-Creb was evident. These results support a crucial role for phosphorylation of Creb in Hx-induced neuroblastic glut3 DNA binding and gene transcription. Phosphorylated Creb (Ser133) that is necessary for DNA binding also activates various other downstream genes involved in biosynthesis of neurotrophic factors, blocking of cell death pathways, long-term potentiation, and synaptic plasticity (1, 39). Thus, p-Creb activates pathways that mediate adaptive responses toward protection of substrate delivery and against cell injury. Creb is also phosphorylated (Ser142, Ser143) by a Ca++ influx-dependent CaM kinase (23), which is necessary for recruiting CBP to associate with p-Creb in a protein-DNA complex that mediates gene transcription (23, 47).
CBP and p-Creb association.
CBP, an important coactivator protein, acetylates histones and promotes a euchromatin state, thereby making DNA more accessible to activating transcription factors (26, 28). CBP also enhances transcription by bridging transcriptional factors, such as Creb, to the basal transcriptional complex (26, 28). Roles of CBP and p-Creb become evident when mutations of either nuclear factor result in human neurodevelopmental disorders (40, 55). CBP also recruits CREST to the p-Creb-DNA complex (2). Furthermore, Hx increases the amount of CREST in this complex in the presence of CBP. In our present investigation, under Nx, CBP is not involved. However, Hx engaged CBP with the pCreb-AP1 complex, which in turn was associated with increased GLUT3 expression. Although CREST participation in the CBP-p-Creb-glut3-AP-1 complex increased some with hypoxia, a key role played in driving the Hx-induced transcriptional activation via the AP-1 site of the glut3 gene is that of CBP.
CBP/CREST and p-Creb/glut3/AP-1 with HIF-1α/glut3/HRE association.
Hx-induced recruitment of the CBP/CREST nuclear coactivators appears to force an interaction between the HIF-1α-HRE and Creb-p-Creb-AP-1 transcriptional complexes facilitated by chromatin remodeling, thereby increasing the proximity and cooperativity between key transactivators, namely HIF-1α and p-Creb. This cooperativity between key nuclear proteins that bind two enhancer elements of the glut3 gene becomes evident within intact chromatin. Hence, Hx is observed to increase p-Creb's interaction, perhaps indirectly with glut3-HRE (Fig. 6D), and HIF-1α's indirect interaction with glut3-AP1 sites (Fig. 7C). This in turn accounts for the Hx-induced cumulative increase of 200% (HIF-1α-HRE = 100% + p-Creb-AP1 = 100%) in neuroblastic glut3 transcription (Fig. 4A).
Summary.
We conclude that Hx-induced increase in neuroblast GLUT3 expression mediates a compensatory adaptation toward preservation of neuroblastic glucose and energy supply. This process is mediated partially by transcriptional activation of at least two enhancer regions (HRE and AP-1) that require transactivators (HIF-1α and p-Creb) that cooperate toward augmenting neuroblastic glut3 gene transcription. This cooperation between two protein-DNA complexes is accomplished by the recruitment of the critical calcium-dependent CBP/CREST coactivator complex. Our present unraveling of the protective role played by these molecular proteins in increasing neuroblastic glut3 transcription under Hx is novel and translates into increased GLUT3 protein mediating glucose transport (Fig. 9). Contribution toward Hx-induced increase in neuroblastic glut3 gene expression product by additional posttranscriptional processes needs future investigation.
Fig. 9.

Schematic representation of the molecular machinery involved in Hx-induced transcriptional control of the glut3 gene in N2A cells, which results in increased GLUT3 mRNA that translates into increased GLUT3 protein and translocates to the plasma membrane to allow intracellular transport of glucose.
GRANTS
This work was supported by National Institute of Child Health and Human Development Grants HD-33997 and HD-46979.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.T., N.R., M.T., and B.-C.S. performed the experiments; S.T., N.R., M.T., B.-C.S., and S.U.D. analyzed the data; S.T., N.R., and S.U.D. interpreted the results of the experiments; S.T., N.R., M.T., B.-C.S., and S.U.D. approved the final version of the manuscript; N.R., B.-C.S., and S.U.D. prepared the figures; B.-C.S. and S.U.D. drafted the manuscript; S.U.D. contributed to the conception and design of the research; S.U.D. edited and revised the manuscript.
ACKNOWLEDGMENTS
We acknowledge the assistance of Dr. Robert McKnight with some of the gel shift experiments. We thank Dr. Anirvan Ghosh (University of San Diego California, La Jolla, CA) for the anti-CREST antibody, Jerry Cheng and Kathleen Sakamoto (UCLA, Los Angeles, CA) for the Creb siRNA, and Kelle H. Moley (Washington University, St. Louis, MO) for the anti-GLUT8 antibody used in this study.
REFERENCES
- 1.Abel T, Martin KC, Bartsch D, Kandel ER. Memory suppressor genes: inhibitory constraints on the storage of long-term memory. Science 279: 338–341, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303: 197–202, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Arluison M, Quignon M, Thorens B, Leloup C, Penicaud L. Immunocytochemical localization of the glucose transporter 2 (GLUT2) in the adult rat brain. II. Electron microscopic study. J Chem Neuroanat 28: 137–146, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Bell GI, Burant CF, Takeda J, Gould GW. Structure and function of mammalian facilitative sugar transporters. J Biol Chem 268: 19161–19164, 1993 [PubMed] [Google Scholar]
- 5.Boyd KE, Farnham PJ. Myc versus USF: discrimination at the cad gene is determined by core promoter elements. Mol Cell Biol 17: 2529–2537, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976 [DOI] [PubMed] [Google Scholar]
- 7.Bruckner BA, Ammini CV, Otal MP, Raizada MK, Stacpoole PW. Regulation of brain glucose transporters by glucose and oxygen deprivation. Metabolism 48: 422–431, 1999 [DOI] [PubMed] [Google Scholar]
- 8.Carayannopoulos MO, Chi MM, Cui Y, Pingsterhaus JM, McKnight RA, Mueckler M, Devaskar SU, Moley KH. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc Natl Acad Sci USA 97: 7313–7318, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan YL, Gutell R, Noller HF, Wool IG. The nucleotide sequence of a rat 18 S ribosomal ribonucleic acid gene and a proposal for the secondary structure of 18 S ribosomal ribonucleic acid. J Biol Chem 259: 224–230, 1984 [PubMed] [Google Scholar]
- 10.Devaskar S, Zahm DS, Holtzclaw L, Chundu K, Wadzinski BE. Developmental regulation of the distribution of rat brain insulin-insensitive (Glut 1) glucose transporter. Endocrinology 129: 1530–1540, 1991 [DOI] [PubMed] [Google Scholar]
- 11.Devaskar SU, Mueckler MM. The mammalian glucose transporters. Pediatr Res 31: 1–13, 1992 [DOI] [PubMed] [Google Scholar]
- 12.Dierckx RA, Van de Wiele C. FDG uptake, a surrogate of tumour hypoxia? Eur J Nucl Med Mol Imaging 35: 1544–1549, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eid JE, Kung AL, Scully R, Livingston DM. p300 interacts with the nuclear proto-oncoprotein SYT as part of the active control of cell adhesion. Cell 102: 839–848, 2000 [DOI] [PubMed] [Google Scholar]
- 14.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA 94: 4273–4278, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esterman A, Greco MA, Mitani Y, Finlay TH, Ismail-Beigi F, Dancis J. The effect of hypoxia on human trophoblast in culture: morphology, glucose transport and metabolism. Placenta 18: 129–136, 1997 [DOI] [PubMed] [Google Scholar]
- 16.Fields HM, Rinaman L, Devaskar SU. Distribution of glucose transporter isoform-3 and hexokinase I in the postnatal murine brain. Brain Res 846: 260–264, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Freeland K, Boxer LM, Latchman DS. The cyclic AMP response element in the Bcl-2 promoter confers inducibility by hypoxia in neuronal cells. Brain Res Mol Brain Res 92: 98–106, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Gazit V, Ben-Abraham R, Vofsi O, Katz Y. l-cysteine increases glucose uptake in mouse soleus muscle and SH-SY5Y cells. Metab Brain Dis 18: 221–231, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Heather West Greenlee M, Uemura E, Carpenter SL, Doyle RT, Buss JE. Glucose uptake in PC12 cells: GLUT3 vesicle trafficking and fusion as revealed with a novel GLUT3-GFP fusion protein. J Neurosci Res 73: 518–525, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem 277: 26351–26355, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12: 149–162, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joost HG, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, Moley JF, Mueckler M, Rogers S, Schurmann A, Seino S, Thorens B. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab 282: E974–E976, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Kornhauser JM, Cowan CW, Shaywitz AJ, Dolmetsch RE, Griffith EC, Hu LS, Haddad C, Xia Z, Greenberg ME. CREB transcriptional activity in neurons is regulated by multiple, calcium-specific phosphorylation events. Neuron 34: 221–233, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Kushnaryov VM, Sedmak JJ, Markwald RR, Faculjak ML, Loo PM. Actin and lamin comprised filaments in the nuclei of Chinese hamster ovary cells affected with Clostridium difficile enterotoxin A. Cytobios 64: 181–196, 1990 [PubMed] [Google Scholar]
- 25.Leino RL, Gerhart DZ, van Bueren AM, McCall AL, Drewes LR. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J Neurosci Res 49: 617–626, 1997 [DOI] [PubMed] [Google Scholar]
- 26.Liu G, Xia T, Chen X. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J Biol Chem 278: 17557–17565, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Li YM, Tian RF, Liu WP, Fei Z, Long QF, Wang XA, Zhang X. The expression and significance of HIF-1alpha and GLUT-3 in glioma. Brain Res 1304: 149–154, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Lu Q, Hutchins AE, Doyle CM, Lundblad JR, Kwok RP. Acetylation of cAMP-responsive element-binding protein (CREB) by CREB-binding protein enhances CREB-dependent transcription. J Biol Chem 278: 15727–15734, 2003 [DOI] [PubMed] [Google Scholar]
- 29.Maher F, Simpson IA. Modulation of expression of glucose transporters GLUT3 and GLUT1 by potassium and N-methyl-d-aspartate in cultured cerebellar granule neurons. Mol Cell Neurosci 5: 369–375, 1994 [DOI] [PubMed] [Google Scholar]
- 30.Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 15: 2675–2686, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature 386: 403–407, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Marín-Hernández A, Gallardo-Pérez JC, Ralph SJ, Rodríguez-Enríquez S, Moreno-Sánchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem 9: 1084–1101, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Maurer MH, Geomor HK, Burgers HF, Schelshorn DW, Kuschinsky W. Adult neural stem cells express glucose transporters GLUT1 and GLUT3 and regulate GLUT3 expression. FEBS Lett 580: 4430–4434, 2006 [DOI] [PubMed] [Google Scholar]
- 34.McVie-Wylie AJ, Lamson DR, Chen YT. Molecular cloning of a novel member of the GLUT family of transporters, SLC2a10 (GLUT10), localized on chromosome 20q13.1: a candidate gene for NIDDM susceptibility. Genomics 72: 113–117, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Miyata T, Ono Y, Okamoto M, Masaoka M, Sakakibara A, Kawaguchi A, Hashimoto M, Ogawa M. Migration, early axonogenesis, and Reelin-dependent layer-forming behavior of early/posterior-born Purkinje cells in the developing mouse lateral cerebellum. Neural Dev 5: 23, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mobasheri A, Richardson S, Mobasheri R, Shakibaei M, Hoyland JA. Hypoxia inducible factor-1 and facilitative glucose transporters GLUT1 and GLUT3: putative molecular components of the oxygen and glucose sensing apparatus in articular chondrocytes. Histol Histopathol 20: 1327–1338, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Moulder KL, Cormier RJ, Shute AA, Zorumski CF, Mennerick S. Homeostatic effects of depolarization on Ca2+ influx, synaptic signaling, and survival. J Neurosci 23: 1825–1831, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagamatsu S, Sawa H, Inoue N, Nakamichi Y, Takeshima H, Hoshino T. Gene expression of GLUT3 glucose transporter regulated by glucose in vivo in mouse brain and in vitro in neuronal cell cultures from rat embryos. Biochem J 300: 125–131, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Obrietan K, Gao XB, Van Den Pol AN. Excitatory actions of GABA increase BDNF expression via a MAPK-CREB-dependent mechanism—a positive feedback circuit in developing neurons. J Neurophysiol 88: 1005–1015, 2002 [DOI] [PubMed] [Google Scholar]
- 40.Pescucci C, Meloni I, Bruttini M, Ariani F, Longo I, Mari F, Canitano R, Hayek G, Zappella M, Renieri A. Chromosome 2 deletion encompassing the MAP2 gene in a patient with autism and Rett-like features. Clin Genet 64: 497–501, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Phay JE, Hussain HB, Moley JF. Cloning and expression analysis of a novel member of the facilitative glucose transporter family, SLC2A9 (GLUT9). Genomics 66: 217–220, 2000 [DOI] [PubMed] [Google Scholar]
- 42.Rajakumar A, Thamotharan S, Raychaudhuri N, Menon RK, Devaskar SU. Trans-activators regulating neuronal glucose transporter isoform-3 gene expression in mammalian neurons. J Biol Chem 279: 26768–26779, 2004 [DOI] [PubMed] [Google Scholar]
- 43.Rajakumar RA, Thamotharan S, Menon RK, Devaskar SU. Sp1 and Sp3 regulate transcriptional activity of the facilitative glucose transporter isoform-3 gene in mammalian neuroblasts and trophoblasts. J Biol Chem 273: 27474–27483, 1998 [DOI] [PubMed] [Google Scholar]
- 44.Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 34: 999–1010, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Rogers S, Macheda ML, Docherty SE, Carty MD, Henderson MA, Soeller WC, Gibbs EM, James DE, Best JD. Identification of a novel glucose transporter-like protein—GLUT-12. Am J Physiol Endocrinol Metab 282: E733–E738, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol 15: 551–578, 1999 [DOI] [PubMed] [Google Scholar]
- 47.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem 68: 821–861, 1999 [DOI] [PubMed] [Google Scholar]
- 48.Shin BC, McKnight RA, Devaskar SU. Glucose transporter GLUT8 translocation in neurons is not insulin responsive. J Neurosci Res 75: 835–844, 2004 [DOI] [PubMed] [Google Scholar]
- 49.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64: 435–459, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su H, Arakawa-Hoyt J, Kan YW. Adeno-associated viral vector-mediated hypoxia response element-regulated gene expression in mouse ischemic heart model. Proc Natl Acad Sci USA 99: 9480–9485, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tan YW, Zhang SJ, Hoffmann T, Bading H. Increasing levels of wild-type CREB up-regulates several activity-regulated inhibitor of death (AID) genes and promotes neuronal survival. BMC Neurosci 13: 48, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tang Y, Shah K, Messerli SM, Snyder E, Breakefield X, Weissleder R. In vivo tracking of neural progenitor cell migration to glioblastomas. Hum Gene Ther 14: 1247–1254, 2003 [DOI] [PubMed] [Google Scholar]
- 53.Thamotharan M, Shin BC, Suddirikku DT, Thamotharan S, Garg M, Devaskar SU. GLUT4 expression and subcellular localization in the intrauterine growth-restricted adult rat female offspring. Am J Physiol Endocrinol Metab 288: E935–E947, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Thamotharan S, Stout D, Shin BC, Devaskar SU. Temporal and Spatial Distribution of Murine Placental and Brain Glut3-Luciferase Transgene as a Read Out of In-vivo Transcription. Am J Physiol Endocrinol Metab. First published November 27, 2012; doi:10.1152/ajpendo.00214.2012. [DOI] [PMC free article] [PubMed]
- 55.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 8: 1056–1072, 2009 [DOI] [PubMed] [Google Scholar]
- 56.Vannucci SJ, Seaman LB, Vannucci RC. Effects of hypoxia-ischemia on GLUT1 and GLUT3 glucose transporters in immature rat brain. J Cereb Blood Flow Metab 16: 77–81, 1996 [DOI] [PubMed] [Google Scholar]
- 57.Wu X, Freeze HH. GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics 80: 553–557, 2002 [DOI] [PubMed] [Google Scholar]
- 58.Wu X, Li W, Sharma V, Godzik A, Freeze HH. Cloning and characterization of glucose transporter 11, a novel sugar transporter that is alternatively spliced in various tissues. Mol Genet Metab 76: 37–45, 2002 [DOI] [PubMed] [Google Scholar]
- 59.Yang EJ, Yoon JH, Chung KC. Bruton's tyrosine kinase phosphorylates cAMP-responsive element-binding protein at serine 133 during neuronal differentiation in immortalized hippocampal progenitor cells. J Biol Chem 279: 1827–1837, 2004 [DOI] [PubMed] [Google Scholar]
- 60.Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382: 319–324, 1996 [DOI] [PubMed] [Google Scholar]
- 61.Yu J, Li J, Zhang S, Xu X, Zheng M, Jiang G, Li F. IGF-1 induces hypoxia-inducible factor 1alpha-mediated GLUT3 expression through PI3K/Akt/mTOR dependent pathways in PC12 cells. Brain Res 1430: 18–24, 2012 [DOI] [PubMed] [Google Scholar]
- 62.Zaman K, Ryu H, Hall D, O'Donovan K, Lin KI, Miller MP, Marquis JC, Baraban JM, Semenza GL, Ratan RR. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci 19: 9821–9830, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang WW, Zhang L, Hou WK, Xu YX, Xu H, Lou FC, Zhang Y, Wang Q. Dynamic expression of glucose transporters 1 and 3 in the brain of diabetic rats with cerebral ischemia reperfusion. Chin Med J (Engl) 122: 1996–2001, 2009 [PubMed] [Google Scholar]
- 64.Zovein A, Flowers-Ziegler J, Thamotharan S, Shin D, Sankar R, Nguyen K, Gambhir S, Devaskar SU. Postnatal hypoxic-ischemic brain injury alters mechanisms mediating neuronal glucose transport. Am J Physiol Regul Integr Comp Physiol 286: R273–R282, 2004 [DOI] [PubMed] [Google Scholar]