

Abstract
Previous studies show that neuropeptide Y1–36 (NPY1–36) and peptide YY1–36 (PYY1–36), by engaging Y1 receptors, stimulate proliferation of spontaneous hypertensive rat (SHR) preglomerular vascular smooth muscle cells (PGVSMCs). In contrast, these peptides have little effect on proliferation of Wistar-Kyoto (WKY) PGVSMCs. Why SHR and WKY PGVSMCs differ in this regard is unknown. Because receptor for activated C kinase 1 (RACK1) can modulate cell proliferation, we tested the hypothesis that differences in RACK1 levels/localization may explain the differential response of SHR vs. WKY PGVSMCs to NPY1–36 and PYY1–36. Western blotting for RACK1 in subcellular fractions of cultured SHR and WKY PGVSMCs demonstrated increased levels of RACK1 in the membrane and cytoskeletal subcellular fractions of SHR vs. WKY PGVSMCs. NPY1–36 and PYY1–36 stimulated proliferation of SHR PGVSMCs, and siRNA knockdown of RACK1 abrogated this effect. Neither NPY1–36 nor PYY1–36 stimulated the proliferation of WKY PGVSMCs. However, in WKY PGVSMCs treated with a RACK1 plasmid, both NPY1–36 and PYY1–36 stimulated proliferation. In SHR PGVSMCs, inhibitors of the Gi/phospholipase C/PKC pathway (a pathway known to be organized by RACK1) attenuated the ability of NPY1–36 to stimulate the proliferation of SHR PGVSMCs. Our results suggest that RACK1 modulates the ability of PGVSMCs to respond to the proliferative actions of NPY1–36 and PYY1–36 and differences in RACK1 levels/localization account for, in part, differential proliferative responses to NPY1–36 and PYY1–36 in SHR vs. WKY PGVSMCs. Because dipeptidyl peptidase IV inhibitors increase NPY1–36 and PYY1–36 levels, our findings have implications for the use of such drugs in diabetic patients.
Keywords: receptor for activated C kinase 1, cell proliferation, preglomerular vascular smooth muscle cells, spontaneously hypertensive rats, Wistar-Kyoto rats
neuropeptide y1–36 (npy1–36) and peptide YY1–36 (PYY1–36) are members of the pancreatic polypeptide-fold (PP-fold) family (6). Importantly, the kidney is likely exposed to biologically active levels of PP-fold peptides because 1) renal sympathetic varicosities release NPY1–36 in response to CNS-mediated activation of renal sympathetic nerves (13); 2) NPY1–36 is made by renal epithelial cells and released into the renal interstitium (22); and 3) fatty meals stimulate endocrine L-cells in the GI tract to release PYY1–36 into the systemic circulation, producing physiologically active levels of PYY1–36 in plasma (3, 4, 19, 28, 34, 44, 53), and this PYY1–36 would be promptly delivered to the kidney via the bloodstream.
The exposure of the kidney to pharmacologically active levels of NPY1–36 or PYY1–36 may have implications for renovascular structure and health. In this regard, our recent experiments indicate that both NPY1–36 and PYY1–36 stimulate the proliferation of preglomerular microvascular smooth muscle cells (PGVSMCs) obtained from spontaneously hypertensive rats (SHR) via a mechanism involving activation of Y1 receptors (26). In contrast, NPY1–36 and PYY1–36 exert little or no effect on proliferation of PGVSMCs obtained from normotensive Wistar-Kyoto rats (WKY). Thus, endogenous PP-fold peptides may have adverse effects on renovascular structure in genetic hypertension. However, the reason that NPY1–36 and PYY1–36 robustly stimulate proliferation of PGVSMCs only in PGVSMCs obtained from genetically susceptible kidneys is unclear. Thus, the focus of the current study was to investigate why SHR, but not WKY, PGVSMCs are responsive to the proliferative effects of NPY1–36 and PYY1–36.
Our previously published work demonstrates that Y1 receptor expression is not different in preglomerular microvessels from SHR vs. WKY (15), so receptor expression differences are unlikely to account for the differential proliferative responses of SHR vs. WKY to NPY1–36 and PYY1–36. On the other hand, our previously published work does show that signaling via Gi-linked receptors is enhanced in the renal vasculature of SHR vs. WKY (15). Inasmuch as Y1 receptors are Gi-coupled receptors, the most likely reason that NPY1–36 and PYY1–36 stimulate growth more in SHR than WKY PGVSMCs is that postreceptor signaling via the Gi pathway is enhanced in SHR PGVSMCs.
Receptor for activated C kinase 1 (RACK1) is a seven-sided propeller protein with seven WD40 repeats, and RACK1 participates in cell signaling by functioning as a scaffolding protein that interacts with G protein βγ subunits, phospholipase C, and PKC (10, 49) and, therefore, organizes and modulates the Gi/phospholipase C/PKC pathway. Because Y1 receptors are coupled to Gi (38) and because recent studies from our laboratory indicate that RACK1 participates in proliferation of PGVSMCs (11), we hypothesized that differences in RACK1 levels or distribution, via interacting with the Gi/phospholipase C/PKC pathway, may explain why SHR, but not WKY, PGVSMCs are sensitive to the proliferative effects of NPY1–36 and PYY1–36.
METHODS
Animals.
Studies utilized male SHR and WKY obtained from Taconic Farms (Germantown, NY). The Institutional Animal Care and Use Committee approved all procedures. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Isolation and culture of PGVSMCs.
A complete description of our method for culturing and confirming the identity of rat PGVSMCs can be found in Mokkapatti et al. (39). All experiments employed cells in 3rd to 5th passage.
Levels of RACK1 in cellular fractions from SHR vs. WKY PGVSMCs.
Membrane, cytosolic, nuclear, and cytoskeletal fractions were isolated from SHR and WKY PGVSMCs using the BioVision FractionPREP Cell Fractionation System Kit (BioVision Research Products, Mountain View, CA). This cell fractionation system has been widely used in a variety of cell types, including vascular and airway smooth muscle cells and renal cells, and reliably separates membrane, cytosolic, nuclear, and cytoskeletal fractions (2, 32, 43, 54–56). Briefly, four separate batches (∼22.5 million cells per batch) of WKY and SHR PGVSMCs were treated with trypsin and suspended in PBS. The cells were centrifuged, and the supernatant was replaced with cytosol extraction buffer mix containing protease inhibitors, phosphatase inhibitors, and dithiothreitol (DTT). After 20 min, samples were centrifuged, and the supernatant was collected and used as the cytosolic fraction. The pellet was resuspended in membrane extraction buffer-A containing protease inhibitors, phosphatase inhibitors, and DTT and then treated with membrane extraction buffer-B solution. After 1 min, samples were centrifuged, and the supernatant was collected as the membrane fraction. The pellet was resuspended in nuclear extraction buffer mix containing protease inhibitors, phosphatase inhibitors, and DTT, and after 40 min, the samples were centrifuged, and the supernatant was collected as the nuclear fraction. The remaining pellet was dissolved in sample/lysis buffer and collected as the cytoskeletal fraction. Samples were stored at −80°C until analyzed.
Protein in each sample was determined by the BCA assay (Pierce Biotechnology, Rockford, IL), and then diluted with loading buffer containing β-mercaptoethanol, boiled for 10 min, cooled on ice for 5 min, and then centrifuged. SDS-polyacrylamide-gel electrophoresis was performed on each sample (16 μg of protein per sample). Samples were loaded along with Bio-Rad Precision Plus Protein Standard markers (Bio-Rad; Hercules, CA) onto Invitrogen NuPAGE Novex 12% Bis-Tris gels (1.0 mm, 12 wells) (Life Technologies; Carlsbad, CA). Electrophoresis was performed using NuPAGE MOPS SDS running buffer (Life Technologies), and proteins were transferred onto polyvinylidene difluoride membranes in NuPAGE Transfer Buffer (Life Technologies), and membranes were washed briefly in methanol and blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) and incubated in Odyssey blocking buffer overnight with anti-RACK1 mouse monoclonal antibody (1:400 dilution; Santa Cruz Biotechnology; Santa Cruz, CA; expected size of signal band, 36 kDa) and anti-β-actin rabbit polyclonal antibody (1:10,000; Santa Cruz Biotechnology). Membranes were washed, incubated in Odyssey blocking buffer for 50 min in the dark at room temperature with IRDye 800CW goat anti-mouse IgG (1:10,000; LI-COR Biosciences), and IRDye 680 goat anti-rabbit IgG (1:10,000; LI-COR Biosciences). Membranes were again washed and placed on filter paper to air dry in the dark. Blots were analyzed using the Odyssey infrared imager (LI-COR Biosciences). For quantitative data analysis, rectangles were placed around the RACK1 (green) and β-actin (red) bands, and the integrated intensity values for both red and green channels were determined for each band. Each RACK1 band was normalized to the β-actin band. SHR and WKY samples were run on the same gel for each subcellular fraction.
Effect of RACK1 siRNA on cell growth induced by NPY1–36 and PYY1–36 in SHR PGVSMCs.
SHR PGVSMCs were cultured in DMEM/F12 medium containing 10% FCS, 20 units/ml penicillin, 20 μg/ml streptomycin, and 0.05 μg/ml amphotericin at 37°C with 5% CO2. One day before transfection, 100,000 cells were plated in 500 μl of DMEM/F12 medium containing 2.5% FCS without antibiotics in a 24-well plate. On the day of transfection, 40 pmol of RACK1 siRNA pool or nontargeting siRNA pool (Dharmacon, Lafayette, CO) and 1.5 μl of DharmaFECT 1 (Dharmacon) were diluted to 50 μl in Opti-MEM I medium and incubated for 5 min at room temperature. Then the diluted siRNA and DharmaFECT 1 were combined and incubated for 20 min at room temperature to allow transfection complexes to form. DMEM/F12 medium containing platelet-derived growth factor-BB (PDGF-BB; 25 ng/ml) without antibiotics was added to the complexes (transfection mixture). In some experiments, the transfection mixture included NPY1–36 (10 nM) plus sitagliptin (1 μM) or PYY1–36 (10 nM) plus sitagliptin (1 μM). Sitagliptin was included because PGVSMCs express dipeptidyl peptidase IV (DPPIV), an enzyme that metabolizes and inactivates both NPY1–36 and PYY1–36, and sitagliptin, an inhibitor of DPPIV, augments and prolongs the proliferative effects of both NPY1–36 and PYY1–36 by blocking their metabolism (26). The growth medium was removed from the cells and replaced with the transfection mixture, and the cells were incubated with the transfection mixture at 37°C with 5% CO2. After 72 h, the transfection mixture was replaced with fresh DMEM/F12 medium containing PDGF-BB and other treatments as noted, and at 96 h, cells were dislodged and counted on a Coulter counter.
Assessment of siRNA knockdown of RACK1 mRNA.
RNA was isolated (TRIzol Reagent; Life Technologies), and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad). The RACK1 primers were forward, 5′-gtgctcttcgaggtcactcc-3′, reverse, 5′-cggttgtcagaggagaaagc-3′, and 184-bp amplification product. β-actin primers were forward, 5′-actcttccagccttccttc-3′, reverse, 5′-atctccttctgcatcctgtc-3′, and 171-bp amplification product. Real-time PCR analysis was performed using SYBR Green PCR Master Mix (Applied Biosystems; Foster City, CA) in the AB 7300 real-time PCR System (Applied Biosystems). Threshold cycle (Ct) for β-actin was subtracted from Ct for target to calculate 2ΔCt.
Assessment of siRNA knockdown of RACK1 protein.
Protein was extracted (mammalian protein extraction reagent; Pierce Biotechnology), measured (BCA assay; Pierce) and boiled (5 min in Laemmli buffer). SDS-PAGE was performed on polyacrylamide gels (8–16%) with 40 μg of protein per lane. Proteins were transferred to PDVF membranes. Membranes were blocked in TBS-Tween-20 containing 5% milk and probed with an anti-RACK1 mouse monoclonal primary antibody (1:400; Santa Cruz Biotechnology). Membranes were exposed to horseradish-peroxidase-conjugated-goat anti-mouse antibody (1:4,000; Pierce) and visualized with the Bio-Rad VersaDoc Imaging System using luminal-based enhanced chemiluminescence substrate (Supersignal West Dura Extended Duration Substrate; Pierce).
Cloning of RACK1.
The cDNA fragment encoding rat RACK1 was obtained by PCR using rat PGVSMC cDNA as a template with primer set: sense, 5′-ctaagctatccggtgccatc-3′; antisense, 5′-gcgggtaccaatagtcacctg-3′. The RACK1 cDNA was then cloned into pcDNA 3.1/V5-His-TOPO vector by using pcDNA3.1/V5-His TOPO TA expression kit (Life Technologies) as per the manufacturer's instructions. The clone was verified by DNA sequencing.
Effect of RACK1 plasmid on cell growth induced by NPY1–36 and PYY1–36 in WKY PGVSMCs.
WKY PGVSMCs were cultured in DMEM/F12 medium containing 10% FCS, 20 unit/ml penicillin, 20 μg/ml streptomycin, and 0.05 μg/ml amphotericin at 37°C with 5% CO2. On the day of transfection, 100,000 cells were plated in 500 μl of growth medium without antibiotics in a 24-well plate so that cells would be 60–80% confluent. After cells were attached (2 h), 0.45 μg of RACK1 plasmid DNA or vector control and 2 μl of Plus Reagent (Life Technologies) were diluted to 25 μl in Opti-MEM I medium and incubated for 15 min at room temperature. Two microliters of Lipofectamine (Life Technologies) were diluted to 25 μl in Opti-MEM I medium. Then, the diluted precomplexed plasmid DNA and diluted Lipofectamine were combined and incubated for 30 min at room temperature to allow transfection complexes to form. The complexes (brought to 250 μl with Opti-MEM I) were added into the wells of WKY PGVSMCs, and the cells were incubated for 3.5 h at 37°C with 5% CO2. An equal volume (250 μl) of DMEM/F12 containing 2.5% FCS was added to each well, and the cells were incubated for another 18 h. Next, the transfection complexes were removed and replaced with 500 μl of DMEM/F12 containing PDGF-BB (25 ng/ml), and the cells were incubated for another 96 h in the absence or presence of NPY1–36 (10 nM) plus sitagliptin (1 μM) or PYY1–36 (10 nM) plus sitagliptin (1 μM). Cells were then dislodged and counted on a Coulter counter. RACK1 protein expression level in the transfected WKY PGVSMCs was evaluated by Western blotting (luminal based enhanced chemiluminescence substrate as described above) using a mouse anti-V5 primary antibody (1:1,000; Life Technologies) and a goat anti-mouse IgG, secondary antibody (1:2,500; Thermo Fisher Scientific, Waltham, MA).
Determination of the effects of inhibitors of the Gi/phospholipase C/PKC pathway and downstream signaling molecules on [3H]thymidine incorporation induced by NPY1–36 in SHR PGVSMCs.
SHR PGVSMCs were plated at a density of 5,000 cells per well in 24-well tissue culture dishes and allowed to grow to subconfluence. Cells were growth arrested by feeding DMEM containing 0.4% albumin for 48 h. Next, PGVSMCs were stimulated with DMEM supplemented with PDGF-BB (25 ng/ml) in the presence or absence of various treatments as summarized below. After 20 h of incubation, treatments were repeated with freshly prepared solutions but supplemented with [3H]thymidine (1 μCi/ml). Four hours later, experiments were terminated by washing the cells twice with Dulbecco's PBS and twice with ice-cold trichloroacetic acid (10%). The precipitate was solubilized in 500 μl of 0.3 N NaOH and 0.1% sodium dodecylsulfate by incubating at 50°C for 2 h. Samples were mixed with 10 ml scintillation fluid and counted in a liquid scintillation counter. Treatment groups were as follows: No treatment, NPY1–36 (10 nM), pertussis toxin (100 ng/ml; starting 24 h before the experiment), pertussis toxin (100 ng/ml; starting 24 h before the experiment) + NPY1–36 (10 nM), U73122 (10 μM), U73122 (10 μM) + NPY1–36 (10 nM), GF109203X (10 μM), GF109203X (10 μM) + NPY1–36 (10 nM), PP1 (0.1 μM), PP1 (0.1 μM) + NPY1–36 (10 nM), diphenyleneiodonium (DPI, 10 μM), DPI (10 μM) + NPY1–36 (10 nM), Y27623 (10 μM), Y27623 (10 μM) + NPY1–36 (10 nM), rapamycin (0.2 μM), rapamycin (0.2 μM) + NPY1–36 (10 nM), PD98059 (10 μM), PD98059 (10 μM) + NPY1–36 (10 nM), LY294002 (10 μM), LY294003 (10 μM) + NPY1–36 (10 nM), SB203580 (10 μM), and SB203580 (10 μM) + NPY1–36 (10 nM). Cells treated with NPY1–36 were also cotreated with sitagliptin (1 μM) to block the metabolism and prolong the action of NPY1–36.
Statistical analysis.
Data were analyzed either by two-factor analysis of variance (ANOVA; in which case if and only if the interaction term was statistically significant, post hoc tests were performed with a Fisher's LSD test to characterize the nature of the interaction) or by unpaired Student's t-tests. The criterion of significance was P < 0.05. Data are presented as means ± SE.
RESULTS
Levels of RACK1 in PGVSMC subcellular compartments.
To determine whether subcellular levels of RACK1 in PGVSMCs differ between SHR vs. WKY, subcellular compartments of SHR and WKY PGVSMCs were fractionated and examined for RACK1 expression. Western blotting for RACK1 revealed signals at 36 kDa in the membrane, cytosolic, nuclear and cytoskeletal fractions of PGVSMCs, indicating the presence of RACK1 in all of these subcellular compartments (Fig. 1A). The β-actin signal was not different between SHR and WKY for any of the subcellular fractions. Importantly, RACK1 levels were significantly greater in both SHR membrane fractions (2.5-fold; P < 0.0001) and SHR cytoskeletal fractions (1.8-fold; P = 0.0046) compared with the corresponding WKY fractions (Fig. 1, B and C, respectively); however, RACK1 levels were similar in the nuclear and cytosolic fractions of SHR vs. WKY PGVSMCs (Fig. 1, D and E, respectively).
Fig. 1.

A: Western blots depict subcellular localization of receptor for activated C kinase 1 (RACK1) (green bands; 36 kDa) expression in preglomerular vascular smooth muscle cells (PGVSMCs) from spontaneously hypertensive rats (SHR) vs. Wistar-Kyoto normotensive rats (WKY). Also shown are the signals for β-actin (red bands), which were used as loading controls. Subcellular fractions from SHR vs. WKY were processed on the same gel with the same amount of total protein. Bar graphs depict RACK1 expression in the membrane (B), cytoskeletal (C), nuclear (D), and cytosolic (E) fractions in SHR vs. WKY PGVSMCs. P values are from Student's unpaired t-test. Values are expressed as means ± SE.
Effects of NPY1–36 and PYY1–36 on cell proliferation in SHR PGVSMCs treated with RACK1 siRNA.
To determine whether RACK1 is a critical determinant of the proliferative response to NPY1–36 and PYY1–36 in SHR PGVSMCs, we next examined whether siRNA knockdown of RACK1 would attenuate the proliferative response to NPY1–36 and PYY1–36 in SHR PGV SMCs. These experiments were not performed in WKY PGVSMCs, since in these cells, there was no response to either NPY1–36 or PYY1–36 and, therefore, no response to inhibit. As reported previously in PGVSMCs (11), RACK1 siRNA decreased the expression of both RACK1 mRNA and RACK1 protein (Fig. 2, A and B, respectively). Two-factor analysis of variance (2F-ANOVA) indicated a significant (P = 0.0044) interaction between RACK1 siRNA and NPY1–36 (10 nM) on proliferation of SHR PGVSMCs (Fig. 3A), and post hoc tests indicated that NPY1–36 increased cell number in SHR PGVSMCs transfected with a nontargeting siRNA (control siRNA) but did not affect cell proliferation in SHR PGVSMCs transfected with targeting RACK1 mRNA. 2F-ANOVA also indicated a significant (P = 0.0328) interaction between RACK1 siRNA and PYY1–36 (10 nM) on SHR PGVSMC proliferation (Fig. 3B), and post hoc tests indicated that PYY1–36 increased cell number in SHR PGVSMCs transfected with a control siRNA, but it did not affect cell proliferation in PGVSMCs transfected with targeting RACK1 siRNA.
Fig. 2.

Bar graphs depict effects of RACK1 siRNA vs. negative control siRNA (control) on RACK1 mRNA (A) and protein (B) expression in PGVSMCs from SHR. For both negative control siRNA-treated cells (control) and RACK1 siRNA-treated cells (RACK1 siRNA), expression levels of RACK1 mRNA and protein were normalized to β-actin mRNA or β-actin protein, respectively. For mRNA, this was performed by subtracting the threshold cycle (Ct) for β-actin mRNA from the Ct for RACK1 mRNA to calculate 2ΔCt. For protein, this was performed by dividing the optical density of the RACK1 band by the optical density of the β-actin band. For both mRNA and protein, the readings in the RACK1 siRNA cells were expressed as a % of the average reading for the control cells. Values are expressed as means ± SE, and P values are from unpaired Student's t-test.
Fig. 3.

Bar graphs illustrate the effects of either neuropeptide Y1–36 (NPY1–36; 10 nmol/l) (A) or peptide YY1–36 (PYY1–36; 10 nmol/l) (B) on cell number in PGVSMCs from SHR in the absence and presence of RACK1 siRNA. The PP-fold peptides were coadministered with sitagliptin (1 μM) to block their metabolism and inactivation. P values in panels are from two-factor ANOVA, and letters above bars (a, b, c, d) indicate significantly different [Fisher's least significant difference (LSD) test] from group without RACK1 siRNA and without NPY1–36 or PYY1–36 (a), group without RACK1 siRNA but with NPY1–36 or PYY1–36 (b), group with RACK1 siRNA but without NPY1–36 or PYY1–36 (c), or group with siRNA and with NPY1–36 or PYY1–36 (d). “Control siRNA” indicates nontargeting siRNA. Values are expressed as means ± SE.
Effects of NPY1–36 and PYY1–36 on cell proliferation in WKY PGVSMCs transfected with a RACK1 plasmid.
If RACK1 participates in the proliferative effects of NPY1–36 and PYY1–36 in SHR PGVSMCs, and if reduced levels of RACK1 are responsible for the lack of a proliferative response to these peptides in WKY PGVSMCs, then overexpression of RACK1 in WKY PGVSMCs should reproduce the SHR phenotype in WKY PGVSMCs. Western blot analysis confirmed that transfection with RACK1 plasmid was successful in WKY PGVSMCs (Fig. 4). 2F-ANOVA indicated a significant (P = 0.0160) interaction between RACK1 plasmid and NPY1–36 (10 nM) on proliferation of WKY PGVSMCs (Fig. 5A), and post hoc tests indicated that NPY1–36 increased cell number in WKY PGVSMCs transfected with RACK1 plasmid but did not affect cell proliferation in WKY PGVSMCs transfected with an empty plasmid (control plasmid). 2F-ANOVA also indicated a significant (P = 0.0157) interaction between RACK1 plasmid and PYY1–36 (10 nM) on proliferation of WKY PGVSMCs (Fig. 5B), and post hoc tests indicated that PYY1–36 increased cell number in WKY PGVSMCs transfected with RACK1 plasmid but did not affect cell proliferation in WKY PGVSMCs transfected with control plasmid.
Fig. 4.

The cDNA fragment encoding rat RACK1 was obtained by PCR using rat PGVSMC cDNA as a template with primer set: sense, 5′-ctaagctatccggtgccatc-3′ and antisense, 5′-gcgggtaccaatagtcacctg-3′. The RACK1 cDNA was then cloned into pcDNA 3.1/V5-His-TOPO vector by using pcDNA3.1/V5-His TOPO TA expression kit (Life Technologies) as per the manufacturer's instructions. The clone was verified by DNA sequencing. Western blot depicts expression of V5-tagged RACK1 in WKY PGVSMCs treated with RACK1-expressing plasmid vs. and empty (control) plasmid.
Fig. 5.

Bar graphs illustrate the effects of either NPY1–36 (10 nmol/l; A) or peptide YY1–36 (PYY1–36; 10 nmol/l; B) on cell number in PGVSMCs from WKY in the absence and presence of a RACK1-expressing plasmid. The polypeptide (PP)-fold peptides were coadministered with sitagliptin (1 μM) to block their metabolism and inactivation. P values in panels are from two-factor ANOVA, and letters above bars indicate significantly different (Fisher's LSD test) from group without RACK1-expressing plasmid and without NPY1–36 or PYY1–36 (a), group without RACK1-expressing plasmid but with NPY1–36 or PYY1–36 (b), group with RACK1-expressing plasmid but without NPY1–36 or PYY1–36 (c), or group with RACK1-expressing plasmid and with NPY1–36 or PYY1–36 (d). “Control plasmid” indicates empty plasmid not expressing RACK1. Values are expressed as means ± SE.
Effects of NPY1–36 on [3H]thymidine incorporation in SHR PGVSMCs treated with inhibitors of the Gi/phospholipase C/PKC pathway.
The results described above suggest that RACK1 is critically involved in proliferation induced by PP-fold peptides. Since RACK1 modulates the Gi/phospholipase C/PKC pathway (10, 49), if RACK1 participates in the proliferative response to PP-fold peptides in SHR PGVSMCs, then inhibition of this pathway should attenuate the proliferative effects of PP-fold peptides. Therefore, we examined whether inhibition of this pathway would attenuate the ability of NPY1–36 to stimulate [3H]thymidine incorporation in SHR PGVSMCs. Corresponding experiments were not performed in WKY PGVSMCs because in these cells, there was no response to NPY1–36 and, therefore, no response to inhibit. Because the proliferative effects of NPY1–36 and PYY1–36 are similar, we chose to examine NPY1–36 as representative of the PP-fold family. Also, we used [3H]thymidine incorporation as an index of cell proliferation because of the large number of experiments conducted using this protocol, and measurement of [3H]thymidine incorporation can be performed rapidly.
In SHR PGVSMCs, NPY1–36 (10 nM) increased [3H]thymidine incorporation, and this response was attenuated by pretreatment of PGVSMCs with pertussis toxin [100 ng/ml; inhibitor of Gi-proteins (59); Fig. 6A], U73122 [10 μM; inhibitor of phospholipase C (7); Fig. 6B], and GF109203X [10 μM; inhibitor of PKC (29); Fig. 6C] The conclusion that pertussis toxin, U73122 and GF109203X attenuated the ability of NPY1–36 to increase [3H]thymidine incorporation was supported by the highly significant interaction terms in the 2F-ANOVAs (NPY1–36 × pertussis toxin, P < 0.0001; NPY1–36 × U73122, P = 0.0010; NPY1–36 × GF109203X, P < 0.0001). These findings are consistent with the concept that in SHR PGVSMCs RACK1, by organizing the Gi/phospholipase C/PKC pathway, participates in the proliferative response to PP-fold peptides.
Fig. 6.

Bar graphs illustrate the effects of neuropeptide Y1–36 (NPY1–36; 10 nmol/l) on [3H]thymidine incorporation in PGVSMCs from SHR in the absence and presence of pertussis toxin (A; 100 ng/ml), U73122 (B; 10 μmol/l), and GF109203X (C; 10 μmol/l). NPY1–36 was coadministered with sitagliptin (1 μM) to block its metabolism and inactivation. P values in panels are from two-factor ANOVA, and letters above bars indicate significantly different (Fisher's LSD test) from group without inhibitor and without NPY1–36 (a), group without inhibitor but with NPY1–36 (b), group with inhibitor but without NPY1–36 (c), or group with inhibitor and with NPY1–36 (d). Values are expressed as means ± SE.
Effects of NPY1–36 on [3H]thymidine incorporation in SHR PGVSMCs treated with inhibitors of signal transduction downstream of PKC.
PKC activates an array of downstream signal transduction pathways (either directly or indirectly), including phosphatidylinositol 3-kinases (PI3Ks; Ref. 52), extracellular signal-regulated kinases (ERKs; Ref. 61), mammalian target of rapamycin (mTOR; Refs. 58, 60), and src family kinases (40). Therefore, we also examined the effects of inhibitors of these signal transduction pathways on the ability of NPY1–36 to increase [3H]thymidine incorporation. In SHR PGVSMCs, NPY1–36 (10 nM) increased [3H]thymidine incorporation, and this response was attenuated by pretreatment of PGVSMCs with LY294002 [10 μM; inhibitor of PI3Ks (9); Fig. 7A], PD98059 [10 μM; inhibitor of activation of ERKs (1); Fig. 7B], rapamycin [0.2 μM; mTOR inhibitor (24); Fig. 7C], and PP1 [0.1 μM; src family kinase inhibitor (23); Fig. 7D]. The conclusion that LY294002, PD98059, rapamycin, and PPI attenuated the ability of NPY1–36 to increase [3H]thymidine incorporation was supported by the highly significant interaction terms in the 2F-ANOVAs (NPY1–36 × LY294002, P < 0.0001; NPY1–36 × PD98059, P < 0.0001; NPY1–36 × rapamycin, P < 0.0001; NPY1–36 × PP1, P = 0.0024). These findings are consistent with the concept that RACK1, by organizing the Gi/phospholipase C/PKC pathway, participates in the proliferative response to PP-fold peptides via activation of downstream mediators of PKC.
Fig. 7.

Bar graphs illustrate the effects of neuropeptide Y1–36 (NPY1–36; 10 nmol/l) on [3H]thymidine incorporation in PGVSMCs from SHR in the absence and presence of LY294002 (A; 10 μmol/l), PD98059 (B; 10 μmol/l), rapamycin (C; 0.2 μmol/l), and PP1 (D; 0.1 μmol/l). NPY1–36 was coadministered with sitagliptin (1 μM) to block its metabolism and inactivation. P values in panels are from two-factor ANOVA, and letters above bars indicate significantly different (Fisher's LSD test) from group without inhibitor and without NPY1–36 (a), group without inhibitor but with NPY1–36 (b), group with inhibitor but without NPY1–36 (c), or group with inhibitor and with NPY1–36 (d). Values are expressed as means ± SE.
Effects of NPY1–36 on [3H]thymidine incorporation in SHR PGVSMCs treated with other inhibitors of signal transduction.
Importantly, not all inhibitors altered the ability of NPY1–36 to increase [3H]thymidine incorporation. For example, neither diphenyleneiodonium [10 μM; inhibitor of NADPH oxidases (25)] nor SB203580 [10 μM; inhibitor p38 MAPK (12)] affected the response to NPY1–36 (Fig. 8, A and B, respectively), and although Y27623 [10 μM; inhibitor of Rho-associated protein kinases (57)] statistically reduced the response to NPY1–36, the attenuation was barely detectable (Fig. 8C).
Fig. 8.
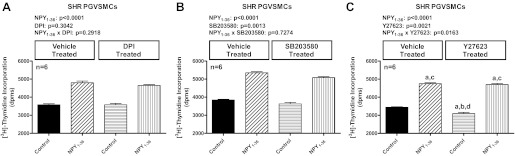
Bar graphs illustrate the effects of NPY1–36 (10 nmol/l) on [3H]thymidine incorporation in PGVSMCs from SHR in the absence and presence of diphenyleneiodonium (A; DPI, 10 μmol/l), SB203580 (B; 10 μmol/l), and Y27623 (C; 10 μmol/l). NPY1–36 was coadministered with sitagliptin (1 μM) to block its metabolism and inactivation. P values in panels are from two-factor ANOVA, and letters above bars indicate significantly different (Fisher's LSD test) from group without inhibitor and without NPY1–36 (a), group without inhibitor but with NPY1–36 (b), group with inhibitor but without NPY1–36 (c), or group with inhibitor and with NPY1–36 (d). Values are expressed as means ± SE.
DISCUSSION
As demonstrated previously (26) and as confirmed in this study, the Y1 receptor agonists NPY1–36 and PYY1–36 stimulate proliferation of SHR PGVSMCs yet do not stimulate proliferation of WKY PGVSMCs. In the present study, we focused our attention on the possible role of RACK1 as a cause for the greater effects of Y1-receptor activation on proliferation of SHR vs. WKY PGVSMCs. The rationale for this hypothesis was that 1) Y1 receptors signal via Gi (38); 2) Gi releases βγ subunits (46); 3) βγ subunits are well known to activate phospholipase C (46); 4) the classical effect of phospholipase C is to activate PKC via DAG and calcium (42); 5) the scaffolding protein RACK1 is well known to scaffold and localize/organize all of these signal transduction components [βγ subunits, phospholipase C and PKC (10, 49)]; and 6) our previous studies suggest a role for RACK1 in regulating the proliferation of PGVSMCs (11). The current study corroborates a role for RACK1 with regard to participating in the differential response of SHR vs. WKY PGVSMCs to Y1 receptor agonists, and this is a novel finding.
We reasoned that increased localization of RACK1 to cell membranes could be involved in the greater response of SHR vs. WKY PGVSMCs to the proliferative effects of Y1-receptor activation. This reasoning was based on the concept that activation of phospholipase C by βγ subunits occurs mainly in the membrane domain, and localization of PKC is known to facilitate the biological effects of PKC (42). Thus, increased localization of RACK1 to cell membranes could be involved in the greater response of SHR vs. WKY PGVSMCs to the proliferative effects of Y1-receptor activation. Another reason to entertain the hypothesis that RACK1 localization is important is that our previous study shows that in PGVSMCs, RACK1 binds to the inositol-1,4,5-trisphosphate receptor and enhances the release of calcium from the sarcoplasmic reticulum (11), and intracellular calcium activates PKC (42).
A novel finding of the present study is that RACK1 levels in the membrane fraction are 2.5-fold higher in SHR PGVSMCs compared with WKY PGVSMCs. This is in marked contrast to the equal levels of RACK1 in the nuclear and cytosolic fractions of SHR vs. WKY PGVSMCs. Although RACK1 levels are also higher in the cytoskeletal fraction of SHR PGVSMCs, the difference between SHR and WKY PGVSMCs in this regard is not as striking compared with the increased RACK1 levels in the membrane fraction. Therefore, it appears that RACK1 expression in SHR PGVSMCs is increased in precisely the location to most efficiently influence cell proliferation.
Besides using identical amounts of total protein in our Western blot experiments, we also employed β-actin as a loading control. Although β-actin is well known to exist in the cytosolic and membrane compartments, β-actin is also connected to the cytoskeletal compartment (50) and is transported into the nucleus (14). Therefore, employing β-actin as a loading control allows for a uniform method of normalizing protein expression across cell types and between compartments. The levels of β-actin did not differ between SHR vs. WKY PGVSMCs, thus indicating that β-actin served as a reliable loading control to normalize between strains.
Our molecular studies corroborate the hypothesis that RACK1 importantly contributes to the sensitivity of SHR PGVSMCs to the proliferative enhancing effects of Y1-receptor activation. In this regard, siRNA-based knockdown of RACK1 abolishes the ability of Y1-receptor activation to enhance proliferation. Corresponding experiments were not performed in WKY PGVSMCs because in these cells there was no response to either NPY1–36 or PYY1–36 and, therefore, no response to inhibit with siRNA knockdown of RACK1.
In the current study, we used a RACK1 plasmid to determine whether overexpression of RACK1 in WKY PGVSMCs could reproduce the SHR phenotype with regard to the proliferative response to Y1-receptor activation. Importantly, overexpression of RACK1 in WKY PGVSMCs increases the proliferation rate of WKY PGVSMCs and renders WKY PGVSMCs sensitive to the proliferative effects of Y1-receptor activation. In control SHR PGVSMCs Y1-receptor activation increases proliferation by 29%, and in WKY PGVSMCs pretreated with RACK1 plasmid, Y1-receptor activation increases proliferation by 32%. Therefore, pretreatment with RACK1 plasmid reproduces the SHR phenotype with respect to sensitivity to the proliferative effects of Y1-receptor activation.
If increased localization of RACK1 to membranes is the main reason that SHR PGVSMCs are more responsive to the proliferative effects of Y1-receptor activation, then inhibition of the βγ/phospholipase C/PKC pathway also should attenuate the ability of Y1-receptor activation to increase the rate of proliferation of SHR PGVSMCs. The results of the current study are in agreement with this prediction. Blocking the βγ/phospholipase C/PKC pathway at the level of any of these three components significantly attenuates the proproliferative effects of Y1-receptor activation in SHR PGVSMCs. Thus, the involvement of the βγ/phospholipase C/PKC pathway is entirely consistent with what is known about RACK1 scaffolding. Moreover, blocking some key downstream mediators of PKC also attenuates the proliferative effects of Y1-receptor activation in SHR PGVSMCs. Importantly, not all pathway inhibitors were effective in this regard, which suggests that the efficacy of those that were effective was specific. It is important to note that corresponding inhibitor experiments were not performed in WKY PGVSMCs because in these cells, there was no response to NPY1–36 and, therefore, no response to inhibit.
Taken together, our results support the model illustrated in Fig. 9. In SHR PGVSMCs, Y1-receptor activation generates the release of βγ subunits from Gi proteins and facilitates the activation of the βγ/phospholipase C/PKC pathway. Increased localization of RACK1 to membranes in SHR PGVSMCs enhances activation of the βγ/phospholipase C/PKC pathway leading to an enhanced proliferative response to Y1-receptor activation. Several downstream mediators are also involved, including ERKs, PI3Ks, src family kinases, and mTOR, but not including ROCK, p38 MAPKs, or NADPH oxidases.
Fig. 9.

Summary of signaling pathway by which PP-fold peptides increase cell proliferation. Components in green circles indicate sites of interventions (noted in red next to the green circles) conducted in this study. DPPIV, dipeptidyl peptidase IV; NPY1–36, neuropeptide Y1–36; PYY1–36, peptide YY1–36; NPY3–36, neuropeptide Y3–36; PYY3–36, peptide YY3–36; Gi, inhibitory G-protein; βγ, βγ subunits of G protein; PLC, phospholipase C; PKC, protein kinase C; DAG, diacyglycerol; IP3, inositol trisphosphate; Ca2+, calcium; RACK1, receptor for activated C kinase 1; ERKs, extracellular-signal-regulated kinases; PI3Ks, phosphatidylinositol 3-kinases; DPI, diphenyleneiodonium; SFKs, src family kinases; mTOR, mammalian target of rapamycin; ROCK, Rho-associated protein kinase; p38 MAPKs, p38 mitogen-activated protein kinases; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidases.
What is the reason for the difference in the subcellular distributions of RACK1 between SHR and WKY PGVSMCs? The answer to this question is unknown but worth exploring because the mechanism of differential RACK1 localization could provide insights into the molecular mechanisms of genetic hypertension. In this regard, Liu et al. (33) recently identified Ser-146 in the 4th WD domain of RACK1 as a phosphorylation site that results in dimerization of RACK1 followed by binding of the RACK1 dimer to a number of other proteins (33). It is conceivable, therefore, that the phosphorylation state of RACK1 determines the localization of RACK1. Another nonmutually exclusive possibility is that some of the binding partners of RACK1 are differentially localized to membranes in SHR PGVSMCs.
Can the findings in the present study be applied to other cell types that participate in the pathophysiology of hypertension, such as smooth muscle cells in different vascular beds or mesangial cells? This is also an important question, and the answer is likely, yes, because recent studies show that RACK1 regulates proliferation of glomerular mesangial cells (11).
What is the clinical significance of the current findings? DPPIV metabolizes incretin hormones, such as gastric inhibitory polypeptide and glucagon-like peptide-1. Consequently, DPPIV inhibitors raise circulating levels of incretin hormones and, thereby, increase insulin release, which results in sustained reductions in HbA1c in Type 2 diabetics (5, 21). However, because there are at least 35 peptide substrates for DPPIV (20, 37), inhibition of DPPIV increases levels of many other biologically active peptides, in addition to incretins, and this may result in long-term consequences. For example, studies by Brown et al. (8) demonstrate that DPPIV inhibitors increase the risk of angioedema in patients treated with angiotensin I-converting enzyme inhibitors, likely because of blockade of substance P metabolism. Because patients with Type 2 diabetes, particular those with the metabolic syndrome, have increased renal sympathetic tone (16, 17) and often ingest fatty meals (18, 36, 45, 47), they likely would have high renal levels of both NPY1–36 and PYY1–36. In this regard, although NPY1–36 and PYY1–36 are potent agonists of Y1 receptors (6, 38), DPPIV efficiently converts PYY1–36 to PYY3–36 and NPY1–36 to NPY3–36 by cleaving two amino acids from the N-termini of PYY1–36 and NPY1–36 (35, 37) (Fig. 9). PYY3–36 and NPY3–36 are inactive at Y1 receptors but are selective Y2-receptor agonists (6, 38). Thus, DPPIV inhibitors chronically increase Y1-receptor activation. DPPIV inhibitors are becoming widely employed to treat patients with Type 2 diabetes, many of whom have hypertension, vascular disease, and renal diseases as comorbidities. Studies by Kirino et al. (27) show that in DPPIV-deficient rats, more so than in DPPIV-expressing rats, streptozotocin-induced diabetes profoundly and chronically reduces creatinine clearance, and case reports are beginning to link DPPIV inhibitors to renal impairment in patients with Type 2 diabetes (30). These findings suggest that in some patients, DPPIV inhibitors may in the long term adversely influence renal structure and function. The present study suggests that the mechanism by which DPPIV inhibitors adversely affect the kidney may involve the βγ/phospholipase C/PKC pathway and that patients with increased membrane localization of RACK1 could be most at risk. Our results also suggest that inhibitors of RACK1 could be useful to prevent adverse renal effects of DPPIV inhibitors, could be antihypertensive, and could prevent or treat hypertension-induced or diabetes-induced nephropathy. Finally, some studies implicate RACK1 in the pathology of cancer (31, 41, 48), and a recent analysis suggests that patients with hypertension are at increased risk of cancer (51). It is conceivable, although speculative, that inhibitors of RACK1 could prevent the adverse effects of DPPIV inhibitors, could lower arterial blood pressure, and could decrease the risk of cancer.
GRANTS
The work was supported by the National Institutes of Health Grants HL-109002, DK-091190, HL-069846, DK-068575, and DK-079307.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: D.C., X.Z., and E.K.J. conception and design of research; D.C., X.Z., and D.G.G. performed experiments; D.C., X.Z., D.G.G., and E.K.J. analyzed data; D.C., X.Z., and E.K.J. interpreted results of experiments; D.C., X.Z., and E.K.J. prepared figures; D.C., X.Z., and E.K.J. drafted manuscript; D.C., X.Z., D.G.G., and E.K.J. edited and revised manuscript; D.C., X.Z., D.G.G., and E.K.J. approved final version of manuscript.
REFERENCES
- 1. Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem 270: 27489– 27494, 1995 [DOI] [PubMed] [Google Scholar]
- 2. Ambasta RK, Ai X, Emerson CP., Jr Quail Sulf1 function requires asparagine-linked glycosylation. J Biol Chem 282: 34492– 34499, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Anini Y, Fu-Cheng X, Cuber JC, Kervran A, Chariot J, Roz C. Comparison of the postprandial release of peptide YY and proglucagon-derived peptides in the rat. Pflügers Arch 438: 299– 306, 1999 [DOI] [PubMed] [Google Scholar]
- 4. Armstrong DN, Ballantyne GH, Adrian TE, Bilchik AJ, McMillen MA, Modlin IM. Adaptive increase in peptide YY and enteroglucagon after proctocolectomy and pelvic ileal reservoir construction. Dis Colon Rectum 34: 119– 125, 1991 [DOI] [PubMed] [Google Scholar]
- 5. Barnett A. DPP-4 inhibitors and their potential role in the management of type 2 diabetes. Int J Clin Pract 60: 1454– 1470, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Berglund MM, Hipskind PA, Gehlert DR. Recent developments in our understanding of the physiological role of PP-fold peptide receptor subtypes. Exp Biol Med 228: 217– 244, 2003 [DOI] [PubMed] [Google Scholar]
- 7. Bleasdale JE, Thakur NR, Gremban RS, Bundy GL, Fitzpatrick FA, Smith RJ, Bunting S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J Pharmacol Exp Ther 255: 756– 768, 1990 [PubMed] [Google Scholar]
- 8. Brown NJ, Byiers S, Carr D, Maldonado M, Warner BA. Dipeptidyl peptidase-IV inhibitor use associated with increased risk of ACE inhibitor-associated angioedema. Hypertension 54: 516– 523, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carpenter CL, Cantley LC. Phosphoinositide kinases. Curr Opin Cell Biol 8: 153– 158, 1996 [DOI] [PubMed] [Google Scholar]
- 10. Chen S, Spiegelberg BD, Lin F, Dell EJ, Hamm HE. Interaction of Gbetagamma with RACK1 and other WD40 repeat proteins. J Mol Cell Cardiol 37: 399– 406, 2004 [DOI] [PubMed] [Google Scholar]
- 11. Cheng D, Zhu X, Barchiesi F, Gillespie DG, Dubey RK, Jackson EK. Receptor for activated protein kinase C1 regulates cell proliferation by modulating calcium signaling. Hypertension 58: 689– 695, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95– 105, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev 77: 75– 197, 1997 [DOI] [PubMed] [Google Scholar]
- 14. Dopie J, Skarp KP, Rajakyla EK, Tanhuanpaa K, Vartiainen MK. Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci USA 109: E544– E552, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dubinion JH, Mi Z, Zhu C, Gao L, Jackson EK. Pancreatic polypeptide-fold peptide receptors and angiotensin II-induced renal vasoconstriction. Hypertension 47: 545– 551, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Esler M, Lambert G, Brunner-La Rocca HP, Vaddadi G, Kaye D. Sympathetic nerve activity and neurotransmitter release in humans: translation from pathophysiology into clinical practice. Acta Physiol Scand 177: 275– 284, 2003 [DOI] [PubMed] [Google Scholar]
- 17. Esler M, Rumantir M, Wiesner G, Kaye D, Hastings J, Lambert G. Sympathetic nervous system and insulin resistance: from obesity to diabetes. Am J Hypertens 14: 304S– 309S, 2001 [DOI] [PubMed] [Google Scholar]
- 18. Freire RD, Cardoso MA, Gimeno SGA, Ferreira SR, Japanese-Brazilian Diabetes Study Dietary fat is associated with metabolic syndrome in Japanese Brazilians. Diabetes Care 28: 1779– 1785, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Fu-Cheng X, Anini Y, Chariot J, Castex N, Galmiche JP, Roze C. Mechanisms of peptide YY release induced by an intraduodenal meal in rats: neural regulation by proximal gut. Pflügers Arch 433: 571– 579, 1997 [DOI] [PubMed] [Google Scholar]
- 20. Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci 108: 277– 292, 2005 [DOI] [PubMed] [Google Scholar]
- 21. Grouzmann E, Livio F, Buclin T. Angiotensin-converting enzyme and dipeptidyl peptidase IV inhibitors: an increased risk of angioedema. Hypertension 54: 468– 470, 2009 [DOI] [PubMed] [Google Scholar]
- 22. Haefliger JA, Waeber B, Grouzmann E, Braissant O, Nussberger J, Nicod P, Waeber G. Cellular localization, expression and regulation of neuropeptide Y in kidneys of hypertensive rats. Regul Pept 82: 35– 43, 1999 [DOI] [PubMed] [Google Scholar]
- 23. Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T-cell activation. J Biol Chem 271: 695– 701, 1996 [DOI] [PubMed] [Google Scholar]
- 24. Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther 2: 222– 232, 2003 [DOI] [PubMed] [Google Scholar]
- 25. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49: 1939– 1945, 2000 [DOI] [PubMed] [Google Scholar]
- 26. Jackson EK, Kochanek SJ, Gillespie DG. Dipeptidyl peptidase IV regulates proliferation of preglomerular vascular smooth muscle and mesangial cells. Hypertension 60: 757– 764, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kirino Y, Sato Y, Kamimoto T, Kawazoe K, Minakuchi K, Nakahori Y. Interrelationship of dipeptidyl peptidase IV (DPP IV) with the development of diabetes, dyslipidaemia and nephropathy: A streptozotocin-induced model using wild-type and DPP IV-deficient rats. J Endocrinol 200: 53– 61, 2008 [DOI] [PubMed] [Google Scholar]
- 28. Korner J, Bessler M, Cirilo LJ, Conwell IM, Daud A, Restuccia NL, Wardlaw SL. Effects of Roux-en-Y gastric bypass surgery on fasting and postprandial concentrations of plasma ghrelin, peptide YY, and insulin. J Clin Endocrinol Metab 90: 359– 365, 2005 [DOI] [PubMed] [Google Scholar]
- 29. Ku WC, Cheng AJ, Wang TC. Inhibition of telomerase activity by PKC inhibitors in human nasopharyngeal cancer cells in culture. Biochem Biophys Res Commun 241: 730– 736, 1997 [DOI] [PubMed] [Google Scholar]
- 30. Lestner JM, Baburaj R, Edwards CMB. Renal impairment with sitagliptin: is there a need for active monitoring of potential renal toxicity? Br J Hosp Med 72: 412– 413, 2011 [DOI] [PubMed] [Google Scholar]
- 31. Li J, Guo Y, Feng X, Wang Z, Wang Y, Deng P, Zhang D, Wang R, Xie L, Xu X, Zhou Y, Ji N, Hu J, Zhou M, Liao G, Geng N, Jiang L, Wang Z, Chen Q. Receptor for activated C kinase 1 (RACK1): a regulator for migration and invasion in oral squamous cell carcinoma cells. J Cancer Res Clin Oncol 138: 563– 571, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li W, Febbraio M, Reddy SP, Yu DY, Yamamoto M, Silverstein RL. CD36 participates in a signaling pathway that regulates ROS formation in murine VSMCs. J Clin Invest 120: 3996– 4006, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu YV, Hubbi ME, Pan F, McDonald KR, Mansharamani M, Cole RN, Liu JO, Semenza GL. Calcineurin promotes hypoxia-inducible factor 1α expression by dephosphorylating RACK1 and blocking RACK1 dimerization. J Biol Chem 282: 37064– 37073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. MacIntosh CG, Andrews JM, Jones KL, Wishart JM, Morris HA, Jansen JB, Morley JE, Horowitz M, Chapman IM. Effects of age on concentrations of plasma cholecystokinin, glucagon-like peptide 1, and peptide YY and their relation to appetite and pyloric motility. Am J Clin Nutr 69: 999– 1006, 1999 [DOI] [PubMed] [Google Scholar]
- 35. McIntosh CH, Demuth HU, Pospisilik JA, Pederson R. Dipeptidyl peptidase IV inhibitors: how do they work as new antidiabetic agents? Regul Pept 128: 159– 165, 2005 [DOI] [PubMed] [Google Scholar]
- 36. Melanson EL, Astrup A, Donahoo WT. The relationship between dietary fat and fatty acid intake and body weight, diabetes, and the metabolic syndrome. Ann Nutr Metab 55: 229– 243, 2009 [DOI] [PubMed] [Google Scholar]
- 37. Mentlein R. Dipeptidyl-peptidase IV (CD26)—role in the inactivation of regulatory peptides. Regul Pept 85: 9– 24, 1999 [DOI] [PubMed] [Google Scholar]
- 38. Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall TXVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol Rev 50: 143– 150, 1998 [PubMed] [Google Scholar]
- 39. Mokkapatti R, Vyas SJ, Romero GG, Mi Z, Inoue T, Dubey RK, Gillespie DG, Stout AK, Jackson EK. Modulation by angiotensin II of isoproterenol-induced cAMP production in preglomerular microvascular smooth muscle cells from normotensive and genetically hypertensive rats. J Pharmacol Exp Ther 287: 223– 231, 1998 [PubMed] [Google Scholar]
- 40. Moyers JS, Bouton AH, Parsons SJ. The sites of phosphorylation by protein kinase C and an intact SH2 domain are required for the enhanced response to beta-adrenergic agonists in cells overexpressing c-src. Mol Cell Biol 13: 2391– 2400, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Myklebust LM, Akslen LA, Varhaug JE, Lillehaug JR. Receptor for activated protein C kinase 1 (RACK1) is overexpressed in papillary thyroid carcinoma. Thyroid 21: 1217– 1225, 2011 [DOI] [PubMed] [Google Scholar]
- 42. Newton AC. Protein kinase C: structure, function, and regulation. J Biol Chem 270: 28495– 28498, 1995 [DOI] [PubMed] [Google Scholar]
- 43. Ohnishi T, Ohba H, Seo KC, Im J, Sato Y, Iwayama Y, Furuichi T, Chung SK, Yoshikawa T. Spatial expression patterns and biochemical properties distinguish a second myo-inositol monophosphatase IMPA2 from IMPA1. J Biol Chem 282: 637– 646, 2007 [DOI] [PubMed] [Google Scholar]
- 44. Pappas TN, Debas HT, Chang AM, Taylor IL. Peptide YY release by fatty acids is sufficient to inhibit gastric emptying in dogs. Gastroenterology 91: 1386– 1389, 1986 [DOI] [PubMed] [Google Scholar]
- 45. Riccardi G, Giacco R, Rivellese AA. Dietary fat, insulin sensitivity and the metabolic syndrome. Clin Nutr 23: 447– 456, 2004 [DOI] [PubMed] [Google Scholar]
- 46. Selbie LA, Hill SJ. G protein-coupled-receptor cross-talk: the fine-tuning of multiple receptor-signalling pathways. Trends Pharmacol Sci 19: 87– 93, 1998 [DOI] [PubMed] [Google Scholar]
- 47. Shaw DI, Hall WL, Williams CM. Metabolic syndrome: what is it and what are the implications? Proc Nutr Soc 64: 349– 357, 2005 [DOI] [PubMed] [Google Scholar]
- 48. Shi S, Deng YZ, Zhao JS, Ji XD, Shi J, Feng YX, Li G, Li JJ, Zhu D, Koeffler HP, Zhao Y, Xie D. RACK1 promotes non-small-cell lung cancer tumorigenicity through activating sonic hedgehog signaling pathway. J Biol Chem 287: 7845– 7858, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sklan EH, Podoly E, Soreq H. RACK1 has the nerve to act: structure meets function in the nervous system. Prog Neurobiol 78: 117– 134, 2006 [DOI] [PubMed] [Google Scholar]
- 50. Song J, Rolfe BE, Campbell JH, Campbell GR. Changes in three-dimensional architecture of microfilaments in cultured vascular smooth muscle cells during phenotypic modulation. Tissue Cell 30: 324– 333, 1998 [DOI] [PubMed] [Google Scholar]
- 51. Stocks T, Van Hemelrijck M, Manjer J, Bjorge T, Ulmer H, Hallmans G, Lindkvist B, Selmer R, Nagel G, Tretli S, Concin H, Engeland A, Jonsson H, Stattin P. Blood pressure and risk of cancer incidence and mortality in the Metabolic Syndrome and Cancer Project. Hypertension 59: 802– 810, 2012 [DOI] [PubMed] [Google Scholar]
- 52. Strouch MB, Jackson EK, Mi Z, Metes NA, Carey GB. Extracellular cyclic AMP-adenosine pathway in isolated adipocytes and adipose tissue. Obes Res 13: 974– 981, 2005 [DOI] [PubMed] [Google Scholar]
- 53. Teixeira FV, Pera M, Kelly KA. Enhancing release of peptide YY after near-total proctocolectomy: jejunal pouch vs. ileal pouch-distal rectal anastomosis. J Gastrointest Surg 5: 108– 112, 2001 [DOI] [PubMed] [Google Scholar]
- 54. Thamilselvan V, Menon M, Thamilselvan S. Oxalate-induced activation of PKC-alpha and -delta regulates NADPH oxidase-mediated oxidative injury in renal tubular epithelial cells. Am J Physiol Renal Physiol 297: F1399– F1410, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Townsend EA, Thompson MA, Pabelick CM, Prakash YS. Rapid effects of estrogen on intracellular Ca2+ regulation in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 298: L521– L530, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Trapasso F, Pichiorri F, Gaspari M, Palumbo T, Aqeilan RI, Gaudio E, Okumura H, Iuliano R, Di Leva G, Fabbri M, Birk DE, Raso C, Green-Church K, Spagnoli LG, Venuta S, Huebner K, Croce CM. Fhit interaction with ferredoxin reductase triggers generation of reactive oxygen species and apoptosis of cancer cells. J Biol Chem 283: 13736– 13744, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389: 990– 994, 1997 [DOI] [PubMed] [Google Scholar]
- 58. Velazquez-Garcia S, Valle S, Rosa TC, Takane KK, Demirci C, Alvarez-Perez JC, Mellado-Gil JM, Ernst S, Scott DK, Vasavada RC, Alonso LC, Garcia-Ocana A. Activation of protein kinase C-zeta in pancreatic β-cells in vivo improves glucose tolerance and induces β-cell expansion via mTOR activation. Diabetes 60: 2546– 2559, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vyas SJ, Mokkapatti R, Dubey RK, Chinoy MR, Jackson EK. Guanine nucleotide-binding inhibitory protein-mediated inhibition of adenylyl cyclase is enhanced in spontaneously hypertensive rat preglomerular arteriolar smooth muscle cells. J Pharmacol Exp Ther 285: 828– 834, 1998 [PubMed] [Google Scholar]
- 60. Wang X, Yu W, Nawaz A, Guan F, Sun S, Wang C. Palmitate induced insulin resistance by PKCtheta-dependent activation of mTOR/S6K pathway in C2C12 myotubes. Exp Clin Endocrinol Diabetes 118: 657– 661, 2010 [DOI] [PubMed] [Google Scholar]
- 61. Wu X, Zhu M, Fletcher JA, Giobbie-Hurder A, Hodi FS. The protein kinase C inhibitor enzastaurin exhibits antitumor activity against uveal melanoma. PLoS One 7: e29622, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]