

Abstract
Diabetes is a risk factor for the development of acute kidney injury (AKI) in humans and rodents. However, the mechanistic basis for this observation is unknown. The present studies evaluated the role of inflammation and TNF-α in ischemic AKI in a model of type 2 diabetes mellitus (DM). Diabetic (db/db) and nondiabetic (db/+) littermates were subjected to 20 min of bilateral renal ischemia. The nondiabetic mice developed only mild and transient renal dysfunction. In contrast, the equivalent ischemic insult provoked severe and sustained renal dysfunction in the db/db mice. The expression of TNF-α and Toll-like receptor 4 (TLR4) mRNA was measured in the kidneys of diabetic mice before and after renal ischemia; db/db mice exhibited greater increases in TNF-α and TLR4 mRNA expression following ischemia than did db/+. In addition, urinary excretion of TNF-α after ischemia was higher in db/db mice than in db/+ mice. To determine the possible role of TNF-α in mediating the enhanced susceptibility of diabetic mice to ischemic injury, db/db mice were injected with either a neutralizing anti-mouse TNF-α antibody or nonimmune globulin and then subjected to 20 min of bilateral renal ischemia. Treatment of the db/db mice with the TNF-α antibody provided significant protection against the ischemic injury. These data support the view that diabetes increases the susceptibility to ischemia-induced renal dysfunction. This increased susceptibility derives from a heightened inflammatory response involving TNF-α and perhaps TLR4 signaling.
Keywords: diabetes, ischemia, acute kidney injury, TNF
an increased susceptibility of the diabetic kidney to ischemic insults has been reported in experimental models of diabetes and in humans with diabetes (17, 25, 31, 47). Studies in rats with streptozotocin (STZ)-induced type 1 diabetes indicated that diabetic rats had significantly greater histological damage, tubular apoptosis, and long-term fibrosis after ischemia than nondiabetic rats (31, 32). Treatment with insulin prior to the ischemic event reduced ischemic injury whereas administration of insulin after the ischemic insult had no beneficial effect (32). A study in a murine model of type 2 diabetes mellitus (DM) (db/db) also demonstrated an increased risk for ischemic acute renal failure (47). In that study, a delayed recovery of renal blood flow after the ischemic event was noted in the diabetic mice. However, the mechanisms by which diabetes increases the risk of ischemic acute kidney injury (AKI) are unclear.
AKI is associated with the production of proinflammmatory cytokines and chemokines within the kidney (27, 43), upregulation of leukocyte adhesion molecules, and infiltration of a variety of inflammatory cells into the kidney (2, 42). Although infiltrating leukocytes can elaborate inflammatory cytokines, renal epithelial cells are also capable of producing a variety of cytokines in response to ischemic or toxic injury. Several of these chemokines and cytokines, e.g., IL-6, TNF-α, CCL2/MCP-1, CXCL1, and CXCL10/IP-10, have themselves been shown to contribute to either ischemic or toxic acute renal failure (9, 10, 20, 33, 43).
A growing literature supports a link between inflammation and obesity and insulin resistance (reviewed in Ref. 22). A number of inflammatory cytokines, such as TNF-α and IL-6, are produced by adipocytes and cause insulin resistance (21, 24). Toll-like receptor 4 (TLR4) may be a key mediator of cytokine production in adipocytes (46, 48). Elevated levels of TNF-α and IL-6 have also been noted in the kidney in rodent models of diabetes (16, 37) and in the urine of diabetic animals (7, 37) and diabetic humans (38, 39). Elevated inflammatory cytokines have been implicated in the long-term renal complications of diabetes such as proteinuria and renal hypertrophy (8, 39), but their role in acute diabetic kidney diseases has not been examined. The present studies addressed the hypothesis that diabetes increases the susceptibility to ischemic AKI by enhancing the production of proinflammatory cytokines within the kidney.
METHODS
Renal ischemia-reperfusion.
Experiments were performed using 8-wk-old male diabetic db/db (BKS.Cg-m Leprdb/+ +/J, Jackson Labs) and nondiabetic db/+ littermates. Before conducting experiments, mice were examined for hyperglycemia (VITROS GLU slides, Ortho-clinical Diagnostics). Renal ischemia-reperfusion injury (IRI) was induced by subjecting mice to 20 min of bilateral renal IRI. Briefly, db/db and db/+ mice were anesthetized with pentobarbital sodium and were placed on a heating pad to maintain core temperature at 37°C. Both renal pedicles were identified through dorsal incisions and clamped for 20 min. Reperfusion was confirmed visually upon release of the clamps. Sham-operated mice were subjected to the same surgical procedure except the renal pedicles were not clamped. Surgical wounds were closed and mice were given 1 ml of warm saline intraperitoneally and kept in a warm incubator until they regained consciousness. When indicated, mice were administered 800 μg/kg body wt of goat anti-TNF antibody or normal goat IgG (R and D Systems) intraperitoneally 2 h before surgery. All animal protocols were approved by the Penn State University Animal Care and Use Committee.
Renal function.
Renal function was assessed by measurements of blood urea nitrogen (VITROS DT60II Chemistry slides, Ortho-clinical Diagnostics) and serum creatinine (cat. no. DZ072B, Diazyme Labs).
Quantitation of mRNA by real-time RT-PCR.
Real-time RT-PCR was performed in an Applied Biosystems 7700 Sequence Detection System (Foster City, CA). RNA (1.5 μg total) was reverse transcribed in a reaction volume of 20 μl using Omniscript RT kit and random primers. The product was diluted to a volume of 150 μl and either 2-μl (actin) or 10-μl (all others) aliquots were used as templates for amplification using the SYBR Green PCR amplification reagent (Qiagen) and gene-specific primers. The primer sets used were mouse TNF-α (forward: GCATGATCCGCGACGTGGAA; reverse: AGATCCATGCCGTTG GCCAG); TNF-α receptor 1 (TNFR1) (forward: CCGGGCCACCTGGTCCG; reverse: CAAGTAGGTTCCTTTGTG); TNFR2 (forward: GTCGCGCTGGTCTTCGAACTG; reverse: GGTATACATGCTTGCCTCACAGTC); TLR4 (forward: CCTCTGCCTTCACTACAGAGACTTT; reverse: TGTGGAAGCCTTCCTGGAG); heme oxygenase-1 (forward: AGCATGCCCCAGGATTTG; reverse: AGCTCAATGTTGAGCAGGA); CXCL1 (forward: GCTGGGATTCACCTCAAGAA; reverse: TGGGGACACCTTTTAGCATC); CCL2 (forward: ATGCAGGTCCCTGTCATG; reverse: GCTTGAGGTGGTTGTGGA); G-CSF (forward: CCAGGCCCTGCAGCAGACAC; reverse: TGGTGCAGAGCAAGGCGAGC); IL-6 (forward: GATGCTACCAAACTGGATATAATC; reverse: GGTCCTTAGCCACTCCTTCTG TG); CXCL10/IP-10 (forward: GGTCTGAGTGGGACTCAAGG; reverse: CGTGGCAATGATCTCAACAC); and ICAM-1 (forward: AGATCACATTCACGGTGCTG; reverse: CTTCAGAGGCAGGAAACAGG). The amount of DNA was normalized to the β-actin signal amplified in a separate reaction (forward: AGAGGGAAATCGTGCGTGAC; reverse: CAATAGTGATGACCTGGCCGT).
Cytokine/chemokine quantitation.
Urine cytokines and chemokines (TNF-α, IL-1β, IL-2, IL-6, G-CSF, CXCL1, CXCL10/IP-10, MCP-1/CCL2, and RANTES) were measured using a bead-based multiplexed cytokine analysis kit (Linco Research, St. Charles, MO) using a Luminex-100 system (Luminex, Austin, TX). Assays were run in duplicate according to the manufacturer's protocol, and data were collected and analyzed using MasterPlex QT 2.5 software (MiraiBio, Alameda, CA). Amounts of cytokines in the urine were normalized to the creatinine concentration in the urine.
Histology and immunohistochemistry.
Kidney tissue was fixed in buffered formalin for 24 h and then embedded in paraffin wax. Five-micrometer sections were stained with periodic acid-Schiff (PAS) for assessment of tubular injury or with rat anti-mouse neutrophil antibody. Tubular injury was assessed in PAS-stained sections using a semiquantitative scale in which the percentage of cortical tubules showing epithelial necrosis was assigned a score: 0 = normal; 1 = <10%; 2 = 10–25%; 3 = 26–75%; 4 = >75%. Immunohistochemistry for neutrophils was performed with a rat anti-murine neutrophil primary antibody (MCA 771 GA, SeroTec, Raleigh, NC) followed by a biotinylated anti-rat IgG secondary antibody. Signal was developed using the avidin/biotin complex peroxidase method (Pierce, Rockford, IL). Thirty ×40 fields from each kidney were examined for quantitation of neutrophils. The individual scoring the slides was blinded to the treatment and strain of the animal.
Statistical methods.
All assays were performed in duplicate. The data are reported as means ± SE. Statistical significance was assessed by an unpaired, two-tailed Student's t-test for single comparison or ANOVA for multiple comparisons.
RESULTS
Renal ischemia was induced in male db/db (BKS.Cg-m Leprdb/+ +/J, Jackson Labs) and nondiabetic db/+ littermates (8 wk old). The db/db mice typically develop hyperglycemia between 4 and 8 wk of age (15); 8 wk of age was chosen for this study to allow for the development of hyperglycemia but not chronic changes of diabetic nephropathy. Blood glucose values in the db/db mice at the time of ischemia were 448 ± 66 mg/dl compared with 192 ± 32 mg/dl in the db/+ mice. Both renal pedicles were clamped for 20 min followed by reperfusion. The ischemic interval of 20 min was selected as it causes little renal dysfunction in nondiabetic mice. The blood urea nitrogen (BUN) and plasma creatinine were measured over the next 24 h as a measure of renal function. As seen in Fig. 1, the db/db mice subjected to 20 min of ischemia developed marked renal dysfunction while db/+ mice maintained relatively normal renal function.
Fig. 1.

Diabetic mice are more susceptible to ischemic acute kidney injury (AKI). The db/db and db/+ mice were subjected to 20 min of bilateral renal ischemia or sham surgery. Blood collected at various time points with respect to surgery was analyzed for blood urea nitrogen (BUN; A) and serum creatinine (B) as measures of renal function. I/R, ischemia-reperfusion. Values are means ± SE. *P < 0.025 vs. other groups; n = 4–19.
Kidneys from db/+ and db/db mice subjected to sham surgery were histologically normal (Fig. 2A). Kidneys from db/+ mice subjected to ischemia revealed occasional dilated tubules and casts. Kidneys from the db/db mice subjected to ischemia demonstrated significantly greater injury including frequent sloughing of tubular epithelial cells and cast formation. The tubular necrosis scores for the ischemic db/+ mice (0.8 ± 0.2) were significantly lower than for the ischemic db/db mice (2.5 ± 0.2, P < 0.001, n = 6–7, Fig. 2B). Ischemia induces apoptosis of renal epithelial cells (13). As shown in Fig. 3A, the activation of caspase 3, as reflected by cleavage to its active form, was greater in ischemic db/db kidneys than in ischemic db/+ kidneys. Likewise, the number of apoptotic cells was much greater in kidneys from db/db mice than db/+ mice (Fig. 3B).
Fig. 2.

Ischemia induces significantly more kidney injury in db/db mice than db/+ mice. A: periodic acid-Schiff (PAS)-stained kidney tissue sections from db/+ and db/db mice subjected to bilateral renal ischemia or sham surgery. B: quantitation of tubular injury. *P < 0.005 vs. db/+; n = 3–7.
Fig. 3.
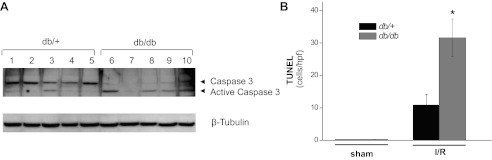
Diabetes increases caspase 3 activation and apoptosis of renal cells in ischemic AKI. The db/db and db/+ mice were subjected to 20 min of bilateral renal ischemia or sham surgery. Kidneys harvested at 24 h of reperfusion were examined for caspase 3 activation by Western blot analysis (A) and for apoptotic cells by TUNEL staining (B). *P = 0.002 vs. db/+; n = 3–9.
Since inflammation is an important mediator of ischemic injury (2, 42), and since diabetes is characterized by an increase in inflammatory mediators, we examined the infiltration of neutrophils and the expression of certain inflammatory mediators in the kidneys of diabetic mice before and after renal ischemia. Immunohistochemical staining for neutrophils revealed few neutrophils in sham-operated kidneys of either strain (Fig. 4). Renal ischemia induced significantly greater accumulation of neutrophils in the peritubular capillaries in db/db kidneys compared with db/+ kidneys. Likewise, db/db mice exhibited greater increases in the expression of TNF-α, TNFR2, CXCL1 (a neutrophil chemokine), G-CSF, CXCL10/IP-10, IL-6, and TLR4 expression following ischemia (Fig. 5) than did db/+ littermates. In addition, the urinary excretion of several chemokines and cytokines, including TNF-α, MCP-1/CCL2, CXCL10/IP-10, IL-6, and G-CSF, after ischemia was higher in db/db mice than in db/+ mice, particularly at later time points (Fig. 6). The levels of urine cytokines did not always parallel renal cytokine gene expression. This discrepancy could be due to nonrenal sources of urine cytokines or posttranscriptional regulation of cytokine production.
Fig. 4.

Increased neutrophil infiltration into diabetic kidneys after ischemia. The db/db and db/+ mice were subjected to 20 min of bilateral renal ischemia or sham surgery. A: immunohistochemical staining of neutrophils in kidneys harvested 24 h after surgery. B: summary data of neutrophil infiltration in kidney cortex. *P < 0.005 vs. db/+; n = 3–7.
Fig. 5.

Diabetes increases expression of proinflammatory proteins in ischemic AKI. The db/db and db/+ mice were subjected to 20 min of bilateral renal ischemia or sham surgery. Kidneys harvested 24 h after surgery were used for real-time RT-PCR quantitation of mRNA. Data are expressed as fold changes relative to control kidneys from mice of the same strain. *P < 0.05 vs. sham surgery; n = 3–5.
Fig. 6.
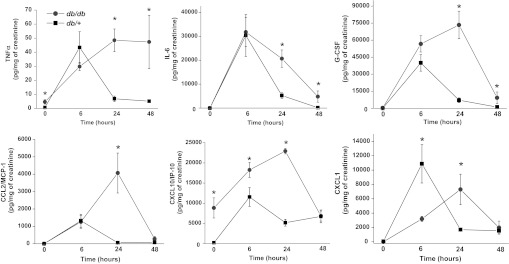
Effect of ischemia in diabetic mice on urinary cytokines and chemokines. The db/db (gray) and db/+ (black) mice were subjected to 20 min of bilateral renal ischemia. Urine cytokines and chemokines were determined by Luminex assay. *P < 0.05 vs. db/+; n = 4.
We had previously demonstrated that TNF-α, acting via TNFR2 expressed on kidney cells, is a key mediator of drug-induced renal injury (43, 44). Increased levels of TNF-α have also been reported in the diabetic kidney (16, 37) and in urine of diabetic individuals (38, 39). As shown above, db/db mice exhibited higher renal expression of TNF-α and higher urinary levels of TNF-α. Therefore, we examined the possible role of TNF-α in mediating the enhanced susceptibility of diabetic mice to ischemic injury; db/db mice were injected with either a neutralizing anti-mouse TNF-α antibody or nonimmune globulin and then subjected to 20 min of bilateral renal ischemia. As shown in Fig. 7, the db/db mice injected with nonimmune globulin developed severe renal failure while the db/db mice injected with neutralizing antibody had minimal renal dysfunction. As demonstrated earlier, db/+ mice subjected to 20 min of ischemia developed minimal renal dysfunction.
Fig. 7.

TNF-α neutralization reduces renal ischemic injury in diabetic mice. The db/db mice were injected with goat IgG or goat anti-mouse TNF-α antibody and then subjected to 20 min of bilateral renal ischemia. +P < 0.01 vs. db/+; *P < 0.01 vs. db/db + IgG; n = 4–5.
DISCUSSION
Increased susceptibility of the kidney to ischemic insults has been reported in experimental models of diabetes and in clinical studies involving patients with diabetes (17, 25, 31, 32, 47). The mechanisms underlying this susceptibility remain unclear. Previous work has demonstrated an increase in the expression of certain inflammatory cytokines in the diabetic kidney (37, 49). In nondiabetic animals, renal ischemia results in the upregulation of a number of proinflammatory chemokines and cytokines and infiltration of the kidney by certain leukocyte populations, including T cells, neutrophils, macrophages, and NK cells (1, 27, 28, 51). This inflammatory response plays a key role in producing renal dysfunction since deletion or inhibition of certain cytokines or depletion of leukocyte subtypes prevents or mitigates ischemic AKI (9, 10, 20, 33, 43). The results reported here in a mouse model of type 2 diabetes confirm that diabetes increases the susceptibility to ischemic AKI and implicate a hyperresponsive inflammatory response as a cause of this susceptibility. Specifically, we found that modest ischemia markedly enhanced the renal expression and/or urinary excretion of a number of proinflammatory chemokines and cytokines in diabetic but not nondiabetic mice. Several of these chemokines and cytokines, e.g., TNF-α, IL-6, CCL2/MCP-1, CXCL1, and CXCl10/IP-10, have been shown to contribute to either ischemic or toxic AKI (9, 10, 20, 33, 43), although their role in ischemic AKI in the setting of diabetes has not been examined.
Our previous studies in mice lacking TNF-α or its receptors established that the TNF-α mediates chemokine and cytokine production and renal injury in drug-induced nephrotoxicity (43, 44). Likewise, neutralizing antibodies to TNF-α reduce renal ischemic injury in mice (4). Kalantarinia et al. (16) demonstrated increases in renal interstitial TNF-α levels within weeks after the induction of diabetes in rats and which preceded the onset of albuminuria. In diabetic rats, levels of kidney TNF-α mRNA expression correlate with urine albumin excretion (36). In humans, urinary levels of TNF-α are elevated in diabetes and correlate with markers of diabetic nephropathy (40). In addition, elevated levels of soluble TNFR1 and TNFR2 in the blood are associated with increased risk of progressive diabetic kidney disease (11, 41). These observations suggest that TNF-α may have a pathogenic role in chronic diabetic nephropathy (35). They also raise the possibility that increased levels of TNF-α in the diabetic kidney predispose to acute ischemic injury. Indeed, our results support this view. Modest durations of ischemia induced higher expression of TNF-α mRNA and resulted in higher levels of TNF-α in the urine of diabetic mice compared with nondiabetic littermates. Moreover, neutralization of TNF-α largely negated the increased susceptibility of the diabetic mice to ischemic kidney injury, establishing the pathogenic significance of TNF-α in diabetic renal ischemia.
The mechanism for the observed increase in TNF-α production in the diabetic kidneys is not known. Activation of TLR2 and/or TLR4 expressed on renal epithelial cells contributes to TNF-α production in both ischemic and nephrotoxic AKI (26, 50, 52). Circulating monocytes from diabetic humans express higher levels of TLR2 and TLR4 and secrete more TNF-α than monocytes from nondiabetic subjects (5, 6). In mice with type 1 DM, bone marrow-derived macrophages show increased TLR2 and TLR4 expression and increased cytokine production in response to TLR4 ligands (34). Renal epithelial cells from diabetic kidneys were recently reported to express higher levels of TLR4 than nondiabetic renal epithelial cells (29). Likewise, in the present study we found that renal TLR4 expression was upregulated to a greater extent in diabetic mice than nondiabetic mice. Thus upregulation of TLRs in kidney and/or hematopoietic cells might account for the enhanced inflammatory response of the diabetic kidney to ischemia.
An enhanced inflammatory response may be a common mechanism for susceptibility to ischemic injury in diabetes. In addition to renal ischemic injury, diabetic mice are also more susceptible to cardiac (12, 30) and cerebral IRI (23). A role for TLR4 and TNF-α in the increased susceptibility to myocardial ischemia has been proposed. Thus expression of TLR4 and TNF-α is increased in the ischemic myocardium of diabetic animals compared with nondiabetic animals (14, 30). In addition, elevated plasma TNF-α levels after myocardial infarction are associated with increased risk of recurrent myocardial ischemia in humans (45).
In addition to the substantial morbidity and mortality associated with acute ischemic kidney injury, there is increasing recognition that ischemic AKI may have long-term deleterious effects on renal function (3). This relationship may be particularly strong in diabetes. Kelly et al. (18) determined that diabetic rats subjected to renal ischemia develop progressive chronic renal failure and proteinuria. Of note, ischemia resulted in persistent renal expression of inflammatory mediators and infiltration of leukocytes. The importance of this inflammatory response to the chronic injury was demonstrated using anti-inflammatory drugs which reduced fibrosis and renal dysfunction (19). Thus inflammation may increase the risk of the diabetic individual to both acute ischemic injury and its progression to chronic kidney disease.
In summary, we have determined the impact of diabetes on ischemic kidney injury and examined the role of endogenous TNF-α in ischemia-induced acute kidney injury in diabetic mice. Diabetes increases the susceptibility to ischemic kidney injury, and endogenous TNF-α produced in response to ischemia exacerbates renal dysfunction in diabetes. These results demonstrate an important role for TNF-α in ischemic kidney injury in diabetes. Given the availability of TNF-α inhibitors and neutralizing antibodies, these findings are potentially of clinical importance to both the acute and chronic renal complications of diabetes. Additional studies to investigate the role of TNF-α in human diabetic nephropathy are warranted.
GRANTS
This work was supported by funding from the Penn State Institute for Obesity and Diabetes and the National Institute of Diabetes and Digestive and Kidney Diseases (RO1-DK-063120, R01-DK-083379 and DK-081876) and the Pennsylvania Department of Health using Tobacco Settlement Funds.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: G.G., B.Z., G.R., D.B., R.T., and W.W. performed experiments; G.G., R.T., and W.B.R. analyzed data; G.G., R.T., and W.B.R. prepared figures; G.G., G.R., and W.B.R. edited and revised manuscript; G.G., B.Z., G.R., D.B., R.T., W.W., and W.B.R. approved final version of manuscript; G.R. and W.B.R. conception and design of research; W.B.R. interpreted results of experiments; W.B.R. drafted manuscript.
ACKNOWLEDGMENTS
Portions of this project were completed in satisfaction of the Medical Student Research Project requirement at the Penn State College of Medicine (D. Betterly).
Present address of G. Ramesh: Dept. of Medicine, Georgia Health Sciences Univ., Augusta, GA.
REFERENCES
- 1.Ascon DB, Lopez-Briones S, Liu M, Ascon M, Savransky V, Colvin RB, Soloski MJ, Rabb H. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J Immunol 177: 3380–3387, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int 66: 480–485, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Chawla LS, Kimmel PL. Acute kidney injury and chronic kidney disease: an integrated clinical syndrome. Kidney Int 82: 516–524, 2012 [DOI] [PubMed] [Google Scholar]
- 4.Daemen M, Ven M, Heineman E, Buurman W. Involvement of endogenous interleukin-10 and tumor necrosis factor-α in renal ischemia-reperfusion injury. Transplantation 67: 792–799, 1999 [DOI] [PubMed] [Google Scholar]
- 5.Dasu MR, Devaraj S, Park S, Jialal I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 33: 861–868, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, Jialal I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: further evidence of a proinflammatory state. J Clin Endocrinol Metab 93: 578–583, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiPetrillo K, Coutermarsh B, Gesek FA. Urinary tumor necrosis factor contributes to sodium retention and renal hypertrophy during diabetes. Am J Physiol Renal Physiol 284: F113–F121, 2003 [DOI] [PubMed] [Google Scholar]
- 8.DiPetrillo K, Gesek FA. Pentoxifylline ameliorates renal tumor necrosis factor expression, sodium retention, and renal hypertrophy in diabetic rats. Am J Nephrol 24: 352–359, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Fiorina P, Ansari MJ, Jurewicz M, Barry M, Ricchiuti V, Smith RN, Shea S, Means TK, Auchincloss H, Jr, Luster AD, Sayegh MH, Abdi R. Role of CXC chemokine receptor 3 pathway in renal ischemic injury. J Am Soc Nephrol 17: 716–723, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Furuichi K, Wada T, Iwata Y, Kitagawa K, Kobayashi Ki, Hashimoto H, Ishiwata Y, Tomosugi N, Mukaida N, Matsushima K, Egashira K, Yokoyama H. Gene therapy expressing amino-terminal truncated monocyte chemoattractant protein-1 prevents renal ischemia-reperfusion injury. J Am Soc Nephrol 14: 1066–1071, 2003 [DOI] [PubMed] [Google Scholar]
- 11.Gohda T, Niewczas MA, Ficociello LH, Walker WH, Skupien J, Rosetti F, Cullere X, Johnson AC, Crabtree G, Smiles AM, Mayadas TN, Warram JH, Krolewski AS. Circulating TNF receptors 1 and 2 predict stage 3 CKD in Type 1 diabetes. J Am Soc Nephrol 23: 516–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greer JJ, Ware DP, Lefer DJ. Myocardial infarction and heart failure in the db/db diabetic mouse. Am J Physiol Heart Circ Physiol 290: H146–H153, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int 80: 29–40, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu ZC, Chen YD, Ren YH. Methylprednisolone improves microcirculation in streptozotocin-induced diabetic rats after myocardial ischemia/reperfusion. Chin Med J (Engl) 124: 923–929, 2011 [PubMed] [Google Scholar]
- 15.Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science 153: 1127–1128, 1966 [DOI] [PubMed] [Google Scholar]
- 16.Kalantarinia K, Awad AS, Siragy HM. Urinary and renal interstitial concentrations of TNF-a increase prior to the rise in albuminuria in diabetic rats. Kidney Int 64: 1208–1213, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Katzberg RW, Barrett BJ. Risk of iodinated contrast material-induced nephropathy with intravenous administration. Radiology 243: 622–628, 2007 [DOI] [PubMed] [Google Scholar]
- 18.Kelly KJ, Burford JL, Dominguez JH. Postischemic inflammatory syndrome: a critical mechanism of progression in diabetic nephropathy. Am J Physiol Renal Physiol 297: F923–F931, 2009 [DOI] [PubMed] [Google Scholar]
- 19.Kelly KJ, Dominguez JH. Treatment of the post-ischaemic inflammatory syndrome of diabetic nephropathy. Nephrol Dial Transplant 25: 3204–3212, 2010 [DOI] [PubMed] [Google Scholar]
- 20.Kielar ML, John R, Bennett M, Richardson JA, Shelton JM, Chen L, Jeyarajah DR, Zhou XJ, Zhou H, Chiquett B, Nagami GT, Lu CY. Maladaptive role of IL-6 in ischemic acute renal failure. J Am Soc Nephrol 16: 3315–3325, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Kim HJ, Higashimori T, Park SY, Choi H, Dong J, Kim YJ, Noh HL, Cho YR, Cline G, Kim YB, Kim JK. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 53: 1060–1067, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Kim JK. Fat uses a TOLL-road to connect inflammation and diabetes. Cell Metab 4: 417–419, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Kumari R, Willing LB, Patel SD, Baskerville KA, Simpson IA. Increased cerebral matrix metalloprotease-9 activity is associated with compromised recovery in the diabetic db/db mouse following a stroke. J Neurochem 119: 1029–1040, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lang CH, Dobrescu C, Bagby GJ. Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 130: 43–52, 1992 [DOI] [PubMed] [Google Scholar]
- 25.Leblanc M, Kellum JA, Gibney RT, Lieberthal W, Tumlin J, Mehta R. Risk factors for acute renal failure: inherent and modifiable risks. Curr Opin Crit Care 11: 533–536, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Leemans JC, Stokman G, Claessen N, Rouschop KM, Teske GJD, Kirschning CJ, Akira S, van der Poll T, Weening JJ, Florquin S. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest 115: 2894–2903, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lemay S, Rabb H, Postler G, Singh A. Prominent and sustained up-regulation of GP130-signaling cytokines and of the chemokine MIP-2 in murine renal inschemia-reperfusion injury. Transplantation 69: 959–963, 2000 [DOI] [PubMed] [Google Scholar]
- 28.Li L, Huang L, Sung SsJ, Lobo PI, Brown MG, Gregg RK, Engelhard VH, Okusa MD. NKT Cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol 178: 5899–5911, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC, Au WS, Chan KW, Lai KN, Tang SC. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol 23: 86–102, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marfella R, Di Filippo C, Portoghese M, Siniscalchi M, Martis S, Ferraraccio F, Guastafierro S, Nicoletti G, Barbieri M, Coppola A, Rossi F, Paolisso G, D'Amico M. The ubiquitin-proteasome system contributes to the inflammatory injury in ischemic diabetic myocardium: the role of glycemic control. Cardiovasc Pathol 18: 332–345, 2009 [DOI] [PubMed] [Google Scholar]
- 31.Melin J, Hellberg O, Akyurek LM, Kallskog O, Larsson E, Fellstrom BC. Ischemia causes rapidly progressive nephropathy in the diabetic rat. Kidney Int 52: 985–991, 1997 [DOI] [PubMed] [Google Scholar]
- 32.Melin J, Hellberg O, Larsson E, Zezina L, Fellstrom BC. Protective effect of insulin on ischemic renal injury in diabetes mellitus. Kidney Int 61: 1383–1392, 2002 [DOI] [PubMed] [Google Scholar]
- 33.Miura M, Fu X, Zhang QW, Remick DG, Fairchild RL. Neutralization of Gro-alpha and macrophage inflammatory protein-2 attenuates renal ischemia/reperfusion injury. Am J Pathol 159: 2137–2145, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mohammad MK, Morran M, Slotterbeck B, Leaman DW, Sun Y, Grafenstein H, Hong SC, McInerney MF. Dysregulated Toll-like receptor expression and signaling in bone marrow-derived macrophages at the onset of diabetes in the non-obese diabetic mouse. Int Immunol 18: 1101–1113, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol 19: 433–442, 2008 [DOI] [PubMed] [Google Scholar]
- 36.Navarro JF, Milena FJ, Mora C, Leon C, Claverie F, Flores C, Garcia J. Tumor necrosis factor-alpha gene expression in diabetic nephropathy: relationship with urinary albumin excretion and effect of angiotensin-converting enzyme inhibition. Kidney Int Suppl S98–S102, 2005 [DOI] [PubMed] [Google Scholar]
- 37.Navarro JF, Milena FJ, Mora C, Leon C, Garcia J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol 26: 562–570, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Navarro JF, Mora C, Maca M, Garca J. Inflammatory parameters are independently associated with urinary albumin in type 2 diabetes mellitus. Am J Kidney Dis 42: 53–61, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Navarro JF, Mora C, Muros M, Garcia J. Additive antiproteinuric effect of pentoxifylline in patients with type 2 diabetes under angiotensin II receptor blockade: a short-term, randomized, controlled trial. J Am Soc Nephrol 16: 2119–2126, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Navarro JF, Mora C, Muros M, Garcia J. Urinary tumour necrosis factor-alpha excretion independently correlates with clinical markers of glomerular and tubulointerstitial injury in type 2 diabetic patients. Nephrol Dial Transplant 21: 3428–3434, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Niewczas MA, Gohda T, Skupien J, Smiles AM, Walker WH, Rosetti F, Cullere X, Eckfeldt JH, Doria A, Mayadas TN, Warram JH, Krolewski AS. Circulating TNF receptors 1 and 2 predict ESRD in Type 2 diabetes. J Am Soc Nephrol 23: 507–515, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okusa M. The inflammatory cascade in acute ischemic renal failure. Nephron 90: 133–138, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Ramesh G, Reeves WB. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramesh G, Reeves WB. TNFR2-mediated apoptosis and necrosis in cisplatin-induced acute renal failure. Am J Physiol Renal Physiol 285: F610–F618, 2003 [DOI] [PubMed] [Google Scholar]
- 45.Ridker PM, Rifai N, Pfeffer M, Sacks F, Lepage S, Braunwald E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation 101: 2149–2153, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116: 3015–3025, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi H, Patschan D, Epstein T, Goligorsky MS, Winaver J. Delayed recovery of renal regional blood flow in diabetic mice subjected to acute ischemic kidney injury. Am J Physiol Renal Physiol 293: F1512–F1517, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Song MJ, Kim KH, Yoon JM, Kim JB. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun 346: 739–745, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Wada T, Yokoyama H, Matsushima K, Kobayashi Ki. Monocyte chemoattractant protein-1: does it play a role in diabetic nephropathy? Nephrol Dial Transplant 18: 457–459, 2003 [DOI] [PubMed] [Google Scholar]
- 50.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest 117: 2847–2859, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischemia/reperfusion renal injury. Nephrol Dial Transplant 15: 1562–1574, 2000 [DOI] [PubMed] [Google Scholar]
- 52.Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol 19: 923–932, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]