

Abstract
Transforming growth factor-β1 (TGF-β1)-induced expression of plasminogen activator inhibitor-1 (PAI-1) and p21 in renal mesangial cells (MCs) plays a major role in glomerulosclerosis and hypertrophy, key events in the pathogenesis of diabetic nephropathy. However, the involvement of histone acetyl transferases (HATs) and histone deacetylases (HDACs) that regulate epigenetic histone lysine acetylation, and their interaction with TGF-β1-responsive transcription factors, are not clear. We evaluated the roles of histone acetylation, specific HATs, and HDACs in TGF-β1-induced gene expression in rat mesangial cells (RMCs) and in glomeruli from diabetic mice. Overexpression of HATs CREB binding protein (CBP) or p300, but not p300/CBP-activating factor, significantly enhanced TGF-β1-induced PAI-1 and p21 mRNA levels as well as transactivation of their promoters in RMCs. Conversely, they were significantly attenuated by HAT domain mutants of CBP and p300 or overexpression of HDAC-1 and HDAC-5. Chromatin immunoprecipitation assays showed that TGF-β1 treatment led to a time-dependent enrichment of histone H3-lysine9/14-acetylation (H3K9/14Ac) and p300/CBP occupancies around Smad and Sp1 binding sites at the PAI-1 and p21 promoters. TGF-β1 also enhanced the interaction of p300 with Smad2/3 and Sp1 and increased Smad2/3 acetylation. High glucose-treated RMCs exhibited increased PAI-1 and p21 levels, and promoter H3K9/14Ac, which were blocked by TGF-β1 antibodies. Furthermore, increased PAI-1 and p21 expression was associated with elevated promoter H3K9/14Ac levels in glomeruli from diabetic mice. Thus TGF-β1-induced PAI-1 and p21 expression involves interaction of p300/CBP with Smads and Sp1, and increased promoter access via p300/CBP-induced H3K9/14Ac. This in turn can augment glomerular dysfunction linked to diabetic nephropathy.
Keywords: diabetic nephropathy, histone acetylation, mesangial cell, p300/CBP, TGF-β1
the pathogenesis of diabetic nephropathy (DN) involves hyperglycemia and growth factor-induced cellular hypertrophy, glomerulosclerosis, and interstitial fibrosis (4, 68). Dysregulated expression of cell cycle genes such as p21 play key roles in hypertrophy, whereas the regulation of matrix-degrading proteinases by plasminogen activator inhibitor-1 (PAI-1) combined with increased expression of other profibrotic genes leads to extracellular matrix (ECM) accumulation and fibrosis. Evidence suggests that transforming growth factor-β1 (TGF-β1) is a major mediator of such hypertrophic and profibrotic changes seen in diabetic kidney disease (4, 45, 48, 49, 56, 68).
The ECM is a complex and dynamic meshwork of several proteoglycans and other proteins, including collagens and fibronectin. PAI-1 promotes ECM accumulation by regulating fibrinolysis and plasmin-mediated matrix metalloproteinase activation and is strongly induced in various forms of kidney diseases including DN (36). The induction of PAI-1 by TGF-β1 has been demonstrated in renal mesangial cells (MCs) and epithelial cells (10, 51, 54). In addition, the transcriptional regulation of the cell cycle inhibitor p21 by TGF-β1 is also strongly associated with diabetic glomerular hypertrophy (1, 56, 57).
TGF-β1 signaling through type I and II receptors leads to phosphorylation and nuclear translocation of Smad transcriptional factors (TFs) which are major effectors of TGF-β1-induced gene expression (31, 33). Transcription mediated by Smads involves direct binding to consensus Smad binding elements (SBEs) in the promoters of target genes. In addition, Smads can also interact with other DNA-binding proteins and coactivators to regulate gene expression (12, 33, 58). SBEs have been identified in the PAI-1 and p21 promoters and shown to mediate TGF-β1-induced transcriptional activation (10, 38). Furthermore, Sp1 consensus binding sites in the PAI-1 and p21 promoters have also been implicated in TGF-β1-mediated gene regulation (8, 38).
In addition to the binding of TFs to consensus binding sites at the target gene promoters, transcriptional activation or repression is also controlled by the assembly of nuclear protein complexes that alter chromatin structure via posttranslational modifications (PTMs) of histone tails in the nucleosomes. These PTMs include acetylation, methylation, phosphorylation, and ubiquitylation (25). Histone lysine acetylation (HKAc) mediated by histone acetyltransferases (HATs) is usually associated with gene activation. This is balanced by the removal of acetyl groups by histone deacetylases (HDACs), which are associated with chromatin compaction and transcriptional repression (17, 27). Therefore, the dynamic balance between cellular HAT and HDAC activities can control the expression levels of target genes, while imbalances can result in cellular dysfunction and disease states (2, 27, 41). HATs such as CREB binding protein (CBP), its structural homolog p300, and p300/CBP-activating factor (p/CAF) act as transcriptional coactivators (25, 27). The HAT domain of p300/CBP catalyzes the acetylation of promoter-bound histones, resulting in chromatin relaxation and modulation of transcription (17, 27). In addition, HATs can also regulate gene expression through acetylation of nonhistone proteins such as TFs, including Smads, p53, and NF-κB. Acetylation of TFs in turn regulates their DNA binding activity, nuclear localization, protein stability, and interactions with other transcription regulators (5, 6, 18, 21, 53, 55, 62).
Increasing evidence links the dysregulation of chromatin modifications to the pathogenesis of diabetes and its complications, with important therapeutic implications (11, 30, 39, 43, 44). Reports showed that kidneys from diabetic animals exhibit changes in global histone PTMs as well as H3KAc at the fibrillin 1 promoter and that HDAC-2 may mediate ECM accumulation and epithelial-to-mesenchymal transition in the diabetic kidney and in TGF-β1-treated epithelial cells (14, 37). We recently demonstrated that TGF-β1-induced profibrotic gene expression in MCs was associated with specific alterations in the levels of key active and repressive histone lysine methylation marks at their promoters (51). Studies in fibroblasts showed a key role for the intrinsic HAT activity of p300 in TGF-β-Smad-dependent stimulation of collagen type I-α2 (Col1a2) transcription (16). However, the role of promoter histone lysine acetylation and key HATs in the regulation of other key TGF-β1 target genes in MCs and the specific interplay among HATs, HDACs, and TFs in this process are still unclear. Here, we report the role of these regulatory mechanisms in the expression of two TGF-β1 target genes, PAI-1 and p21, key players in DN. Our results demonstrate that regulation of promoter H3K9/14Ac by p300/CBP and HDACs, as well as direct interaction of p300/CBP with Smad and Sp1 play key roles in TGF-β1-induced PAI-1 and p21 gene expression in MCs. Furthermore, we also demonstrate that increased PAI-1 and p21 gene expression was associated with higher levels of H3K9/14Ac at their promoters under diabetic conditions both in vitro and in vivo.
MATERIALS AND METHODS
Materials.
Recombinant human TGF-β1 and the pan-specific TGF-β1 antibody (MAB1835) were from R&D Systems (Minneapolis, MN); antibodies against acetylated H3K9/14 (catalog no. 06-599), p300 (05-257), Sp1 (07-645), normal mouse IgG (12-371), and normal rabbit IgG (PP64B) were from Millipore (Billerica, MA); Smad2/3 (8685), acetylated-lysine (9441), HDAC1 (2062), and HDAC5 (2082) antibodies were from Cell Signaling (Danvers, MA); the CBP antibody (ab3652) was from Abcam (Cambridge, MA); the β-actin (A5441) antibody was from Sigma (St. Louis, MO).
cDNA kits for reverse transcriptase reactions and SYBR green kits for real-time PCRs were from Applied Biosystems (Foster City, CA). Magnetic Protein A or G Dynabeads were from Invitrogen (Grand Island, NY). Enhanced green fluorescent protein (GFP) plasmid was from Lonza. Luciferase assay reagents and pRL-TK vector were from Promega (Madison, WI), and the NE-PER nuclear protein extraction kit was from Thermo Scientific (Rockford, IL). Plasmids expressing dominant negative (D/N) p300 and p/CAF (29) were from Dr. Michael Stallcup (University of Southern California, Los Angeles, CA). D/N CBP (28) was from Dr. Christopher Glass (University of California, San Diego, CA). Expression vectors for CBP or p300 and p/CAF were from Dr. Barry Forman (Beckman Research Institute, Duarte, CA). HDAC1 and HDAC5 expression vectors were from Dr. Stuart L. Schreiber (Harvard University, Boston, MA). WT PAI-1-luciferase reporter plasmid was from Dr. Satoshi Fujii (Nagoya City University, Nagoya, Japan); and WT p21-luciferase reporter plasmid was from Dr. Ken-ichi Isobe (Nagoya University). RNA-STAT60 reagent was from Tel-Test (Friendswood, TX). Sequences of the PCR primers used in this study are listed in Table 1.
Table 1.
PCR primer sequences
| Primer | Forward Primer | Reverse Primer |
|---|---|---|
| ChIP primers | ||
| PAI-1 P1 (R) | CGGCTATACCAGATGTGGGCCG | GACGACCGACCAGCCAAAG |
| PAI-1 P2 (R) | GACAATATGTGCCCTGTGATTGTC | AGGCTGCTCTACTGGTCCTTGC |
| PAI-1 P3 (R) | GTGCCTTTGAAGGTTGGACAGA | CGGATGATGACAGTTTGTGAATCCG |
| p21 P1 (R) | GTTCAGCCCTGG AACCGAAG | GTACCAAACACCCTTCACCTGGTAC |
| CypA P (R) | CCCGGATGCGTACCTAAGGA | CGGACGTTGCTTCGCTGTCCG |
| PAI-1 P (M) | CGGCCTTTATACCAGATGTGAGCcG | TCCAAACACCAGGCTTTGTAGG |
| p21 P (M) | CAAGAGAATAGCCCAGGTGTGG | CGCGCCTAGACTCTGACACCGcG |
| cDNA primers | ||
| PAI-1 (R) | GCAGCTCTCTGTAGCACAAGCA | CGGCCTCTGTTGGATTGTGCCG |
| p21 (R) | GTGGCCTTGTCGCTGTCTTG | CGATTCTTGCAGAAGACCAATCG |
| PAI-1 (M) | GACGCCTTCATTTGGACGAA | CGGACCTTTTCCCTTCAAGAGTCCG |
| p21 (M) | CGGTGGAACTTTGACTTCGT | CAGGGCAGAGGAAGTACTGG |
| TGF-β1 (R/M) | AGGAAGGACCTGGGTTGGAAG | CGTCTCGACCCACGTAGTAGACG |
| CypA (R/M) | ATGGTCAACCCCACCGTGT | TTCTTGCTGTCTTTGGAACTTTGTC |
| β-Actin (R/M) | CCCTGTATGCCTCTGGTCGT | CGGACGCAGCTCAGTAACAGTCCG |
ChIP, chromatin immunoprecipitation; PAI-1, plasminogen activator inhibitor-1;· CypA, cyclophilin Α; TGF, transforming growth factor; R, rat; M, mouse.
MC culture and treatment with TGF-β1.
All animal studies were carried out in accordance with a protocol approved by our Institutional Animal Care and Use Committee (IACUC). Primary rat MCs (RMCs) from renal glomeruli of Sprague-Dawley rats (9 wk) were isolated and cultured as described earlier (22). RMCs were used between passages 5 and 10. RMCs were treated with TGF-β1 (5 ng/ml) as indicated, while control cells were treated with the vehicle (PBS containing 4 mM HCl and 0.1% BSA).
Isolation of renal glomeruli from mice.
In some experiments, glomeruli were isolated from kidneys of 12-wk-old type 2 diabetic db/db mice (BKS.Cg-m+/+leprdb/J, stock no. 000642; The Jackson Laboratory, Bar Harbor, ME) and age-matched control heterozygote nondiabetic db/+ mice (Jackson) by sequential sieving as described earlier (64). Blood glucose levels were >450 mg/dl in db/db mice compared with <140 mg/dl in db/+ mice. Glomeruli were also studied from a type 1 diabetes model in which 8-wk-old male C57BL/6 mice were injected intraperitoneally with 50 mg/kg of streptozotocin (STZ) for 5 consecutive days, while control mice were injected with normal saline. These STZ-injected mice were killed 16 wk after they became diabetic (blood glucose levels were >300 mg/dl in STZ vs. 145 mg/dl in control mice). Glomeruli preparations from three to four mice in each group were pooled to obtain sufficient material for RNA, protein extraction, and chromatin immunoprecipitation (ChIP) assays.
Transient transfections and luciferase assays.
RMCs at 75% confluence plated in triplicate in 24-well plates (Becton Dickinson Labware) were cotransfected with 0.4 μg each of indicated firefly luciferase reporter plasmids and expression vectors along with an internal control vector of pRL-TK using FuGENE 6 Transfection Reagent (Roche, Indianapolis, IN) as described before (24). The transfected cells were serum starved for 24 h and stimulated with TGF-β1 (5 ng/ml) for the indicated time periods. The cells were lysed, and dual luciferase assays were performed according to the manufacturer's instructions (Promega) in a 96-well plate reader (Turner Biosystems, Promega). Results were normalized to Renilla luciferase activities expressed from pRL-TK and expressed as fold over control.
RNA isolation and quantitative real-time PCR.
Total RNA was isolated from RMCs using RNA-STAT60 reagent as described earlier (64). Total RNA (1 μg) was used for cDNA synthesis using Gene Amp RNA PCR kits, and quantitative real-time PCR (qRT-PCR) was performed using a SYBR Green PCR Master Mix kit with ABI 7300 or 7500 real-time PCR thermal cyclers as described previously (64). Reactions were performed in triplicate in a final volume of 20 μl. Standard curves were generated for all the genes being quantified as well as for β-actin control. Dissociation curves were run to detect nonspecific amplification and to confirm amplification of single products in each reaction. The quantity of each test gene and internal β-actin RNA control were determined from standard curves using Applied Biosystems software. In some experiments, data were analyzed by the 2−ΔΔCt method as described earlier (51).
ChIP assays.
Quiescent RMCs were treated with TGF-β1 (5 ng/ml) for the indicated time periods and then fixed with 1% formaldehyde. Isolated glomeruli from mice were fixed with formaldehyde (final concentration 1% in PBS for 20 min at room temperature) and then quenched with 125 mM glycine (5 min at room temperature). The cross-linked MCs or glomeruli were washed twice with cold PBS containing protease inhibitors and lysed as previously described (51). Lysates were sonicated, and an aliquot was saved to isolate total input DNA. Immunoprecipitation was performed with antibodies to acetylated histone H3K9/14 (H3K9/14Ac), p300, CBP, or IgG (antibody control). Immune complexes were captured using Protein A/G Dynabeads, washed, bound proteins were eluted, and ChIP-enriched DNA was obtained by phenol:chlororm extraction followed by ethanol precipitation. Input DNA samples as well as antibody-enriched ChIP DNA samples were analyzed by qPCR using indicated primers within the PAI-1, p21, and cyclophilin promoters (see Fig. 4A and Table 1). Data were analyzed using the 2−ΔΔCt method and normalized with input samples as described earlier (51).
Fig. 4.

TGF-β1 induces histone H3K9/14Ac at the PAI-1 and p21 promoters. A: upstream promoter regions of the PAI-I, p21, and CypA genes. Location of primers used for the chromatin immunoprecipitation (ChIP)-qPCR assays labeled P1, P2, or P3 are indicated by short arrows. RMCs were treated with TGF-β1 (5 ng/ml) for indicated time periods up to 6 h. The control samples were harvested at 6 h along with samples treated with TGF-β1 for 6 h. B–D: ChIP assays were performed with H3K9/14Ac antibody, and ChIP-enriched DNA was amplified with primers spanning the 3 regions of the PAI-1 promoter. E: ChIP analysis of H3K9/14Ac on the p21 promoter. F: ChIP analysis of H3K9/14Ac on the CypA promoter region. IgG was used as the antibody control. The bar graphs represent relative enrichment levels normalized to the input and expressed as fold over the untreated controls (means ± SE, n = 3). P, ChIP primers; TSS, transcription start site; SBE, Smad Binding Element; Sp1, Sp1 binding element. *P < 0.05, #P < 0.01, $P < 0.001 vs. untreated control.
Coimmunoprecipitation.
Nuclear extracts were isolated from RMCs using an NE-PER nuclear protein extraction kit according to the manufacturer's instructions. Nuclear lysates were sonicated four times for 10 s each at 4°C, and protein concentrations were estimated by the Lowry method (Bio-Rad, Hercules, CA). Cell lysates with equal amounts of protein were incubated with indicated antibodies, and immune complexes were collected on protein G Dynabeads at 4°C according to the manufacturer's instructions. Protein samples were analyzed by Western blotting as previously described earlier (64).
Statistical analyses.
Data were expressed as means ± SE from multiple experiments. Paired Student's t-tests were used to compare two groups or ANOVA with Dunnett's posttests for multiple groups using PRISM software (Graph Pad, San Diego, CA). Statistical significance was detected at the 0.05 level.
RESULTS
CBP and p300, but not p/CAF, enhances TGF-β1-induced expression of PAI-1 and p21.
Coactivator HATs such as CBP and p300 regulate gene expression by increasing histone H3K9/14Ac to promote chromatin relaxation for TF access, as well as by direct acetylation of TFs. However, the role of specific HATs in TGF-β1-induced PAI-1 and p21 gene expression in MCs relevant to DN is not clear. To test this, we first examined PAI-1 and p21 mRNA expression in RMCs treated with either the vehicle (control) or TGF-β1 (5 ng/ml) from 0.5 h to 24 h by qRT-PCR. Results showed that, as anticipated, PAI-1 and p21 mRNA levels were increased in response to TGF-β1 treatment in a time-dependent manner (Fig. 1, A and B).
Fig. 1.

CREB binding protein (CBP) and p300, but not p300/CBP-activating factor (p/CAF), augments transforming growth factor (TGF)-β1-induced plasminogen activator inhibitor-1 (PAI-1) and p21 promoter transactivation. A and B: expression of PAI-1 (A) and p21 (B) mRNAs in rat mesangial cells (RMCs) treated with TGF-β1 relative to control (ctrol) cells. Quiescent RMCs were treated with TGF-β1 (5 ng/ml) from 0.5 to 24 h, and ctrol cells were treated with vehicle. Total RNA was prepared from TGF-β1 treated RMCs at indicated time points while ctrol cells were harvested at the 24-h time point. Gene expression was analyzed by quantitative real-time PCR (qRT-PCR), normalized with internal control gene β-actin and results were expressed as fold over control. Values are means ± SE (n = 3). *P < 0.05, #P < 0.01 vs. control. C and D: effect of histone acetyl transferases (HAT)s on transactivation of PAI-1 and p21 promoters. RMCs were transiently cotransfected with wild-type (WT) CBP, p300, or p/CAF (pCAF) expression vectors, or the respective dominant negative (D/N) mutants lacking HAT domains, or a control enhanced green fluorescence protein (GFP) expression vector; along with the PAI-1 promoter-luciferase (C) or p21 promoter-luciferase construct (D). Transfected cells were then treated without or with TGF-β1 (5 ng/ml) for 6 h, and luciferase activities in cell lysates were determined. Promoter activity was expressed as “relative luciferase activity,” which is relative to the activity of untreated GFP-transfected cells. Values are means ± SE (n = 3). *P < 0.05, #P < 0.01, $P < 0.001 vs. corresponding untreated control. !P < 0.05, !!P < 0.01, !!!P < 0.001 vs. GFP+TGF-β1.
Next, we determined the involvement of specific HATs such as CBP, p300, or p/CAF in the transcriptional regulation of PAI-1 and p21 by TGF-β1. We cotransfected RMCs with luciferase reporter plasmids containing PAI-1 and p21 promoter regions along with plasmid vectors expressing wild-type (WT) CBP, p300, or p/CAF or mutants lacking HAT domains. Luciferase activities were then measured after treatment without or with TGF-β1 (5 ng/ml) for 2 h. Results showed that WT CBP and p300 vectors significantly enhanced TGF-β1-induced activation of the PAI-1 and p21 promoters relative to cells transfected with enhanced GFP control vector (Fig. 1, C and D). Reciprocally, RMCs cotransfected with the HAT dominant-negative (D/N) mutants of CBP and p300 significantly attenuated TGF-β1-induced activation of PAI-1 and p21 promoters. In contrast, both WT and D/N mutants of p/CAF had no significant effects on PAI-1 and p21 promoter activation (Fig. 1, C and D). These results demonstrate that HAT activity of CBP and p300, but not p/CAF, is required for TGF-β1-mediated activation of the PAI-1 and p21 promoters.
Next the effect of these HATs on gene expression was evaluated. Expression vectors for CBP, p300, p/CAF, or control GFP were transfected into RMCs, treated with TGF-β1 (5 ng/ml) for 2 h, and gene expression was analyzed by qRT-PCR. Results showed that overexpression of CBP or p300, but not p/CAF, resulted in a marked augmentation of TGF-β1-induced PAI-1 and p21 mRNA expression compared with control GFP (Fig. 2, A and B). In contrast, no statistically significant changes were seen in CypA expression compared with control under these conditions, suggesting specificity (Fig. 2C).
Fig. 2.
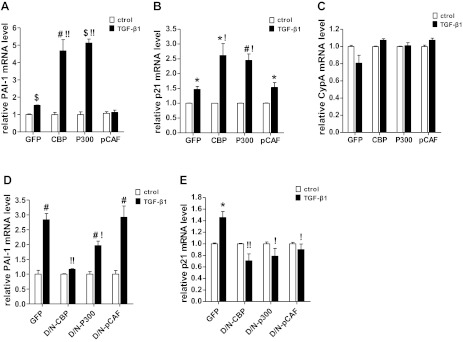
HAT activities of CBP and p300, but not p/CAF, are essential for TGF-β1-induced PAI-1 and p21 gene expression. A–C: quiescent RMCs were transiently transfected with EGFP (GFP), CBP, p300, or p/CAF (pCAF) expression vectors and then treated with TGF-β1 (5 ng/ml) for 2 h. PAI-1, p21, and cyclophilin A (CypA) mRNA expression was analyzed by qRT-PCR. D and E: RMCs were transiently transfected with vectors expressing GFP, D/N mutants of CBP, p300, or p/CAF and then treated with TGF-β1 for 6 h. PAI-1 and p21 mRNA expression was analyzed by qRT-PCR. Values are means ± SE (n = 3). *P < 0.05, #P < 0.01, $P < 0.001 vs. respective untreated control. !P < 0.05, !!P < 0.01 vs. GFP+TGF-β1.
RMCs were next transfected with either the GFP control vector or CBP, p300, and p/CAF mutants lacking HAT domains and then treated with TGF-β1 for 6 h. D/N mutants of p300 and CBP significantly repressed PAI-1 and p21 mRNA induction by TGF-β1 compared with that seen in cells expressing GFP. The D/N mutant of p/CAF had no effect on PAI-1 expression but inhibited p21 expression (Fig. 2, D and E). Together, these results suggest that the HAT activities of p300 and CBP are required for both TGF-β1-induced PAI-1 and p21 promoter activities as well as mRNA expression.
Role of HDACs in TGF-β1-mediated transcriptional activation and gene regulation.
Since our results indicated that HAT activities of CBP and p300 are involved in TGF-β1-mediated gene regulation, we next examined the role of HDACs which can erase H3KAc to promote chromatin condensation and gene repression by reducing chromatin access to TFs. Cotransfection experiments showed that plasmid vectors expressing HDAC1 (class I) or HDAC5 (class II) strongly suppressed TGF-β1-induced PAI-1 and p21 promoter transactivation compared with the control vector expressing GFP (Fig. 3, A and B). Immunoblotting of the transfected cell lysates confirmed the overexpression of HDAC1 and HDAC5 under these conditions (data not shown). Furthermore, overexpression of these HDACs also significantly attenuated TGF-β1-induced PAI-1 and p21 mRNA expression relative to control GFP (Fig. 3, C and D). These results demonstrate the negative regulatory role of HDAC1 and HDAC5 in TGF-β1-induced PAI-1 and p21 gene expression and that a dysregulation of the balance between the actions of HATs and HDACs may be a key mechanism in TGF-β1-mediated gene expression in MCs.
Fig. 3.

HDACs suppress TGF-β1-induced PAI-1 and p21 promoter transactivation and expression. A and B: RMCs were transiently cotransfected with HDAC1, HDAC5, or a control GFP expression vector along with PAI-1-luciferase (A) or p21-luciferase (B) reporter constructs and then stimulated with TGF-β1 (5 ng/ml) for 2 h. The relative luciferase activities were determined by comparison with the activity of untreated GFP. Values are means ± SE (n = 3). #P < 0.01 vs. GFP untreated. !!P < 0.01 vs. GFP+TGF-β1. C and D: RMCs were transiently transfected with GFP, HDAC1, or HDAC5 expression vectors and then treated with TGF-β1 (5 ng/ml) for 2 h. qRT-PCR was used to quantify PAI-1 and p21 mRNA levels. Values are means ± SE (n = 3). *P < 0.05, #P < 0.01 vs. relative untreated control. !P < 0.05, !!P < 0.01 vs. GFP+TGF-β1.
TGF-β1 induces H3K9/14Ac at the PAI-1 and p21 promoters.
Having demonstrated that HATs can modulate PAI-1 and p21 gene expression in response to TGF-β1, we next examined their mechanisms of action. We first examined whether TGF-β1-induced gene regulation is accompanied by alterations in chromatin histone KAc at the PAI-1 and p21 promoters. We performed ChIP assays using anti-H3K9/14Ac antibody, and ChIP DNA was analyzed by qPCR with primers designed to amplify key regions of the PAI-1, p21, and internal control CypA promoter (Fig. 4A). TGF-β1 treatment significantly increased H3K9/14Ac at the PAI-1 promoter P1 region that is close to the transcription start site (TSS) and contains consensus binding sites for TFs Smad (SBE) and Sp1 (Fig. 4B), as well as at the P2 region containing two SBEs (Fig. 4C). In contrast, TGF-β1 did not alter H3K9/14Ac levels at the P3 region (−2094 to −2279), which is much farther away from the TSS (Fig. 4D). Furthermore, the increases in H3K9/14Ac correlated temporally with TGF-β1-induced PAI-1 gene expression (Fig. 1A). TGF-β1 also increased H3K9/14Ac at the −185- to +42-bp region of the p21 promoter (Fig. 4E), which contains binding sites for members of the Sp1 family of TFs (60). However, there was no significant change in H3K9/14Ac at the CypA promoter (−305 to −218 bp) (Fig. 4F), suggesting specificity. Together, these results demonstrate that TGF-β1-mediated H3K9/14Ac might promote chromatin remodeling and confer access to TFs such as Smads and Sp1 to induce PAI-1 and p21 genes relevant to the pathology of DN.
TGF-β1 treatment enhances the occupancies of CBP and p300 at the PAI-1 and p21 promoters.
We next tested whether the enhanced histone KAc triggered by TGF-β1 at the PAI-1 and P21 promoters was due to increased occupancies of CBP and p300. Results of ChIP assays demonstrated that TGF-β1 significantly increased the recruitments of CBP and p300 at the P1 and P2 regions of the PAI-1 promoter (Fig. 5, A and B) as well as at the proximal region of the p21 promoter relative to control (Fig. 5, C and D). In particular, p300 occupancy at the p21 promoter was increased >10-fold at 2 h. The enrichments appeared to be promoter specific because no enrichment of CBP was found at the CypA promoter region in response to TGF-β1 (Fig. 5E). Together, these results further suggest that histone H3 hyperacetylation in response to TGF-β1 at the PAI-1 and p21 promoter regions is due to increased recruitment of CBP and p300 and their associated HAT activities.
Fig. 5.
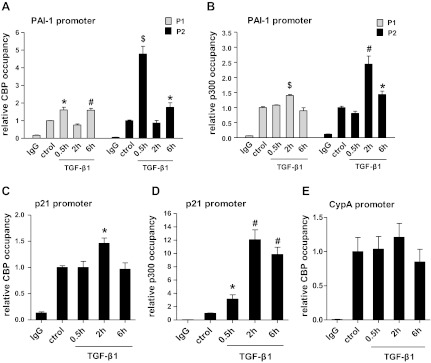
TGF-β1 stimulation leads to an increase in CBP and p300 occupancy at the PAI-1 and p21 promoters. RMCs were treated with TGF-β1 (5 ng/ml) for 0.5, 2, or 6 h and control cells with vehicle (6 h). ChIP assays were performed to evaluate CBP (A) and p300 (B) occupancy at the PAI-1 promoter regions (P1 and P2). C and D: ChIP assays of CBP (C) and p300 (D) occupancy at the p21 promoter. E: ChIP assays of CBP occupancy at the CypA promoter. Bar graphs represent relative enrichment levels normalized to the input and expressed as fold over untreated controls (means ± SE, n = 3). *P < 0.05, #P < 0.01, $P < 0.001 vs. untreated control.
Interactions of p300 with smad and Sp1 proteins in response to TGF-β1.
Next, we speculated that TGF-β1 may also promote the interaction of p300 with Smad and/or Sp1 proteins to regulate PAI-1 and p21 gene expression in RMCs. This was tested by performing coimmunoprecipitation experiments. The nuclear extracts from untreated control and TGF-β1-treated RMCs were first immunoprecipitated with Smad2/3 antibody, and eluted proteins were then immunoblotted with p300 and acetylated lysine antibodies. The input lysate was immunoblotted with Smad2/3 antibody. Results showed that the association of p300 with Smad2/3 was increased in TGF-β1-treated cells (middle bands, Fig. 6A, 2.5-, 1.2-, 0.6-, and 1.4-fold over control at 0.5, 2, 6, and 24 h, respectively, normalized to input). Furthermore, Smad2/3 itself was also acetylated in response to TGF-β1 treatment (top bands, Fig. 6A, 1.7-, 1.3-, 1.2-, and 1.3-fold over control at 0.5, 2, 6, and 24 h, respectively, normalized to input), likely due to these interactions with p300. Second, nuclear extracts were immunoprecipitated with p300 antibody followed by immunoblotting with Smad2/3 and Sp1 antibodies. Results showed that TGF-β1 enhanced the association of p300 with both Smad2/3 (top bands, Fig. 6B, 2.9-, 2.3-, 2.0-, and 2.1-fold over untreated control at 0.5, 2, 6, and 24 h) and Sp1 proteins (middle bands, Fig. 6B, 1.4-, 1.3-, 1.2-, and 1.3-fold over control at 0.5, 2, 6, and 24 h, respectively). These data demonstrate that TGF-β1 enhances the interactions of p300 with Smad2/3 and Sp1 in RMCs. They also suggest that p300 might assist in the recruitment and acetylation of these key TFs to further augment TGF-β1-induced PAI-1 and p21 gene expression in RMC.
Fig. 6.

TGF-β1 enhances the interactions of Smad with the p300 and Sp1 proteins. A: nuclear lysates from RMCs treated either with TGF-β1 (5 ng/ml) for indicated time periods or control cells (treated with vehicle for 6 h) were immunoprecipitated with Smad2/3 antibody. Eluted proteins were immunoblotted with acetylated lysine or p300 antibody. B: nuclear lysates from MCs treated with vehicle control (ctrol) or with TGF-β1 (5 ng/ml) were immunoprecipitated with p300 antibody. Eluted proteins were immunoblotted with Smad2/3 or Sp1 antibody, and the input lysate was blotted with Smad2/3 antibody to normalize protein levels. Data shown are representative of 2 experiments.
The HDAC inhibitor trichostatin A can enhance TGF-β1-mediated transcription.
Since HDAC1 and 5 negatively regulated TGF-β1-induced PAI-1 and p21 gene expression, and inhibition of HDACs with a nonselective inhibitor, trichostatin A (TSA), induced chromatin histone hyperacetylation (52) and p21 expression in RMCs (13), we further tested the effect of TSA on TGF-β1-induced H3K9/14Ac at these gene promoters. Our results showed that inhibition of HDACs with TSA also further increased TGF-β1-induced PAI-1 gene transcriptional activity and expression in RMCs (data not shown). When RMCs were pretreated with 0.3 mM TSA for 24 h and then incubated with or without TGF-β1 for 0.5 h, treatment with TSA not only increased baseline H3K9/14Ac levels but also further augmented TGF-β1-induced H3K9/14Ac levels at the P2 region of the PAI-1 promoter as determined by ChIP assays (Fig. 7A). TSA also increased H3K9/14Ac at the P1 region of the PAI-1 promoter (Fig. 7A) and the p21 promoter (Fig. 7B) but did not augment TGF-β1-mediated effects at these regions. These results further confirm the repressive role of HDACs in TGF-β1-induced PAI-1 and p21 gene expression in RMCs.
Fig. 7.

Effect of trichostatin A (TSA) on basal and TGF-β1-induced H3K9/14Ac at the PAI-1 and p21 promoters. RMCs were pretreated with TSA (0.3 mM) or vehicle for 24 h, washed, and then treated with TGF-β1 (5 ng/ml) for 0.5 h. A: ChIP assays of H3K9/14Ac at PAI-1 promoter regions P1 and P2. B: ChIP analysis of H3K9/14Ac at the p21 promoter region. Bar graphs represent relative enrichment levels normalized to the input and are expressed as fold over untreated controls (means ± SE, n = 3). *P < 0.05, #P < 0.01 vs. control. !!P < 0.01 vs. TGF-β1.
High glucose treatment increases H3K9/14Ac at the PAI-1 and p21 promoters in RMCs, and this is inhibited by a TGF-β1 specific antibody.
Evidence shows that high-glucose (HG) conditions mimicking the diabetic state can increase histone KAc associated with active gene expression in monocytes and endothelial cells (32, 40). However, this has not been examined in MCs. Since HG can increase the expression of PAI-1 and p21 through TGF-β1 in MCs (26, 51), and these genes are also upregulated in the renal cortex of diabetic mice (64), we next tested whether there are changes in histone H3K9/14Ac under these conditions. RMCs were cultured under normal glucose (NG) with mannitol (5.5 mM+20 mM mannitol) or HG (25 mM) for 48 or 72 h, followed by ChIP assays with H3K9/14Ac antibodies. Results showed that HG treatment increased H3K9/14Ac at the P1 region of PAI-1 (Fig. 8A) promoter and the p21 promoter (Fig. 8B), which was statistically significant at 72 h. There were no significant differences in the H3K9/14Ac level at the PAI-1 and p21 promoters between 48 and 72 h under NG conditions. These results demonstrate that HG can increase H3K9/14Ac at TGF-β1-inducible genes in RMCs. To determine the potential functional mediatory role of TGF-β1 in HG-induced H3K9/14Ac, RMCs were preincubated with a TGF-β1-specific antibody (1 μg/ml) or control mouse IgG (1 μg/ml) and then cultured with NG or HG for 72 h. ChIP analysis of these samples showed that HG-induced H3K9/14Ac enrichment around the PAI-1 and p21 promoters was significantly abrogated in RMCs pretreated with the TGF-β1 antibody (Fig. 8, C and D, respectively). These results suggest that HG-induced changes in epigenetic H3K9/14Ac at promoters of these genes relevant to DN pathogenesis are largely mediated by TGF-β1.
Fig. 8.

High glucose (HG) induces H3K9/14Ac at the PAI-1 and p21 promoters in RMCs, and this is blocked by a TGF-β1-specific antibody. A and B: RMCs were cultured in normal glucose (NG; 5.5 mM glucose+20 mM mannitol) or HG (25 mM glucose) for 48 h or 72 h and then subjected to ChIP assays to determine H3K9/14Ac at the PAI-1 (A) and p21 (B) promoter regions. #P < 0.01 vs. NG, n = 3. C and D: RMCs were preincubated with mouse IgG (1 μg/ml) or TGF-β1-specific antibody (1 μg/ml) for 0.5 h and then cultured under NG or HG for 72 h. H3K9/14Ac at PAI-1 (C) and p21 (D) promoters was determined by ChIP assays. $P < 0.001, n = 3.
Increased H3K9/14Ac at the PAI-1 and p21 promoters in vivo in glomeruli from diabetic mice.
We next examined whether H3K9/14Ac is associated with altered gene expression in vivo using animal models of both type 1 and type 2 DN. Gene expression and ChIP assays were performed with glomeruli isolated from kidneys of STZ-induced type 1 diabetic mice, and of type 2 diabetic db/db mice. Vehicle-injected normal C57BL/6 mice and db/+ mice were used as controls for type 1 and type 2 diabetic mice, respectively. Gene expression analysis revealed significant increases in the mRNA levels of TGF-β1, PAI-1, and p21 in the glomeruli from diabetic mice relative to control mice (Fig. 9, A and C). In parallel, ChIP assays showed significant enrichment of H3K9/14Ac at the PAI-1 (P1) and p21 promoter regions (close to TSS) in glomeruli from diabetic mice compared with nondiabetic control mice (Fig. 9, B and D), thus providing in vivo relevance.
Fig. 9.

Increased H3K9/14Ac at the PAI-1 and p21 promoters in glomeruli from diabetic mice in vivo. A and B: glomeruli were isolated from the kidneys of 12-wk-old control db/+ and diabetic db/db mice, and expression of indicated genes was analyzed by qRT-PCR (A), and H3K9/14Ac levels at the PAI-1 and p21 promoters were determined by ChIP assays (B). C and D: glomeruli were isolated from the kidneys of control and streptozotocin-induced type 1 diabetic C57BL/6 mice. Changes in the expression of indicated genes was determined by qRT-PCR (C), and H3K9/14Ac at PAI-1 and p21 promoters was determined by ChIP assays (D). Values are means ± SE (n = 3). *P < 0.05, #P < 0.01, $P < 0.001 vs. db/+ or nondiabetic C57BL/6 control.
DISCUSSION
In this study, we report that TGF-β1-induced dynamic changes in promoter histone modifications such as H3K9/14Ac, as well as acetylation of Smad2/3, play key roles in PAI-1 and p21 gene expression in MC. We found that TGF-β1 treatment increased p300/CBP occupancies at the PAI-1 and p21 promoters, leading to increased H3K9/14Ac. This was accompanied by increased association of p300 with key TFs Sp1 and Smad2/3, as well as Smad2/3 acetylation. These actions of p300/CBP led to increased expression of PAI-1 and p21, key players in ECM accumulation and hypertrophy in DN (Fig. 10). Furthermore, increased expression of PAI-1 and p21 in MCs treated with HG and glomeruli from diabetic mice was also associated with elevated levels of promoter H3K9/14Ac, demonstrating in vivo relevance to DN.
Fig. 10.

Model for TGF-β1-mediated promoter recruitment of p300/CBP and histone acetylation, and their roles in gene expression in mesangial cells. TGF-β1 induces dynamic changes in histone lysine acetylation at target gene promoters such as PAI-1 and p21 through enhanced recruitment of p300/CBP in glomerular MCs. Furthermore, it also promotes interactions of p300/CBP with transcription factors such as Smad2/3 and Sp1, and acetylation of Smad2/3 by p300. These events play key roles in augmenting TGF-β1-induced gene transcription under diabetic conditions. Thus TGF-β1-induced transcriptional factor activation can cooperate with epigenetic mechanisms in chromatin to regulate pathological gene expression associated with diabetic nephropathy (DN).
Several studies have demonstrated the critical role of histone PTMs, including methylation, phosphorylation, and acetylation, in gene transcription (2, 25). These epigenetic mechanisms act in concert with each other and in cooperation with TFs to regulate the expression of genes involved in cellular proliferation, inflammation, and matrix protein synthesis (25, 39, 43, 44). Hyperacetylation of nucleosomal histones including H3K9/14Ac can promote open chromatin formation by reducing interactions of DNA with histones, resulting in increased access to the transcriptional machinery (17, 27). Histone acetylation can provide binding sites for chromatin-remodeling factors and multiprotein components of basal transcription machinery including RNA polymerase II to facilitate gene transcription. In contrast, histone deacetylation by HDACs promotes chromatin compaction and gene repression (17, 25, 27). Evidence shows that TGF-β1 can regulate epigenetic modifications to regulate target genes in diverse cell types, including vascular smooth muscle cells (42), ovarian (63), and mammary epithelial cells (61), immune cells (66), and fibroblasts (15). Reports show that histone modifications including H3K9Ac and H3K4 methylation (H3K4me) play a role in PMA-induced laminin gene expression in rat mesangial cells (35) and in inflammatory gene expression in tubular cells from acute renal injury models (34, 65). Our recent studies demonstrated that TGF-β1 can induce significant changes in promoter histone H3K4me and H3K9me at the promoters of differentially regulated profibrotic genes CTGF, Col1a1, and PAI-1 in MCs (51). In the current study, we show for the first time that TGF-β1 significantly enhanced H3K9/14Ac at the PAI-1 and p21 promoter regions in MCs. The HDAC inhibitor TSA also increased H3K9/14Ac and gene expression, further demonstrating a key role for chromatin H3KAc.
H3KAc is mediated by several HATs including p300/CBP and can promote chromatin relaxation to increase access to key TFs such as Smad2/3 and Sp1 in MCs. Furthermore, H3K9/14Ac can also act in concert with other histone PTMs like H3K4me induced by TGF-β1 (51) to regulate pathological gene expression relevant to DN. We found that overexpression of HATs CBP and p300 increased TGF-β1-induced PAI-1 and p21 gene expression via promoter activation. Conversely, these effects were reversed by D/N mutants of CBP or p300. However, another HAT, p/CAF, did not alter TGF-β1-induced gene expression, suggesting some specificity in the role of HATs under these conditions in MCs. Transcription regulation by CBP/p300 is a complex process since these HATs not only acetylate histones to alter chromatin accessibility, but also regulate multiple aspects of TF function. These include acetylation of TFs to regulate their activity, stability, and nuclear localization, acting as scaffolds for the assembly of multiprotein transcription complexes and as bridges to facilitate interactions of TFs with transcription machinery (5). Our data clearly demonstrate increases in H3K9/14Ac, suggesting that hyperactylation of chromatin histones as one potential mechanism of p300/CBP-mediated gene expression in MCs.
Multiple growth factors and cytokines including TGF-β1 can induce transcription of the PAI-1 gene. It is well known that Smad2/3 TFs are major effectors of TGF-β1 actions in MCs (12). Activated Smad proteins regulate transcription by binding to SBEs on target promoters and forming complexes with other nuclear factors (12, 58). Activity of Smads is regulated by posttranslational modifications, including phosphorylation and acetylation (50, 58). Coactivator HATs including CBP, p300, and p/CAF can form multiprotein complexes with Smads and acetylate Smad2 and Smad3 to regulate TGF-β1-induced gene expression in various cell types, including MCs, where it was shown that phosphatidylinositol 3-kinase/Akt signaling is a critical regulator of Smad3-CBP interaction, Smad acetylation, and PAI-1 expression (7, 21, 55). Interaction of CBP/p300 was also shown in Smad-mediated fibrosis in fibroblasts (15). Our data showed that TGF-β1 treatment enhanced the interaction of Smad2/3 with p300 as well as the occupancy of p300 near two SBEs in the PAI-1 promoter. These SBEs (GTCTAGAC and CAGACAC) were previously shown to be necessary for TGF-β1-induced PAI-1 expression (10). Furthermore, our data also showed for the first time increased H3K9/14Ac levels around these promoter regions, suggesting that the Smad-p300 interactions might have led to increased Smad acetylation as well as promoter acetylation by the intrinsic HAT activity of p300. Thus interaction of CBP or p300 with Smad proteins and their recruitment to the PAI-1 promoter could be a mechanism by which histones are hyperacetylated at the promoters of TGF-β1/Smad target genes, including PAI-1, which can increase chromatin access to Smads and the general transcription apparatus (Fig. 10). Furthermore, since the acetylation of Smad2/3 increases their transactivation potential (21), it can further augment TGF-β1-induced PAI-1 gene expression.
It is widely accepted that the actions of TGF-β1 are dependent in large part on the Smad pathway with contributions also from other TFs, including Sp1 (19, 38). Previous studies have demonstrated that Sp1-dependent p21 promoter activation by TSA requires p300, implicating a role for histone acetylation also in p21 gene expression (59, 60). Our results demonstrated that p300/CBP and TSA can increase p21 gene expression. Therefore, similar to PAI-1, p300/CBP may act as coactivators, promote chromatin remodeling, and increase the access to TFs at the p21 promoter in response to TGF-β1 signaling. We also found that p300 and Sp1 can interact in TGF-β1-treated MCs. This result is in contrast to previous findings showing that p300 does not interact with Sp1 in HeLa cells treated with TSA (60), suggesting cell- and agonist-specific differences. Whether p300 mediates Sp1 acetylation is not clear from these studies and to date no data are available on Sp1 acetylation by HATs.
Our studies have also demonstrated an inhibitory role for HDACs such as HDAC1 and HDAC5 in TGF-β1-induced gene expression. Overexpression of these two HDACs inhibited PAI-1 and p21 promoter activity. Furthermore, TSA, a broad HDAC inhibitor, increased H3K9/14Ac and gene expression, thus reversing the effects of HDACs and mimicking the effects of the HATs p300/CBP. TSA in general can induce histone hyperacetylation to increase gene expression (3, 42, 46). However, TGF-β1 target genes appear to show varied responses. TSA inhibited TGF-β1-induced fibronectin and Col1a2 but reversed the repression of E-cadherin (16). Repression of Col1a2 was reported to be due to inhibition of Sp1 recruitment at the Col1a2 promoter, while increased E-cadherin expression was attributed to increased histone acetylation (16), suggesting gene-specific effects of HDACs and involvement of both histone and non-histone-dependent mechanisms. Furthermore, Smads can also interact with transcription repressors SKI and SnoN, which recruit corepressor complexes containing HDACs (9). Our results showed that TSA treatment increased H3K9/14Ac at the PAI-1 and p21 promoters and also enhanced TGF-β1-induced enrichment of H3K9/14Ac around SBE sites in the PAI-1 promoter. Taken together, these results suggest repressive roles for HDACs through both histone-dependent and possibly independent mechanisms in the regulation of PAI-1 and p21 expression in TGF-β1-treated RMCs.
TGF-β1 is a major mediator of HG (diabetic condition)-induced gene expression in renal cells (4, 20, 23, 45, 49, 67). We recently demonstrated that a TGF-β1 antibody blocked HG-induced changes in histone lysine methylation marks at fibrotic gene promoters in MCs, suggesting TGF-β1 may mediate HG-induced epigenetic mechanisms (51). In this study, we found that HG could also increase H3K9/14Ac at the PAI-1 and p21 promoters in MCs similar to TGF-β1. Furthermore, the stimulatory effects of HG on H3K9/14Ac at the PAI-1 and p21 promoters were significantly inhibited by a TGF-β1 antibody, suggesting the mediatory role of TGF-β1. Limited studies in diabetic animal models showed evidence of changes in global histone modifications in kidneys from diabetic animals (47), and reversal of diabetes induced changes in gene expression by HDAC inhibitors (37). In the current study, we showed for the first time that increased expression of the PAI-1 and p21 genes was associated with increased H3K9/14Ac at their promoters in vivo in glomeruli from mouse models of T1D and T2D. These results suggest that epigenetic changes at pathological gene promoters may lead to their sustained expression in vivo and contribute to long-term uncontrolled complications in diabetes. However, additional studies are needed to determine the time course of these changes, whether they are coregulated with gene expression, and whether they can be reversed by restoring normoglycemia or by other therapies used for DN.
In summary, our study provides extensive evidence that TGF-β1-induced PAI-1 and p21 gene expression in MCs involves the p300/CBP-mediated promoter H3K9/14Ac as well as Smad2/3 acetylation. Furthermore, H3K9/14Ac was also associated with increased expression of these genes induced by HG and in vivo diabetic conditions that are upstream of TGF-β1. Therefore, it is conceivable that histone hyperacetylation and related chromatin events involved in TGF-β1-mediated PAI-1 and p21 expression play important roles in the pathogenesis of DN and could therefore serve as potential therapeutic targets for diabetes-induced renal dysfunction.
GRANTS
This work was supported by National Institutes of Health Grants R01-DK-058191 and R01-DK-081705 (to R. Natarajan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: H.Y., M.A.R., M.K., and R.N. provided conception and design of research; H.Y., G.S., L.L., and M.W. performed experiments; H.Y. and G.S. analyzed data; H.Y., G.S., L.L., M.W., and R.N. interpreted results of experiments; H.Y., M.A.R., and G.S. prepared figures; H.Y. and M.A.R. drafted manuscript; H.Y., M.A.R., M.K., and R.N. edited and revised manuscript; M.A.R. and R.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Feng Miao, Dr. Louisa M. Villeneuve, and Lingxiao Zhang (Beckman Research Institute of City of Hope), for assistance. We are grateful to Drs. Michael Stallcup, Christopher Glass, Barry Forman, Satoshi Fujii, and Ken-ichi Isobe for providing plasmids.
REFERENCES
- 1. Al-Douahji M, Brugarolas J, Brown PA, Stehman-Breen CO, Alpers CE, Shankland SJ. The cyclin kinase inhibitor p21WAF1/CIP1 is required for glomerular hypertrophy in experimental diabetic nephropathy. Kidney Int 56: 1691–1699, 1999 [DOI] [PubMed] [Google Scholar]
- 2. Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol 14: 1008–1016, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Boekhoudt GH, Guo Z, Beresford GW, Boss JM. Communication between NF-kappa B and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J Immunol 170: 4139–4147, 2003 [DOI] [PubMed] [Google Scholar]
- 4. Brosius FC, Khoury CC, Buller CL, Chen S. Abnormalities in signaling pathways in diabetic nephropathy. Expert Rev Endocrinol Metab 5: 51–64, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci 114: 2363–2373, 2001 [DOI] [PubMed] [Google Scholar]
- 6. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 293: 1653–1657, 2001 [DOI] [PubMed] [Google Scholar]
- 7. Das F, Ghosh-Choudhury N, Venkatesan B, Li X, Mahimainathan L, Choudhury GG. Akt kinase targets association of CBP with SMAD 3 to regulate TGFbeta-induced expression of plasminogen activator inhibitor-1. J Cell Physiol 214: 513–527, 2008 [DOI] [PubMed] [Google Scholar]
- 8. Datta PK, Blake MC, Moses HL. Regulation of plasminogen activator inhibitor-1 expression by transforming growth factor-beta -induced physical and functional interactions between smads and Sp1. J Biol Chem 275: 40014–40019, 2000 [DOI] [PubMed] [Google Scholar]
- 9. Deheuninck J, Luo K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res 19: 47–57, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 17: 3091–3100, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dressler GR. Epigenetics, development, and the kidney. J Am Soc Nephrol 19: 2060–2067, 2008 [DOI] [PubMed] [Google Scholar]
- 12. Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693, 2005 [DOI] [PubMed] [Google Scholar]
- 13. Freidkin I, Herman M, Tobar A, Chagnac A, Ori Y, Korzets A, Gafter U. Effects of histone deacetylase inhibitors on rat mesangial cells. Am J Physiol Renal Physiol 298: F426–F434, 2010 [DOI] [PubMed] [Google Scholar]
- 14. Gaikwad AB, Gupta J, Tikoo K. Epigenetic changes and alteration of Fbn1 and Col3A1 gene expression under hyperglycaemic and hyperinsulinaemic conditions. Biochem J 432: 333–341, 2010 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh AK, Varga J. The transcriptional coactivator and acetyltransferase p300 in fibroblast biology and fibrosis. J Cell Physiol 213: 663–671, 2007 [DOI] [PubMed] [Google Scholar]
- 16. Ghosh AK, Yuan W, Mori Y, Varga J. Smad-dependent stimulation of type I collagen gene expression in human skin fibroblasts by TGF-beta involves functional cooperation with p300/CBP transcriptional coactivators. Oncogene 19: 3546–3555, 2000 [DOI] [PubMed] [Google Scholar]
- 17. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 389: 349–352, 1997 [DOI] [PubMed] [Google Scholar]
- 18. Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90: 595–606, 1997 [DOI] [PubMed] [Google Scholar]
- 19. Harding P, Balasubramanian L, Swegan J, Stevens A, Glass WF., 2nd Transforming growth factor beta regulates cyclooxygenase-2 in glomerular mesangial cells. Kidney Int 69: 1578–1585, 2006 [DOI] [PubMed] [Google Scholar]
- 20. Hoffman BB, Sharma K, Zhu Y, Ziyadeh FN. Transcriptional activation of transforming growth factor-beta1 in mesangial cell culture by high glucose concentration. Kidney Int 54: 1107–1116, 1998 [DOI] [PubMed] [Google Scholar]
- 21. Inoue Y, Itoh Y, Abe K, Okamoto T, Daitoku H, Fukamizu A, Onozaki K, Hayashi H. Smad3 is acetylated by p300/CBP to regulate its transactivation activity. Oncogene 26: 500–508, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Kang SW, Adler SG, Nast CC, LaPage J, Gu JL, Nadler JL, Natarajan R. 12-lipoxygenase is increased in glucose-stimulated mesangial cells and in experimental diabetic nephropathy. Kidney Int 59: 1354–1362, 2001 [DOI] [PubMed] [Google Scholar]
- 23. Kato M, Arce L, Wang M, Putta S, Lanting L, Natarajan R. A microRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int 80: 358–368, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi J, Natarajan R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci USA 104: 3432–3437, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kouzarides T. Chromatin modifications and their function. Cell 128: 693–705, 2007 [DOI] [PubMed] [Google Scholar]
- 26. Kuan CJ, al-Douahji M, Shankland SJ. The cyclin kinase inhibitor p21WAF1, CIP1 is increased in experimental diabetic nephropathy: potential role in glomerular hypertrophy. J Am Soc Nephrol 9: 986–993, 1998 [DOI] [PubMed] [Google Scholar]
- 27. Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 20: 615–626, 1998 [DOI] [PubMed] [Google Scholar]
- 28. Kurokawa R, Kalafus D, Ogliastro MH, Kioussi C, Xu L, Torchia J, Rosenfeld MG, Glass CK. Differential use of CREB binding protein-coactivator complexes. Science 279: 700–703, 1998 [DOI] [PubMed] [Google Scholar]
- 29. Lee YH, Koh SS, Zhang X, Cheng X, Stallcup MR. Synergy among nuclear receptor coactivators: selective requirement for protein methyltransferase and acetyltransferase activities. Mol Cell Biol 22: 3621–3632, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ling C, Groop L. Epigenetics: a molecular link between environmental factors and type 2 diabetes. Diabetes 58: 2718–2725, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Massague J. TGFbeta signaling: receptors, transducers, and Mad proteins. Cell 85: 947–950, 1996 [DOI] [PubMed] [Google Scholar]
- 32. Miao F, Gonzalo IG, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem 279: 18091–18097, 2004 [DOI] [PubMed] [Google Scholar]
- 33. Miyazono K, ten Dijke P, Heldin CH. TGF-beta signaling by Smad proteins. Adv Immunol 75: 115–157, 2000 [DOI] [PubMed] [Google Scholar]
- 34. Naito M, Zager RA, Bomsztyk K. BRG1 increases transcription of proinflammatory genes in renal ischemia. J Am Soc Nephrol 20: 1787–1796, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nelson JD, Flanagin S, Kawata Y, Denisenko O, Bomsztyk K. Transcription of laminin gamma1 chain gene in rat mesangial cells: constitutive and inducible RNA polymerase II recruitment and chromatin states. Am J Physiol Renal Physiol 294: F525–F533, 2008 [DOI] [PubMed] [Google Scholar]
- 36. Nicholas SB, Aguiniga E, Ren Y, Kim J, Wong J, Govindarajan N, Noda M, Wang W, Kawano Y, Collins A, Hsueh WA. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int 67: 1297–1307, 2005 [DOI] [PubMed] [Google Scholar]
- 37. Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, Lee HB. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-β1-induced renal injury. Am J Physiol Renal Physiol 297: F729–F739, 2009 [DOI] [PubMed] [Google Scholar]
- 38. Pardali K, Kurisaki A, Moren A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J Biol Chem 275: 29244–29256, 2000 [DOI] [PubMed] [Google Scholar]
- 39. Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol 6: 665–675, 2010 [DOI] [PubMed] [Google Scholar]
- 40. Pirola L, Balcerczyk A, Tothill RW, Haviv I, Kaspi A, Lunke S, Ziemann M, Karagiannis T, Tonna S, Kowalczyk A, Beresford-Smith B, Macintyre G, Kelong M, Hongyu Z, Zhu J, El-Osta A. Genome-wide analysis distinguishes hyperglycemia regulated epigenetic signatures of primary vascular cells. Genome Res 21: 1601–1615, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol 28: 1057–1068, 2010 [DOI] [PubMed] [Google Scholar]
- 42. Qiu P, Ritchie RP, Gong XQ, Hamamori Y, Li L. Dynamic changes in chromatin acetylation and the expression of histone acetyltransferases and histone deacetylases regulate the SM22alpha transcription in response to Smad3-mediated TGFbeta1 signaling. Biochem Biophys Res Commun 348: 351–358, 2006 [DOI] [PubMed] [Google Scholar]
- 43. Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res 90: 421–429, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reddy MA, Natarajan R. Epigenetics in diabetic kidney disease. J Am Soc Nephrol 22: 2182–2185, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reeves WB, Andreoli TE. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc Natl Acad Sci USA 97: 7667–7669, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sahar S, Reddy MA, Wong C, Meng L, Wang M, Natarajan R. Cooperation of SRC-1 and p300 with NF-kappaB and CREB in angiotensin II-induced IL-6 expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 27: 1528–1534, 2007 [DOI] [PubMed] [Google Scholar]
- 47. Sayyed SG, Gaikwad AB, Lichtnekert J, Kulkarni O, Eulberg D, Klussmann S, Tikoo K, Anders HJ. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant 25: 1811–1817, 2010 [DOI] [PubMed] [Google Scholar]
- 48. Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 45: 522–530, 1996 [DOI] [PubMed] [Google Scholar]
- 49. Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes 44: 1139–1146, 1995 [DOI] [PubMed] [Google Scholar]
- 50. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113: 685–700, 2003 [DOI] [PubMed] [Google Scholar]
- 51. Sun G, Reddy MA, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol 21: 2069–2080, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Toth KF, Knoch TA, Wachsmuth M, Frank-Stohr M, Stohr M, Bacher CP, Muller G, Rippe K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J Cell Sci 117: 4277–4287, 2004 [DOI] [PubMed] [Google Scholar]
- 53. Trouche D, Kouzarides T. E2F1 and E1A(12S) have a homologous activation domain regulated by RB and CBP. Proc Natl Acad Sci USA 93: 1439–1442, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tsuchida K, Zhu Y, Siva S, Dunn SR, Sharma K. Role of Smad4 on TGF-beta-induced extracellular matrix stimulation in mesangial cells. Kidney Int 63: 2000–2009, 2003 [DOI] [PubMed] [Google Scholar]
- 55. Tu AW, Luo K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J Biol Chem 282: 21187–21196, 2007 [DOI] [PubMed] [Google Scholar]
- 56. Wolf G. Cell cycle regulation in diabetic nephropathy. Kidney Int Suppl 77: S59–S66, 2000 [DOI] [PubMed] [Google Scholar]
- 57. Wolf G. Molecular mechanisms of diabetic mesangial cell hypertrophy: a proliferation of novel factors. J Am Soc Nephrol 13: 2611–2613, 2002 [DOI] [PubMed] [Google Scholar]
- 58. Wrana JL. Crossing Smads. Sci STKE 2000: re1, 2000 [DOI] [PubMed] [Google Scholar]
- 59. Xiao H, Hasegawa T, Isobe K. Both Sp1 and Sp3 are responsible for p21waf1 promoter activity induced by histone deacetylase inhibitor in NIH3T3 cells. J Cell Biochem 73: 291–302, 1999 [PubMed] [Google Scholar]
- 60. Xiao H, Hasegawa T, Isobe K. p300 collaborates with Sp1 and Sp3 in p21(waf1/cip1) promoter activation induced by histone deacetylase inhibitor. J Biol Chem 275: 1371–1376, 2000 [DOI] [PubMed] [Google Scholar]
- 61. Yang X, Pursell B, Lu S, Chang TK, Mercurio AM. Regulation of beta 4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J Cell Sci 122: 2473–2480, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol 21: 5979–5991, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yeh KT, Chen TH, Yang HW, Chou JL, Chen LY, Yeh CM, Chen YH, Lin RI, Su HY, Chen GC, Deatherage DE, Huang YW, Yan PS, Lin HJ, Nephew KP, Huang TH, Lai HC, Chan MW. Aberrant TGFbeta/SMAD4 signaling contributes to epigenetic silencing of a putative tumor suppressor, RunX1T1 in ovarian cancer. Epigenetics 6: 727–739, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yuan H, Lanting L, Xu ZG, Li SL, Swiderski P, Putta S, Jonnalagadda M, Kato M, Natarajan R. Effects of cholesterol-tagged small interfering RNAs targeting 12/15-lipoxygenase on parameters of diabetic nephropathy in a mouse model of type 1 diabetes. Am J Physiol Renal Physiol 295: F605–F617, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zager RA, Johnson AC. Progressive histone alterations and proinflammatory gene activation: consequences of heme protein/iron-mediated proximal tubule injury. Am J Physiol Renal Physiol 298: F827–F837, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, Guo L, Zhao K, Shevach EM, Paul WE. Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103+ inducible regulatory T cells. J Exp Med 206: 329–341, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ziyadeh FN. Mediators of diabetic renal disease: the case for tgf-Beta as the major mediator. J Am Soc Nephrol 15, Suppl 1: S55–S57, 2004 [DOI] [PubMed] [Google Scholar]
- 68. Ziyadeh FN, Sharma K. Overview: combating diabetic nephropathy. J Am Soc Nephrol 14: 1355–1357, 2003 [DOI] [PubMed] [Google Scholar]