

Abstract
We postulated that prostaglandin E2 (PGE2), which exhibits regulatory functions to control immune-mediated inflammation, fibrosis, oxidative stress, and tissue/cellular regeneration, has the potential to improve the course of nephritis. Therefore, the therapeutic potential of prostanoid on established nephritis in mice was evaluated focusing on its role on renal cellular recovery, with emphasis on its cytoprotecting and growth-promoting effects. Acute nephritis was induced in mice by single injection of nephrotoxic serum (NTS), followed by PGE2 administration with severity of nephritis evaluated over time. Mice injected with PGE2 recovered promptly with normalization of blood urea nitrogen and urine protein levels and histology. Recovery was observed with dosing of prostanoid at day 1, as well as day 4. With the use of selective EP1–4 receptor agonists, EP3 receptor has been identified as important in mediating beneficial effects of PGE2 in our system. PGE2 normalized glomerular cell losses during nephrotoxic serum-induced nephritis, restored synaptopodin distribution and F-actin filaments arrangement in glomeruli. In cell culture, PGE2 reduced nephrotoxim serum (NTS)-induced apoptosis of glomerular cells and promoted cell reproliferation after NTS-mediated injury. In conclusion, PGE2 treatment promotes resolution of glomerular inflammation. Consistent with this observation, the regenerative and cytoprotective effects of prostanoid on glomerular cells in culture were observed, suggesting that PGE2 may be beneficial in the treatment of glomerulonephritis.
Keywords: NTS, PGE2; nephritis, recovery, synaptopodin, apoptosis
much progress has been made in understanding the mechanisms involved in the pathogenesis of glomerulonephritis; however, efforts to understand and promote cellular and tissue recovery have been less forthcoming. In humans, most therapies are directed at alleviating either the underlying systemic disease process or preventing cellular events that lead to fibrosis within the kidney. Success at repair or reversal of disease has been limited. In this context, understanding how renal tissue recovers/regenerates after insult is especially relevant, since it has the potential to provide insights into strategies to improve outcomes.
Recovery from injury is a highly organized process, influenced by an array of cytokines, growth factors, and prostaglandins, produced by damaged cells and infiltrating leukocytes (6). In this study we investigated the efficacy of prostaglandin E2 (PGE2), a potent lipid mediator and major product of arachidonic acid metabolism, in restoration of nephrotoxic serum (NTS)-induced clinical and pathological changes in mouse kidneys and in cultured mouse glomerular cells. In nephrotoxic serum-induced nephritis (NTN), Fc receptor (FcR) and complement-mediated processes initiate inflammation, with subsequent cellular infiltration and glomerular cell injury, which results in proteinuria and reduced glomerular filtration rate (10, 33, 36). NTS targets include α3(IV) collagen and other basement membrane, cell surface, and matrix proteins (e.g., collagens, α3β1-integrin, aminopeptidase A, and laminin; Refs. 26, 28). Whether FcR or complement-mediated events dominate depends on the NTS preparation and context, although both pathways contribute to disease. PGE2 is produced in nearly all cell types in response to various stimuli and is synthesized from membrane lipids via the sequential actions of phospholipase A2, cyclooxygenases (PLA2, COXs), and prostaglandin (PG) terminal synthases (13, 35). PGE2 plays a role in maintaining the physiological homeostasis and is implicated in many physiologic functions (17, 2, 40). Differential expression of four distinct PGE2 receptors (EP1–4) defines the intracellular pathways activated by PGE2, and differential pathway activation results in specific functions that either promote or inhibit inflammation. The initial view that PGE2 promotes inflammation, like other PGs, has been countered, and the evidence supports its anti-inflammatory nature (8, 34, 23). Nevertheless, the mechanisms of PGE2 activation are complex and context dependent.
A wide range of regulatory actions of PGE2, particularly on immune-mediated inflammation, developmental processes, and tissue/cellular regeneration have been demonstrated on different cell types and diseases. Earlier studies have shown beneficial effect of PGE2 related to its general anti-inflammatory activity and effects on leukocytes (7, 32). More recent studies have focused on its role in tissue remodeling and regeneration. For example, renal PGE2-EP4 interactions inhibited the development of tubulointerstitial fibrosis (27). Additionally, PGE2 signaling via EP2 receptors facilitated growth/regeneration of articular cartilage and bone marrow cells (29), whereas COX-2-deficient systems have shown impaired muscle healing, implying a role for PGs in muscle regeneration (26). In other studies, PGE2 promoted blood cell recovery after injury in zebrafish and mice, and the stable analog of PGE2, dimethyl-PGE2 (dmPGE2), significantly increased engraftment of unfractionated and CD34+, human cord blood cells after xenotransplantation (7, 23). Nevertheless, a role of PGE2 in kidney recovery and regeneration has not been yet elucidated.
Given these considerations, we postulated that PGE2 would improve the course of established nephritis by cumulative effects on limiting inflammation, blunting immune responses, alleviating oxidative stress, and enhancing renal cellular recovery. Our main focus was on the role of PGE2 in renal cellular recovery during NTN, with emphasis on its cytoprotecting and growth-promoting effects. The experiment approach involved administration of PGE2 after cellular injury was established, using both whole animal and culture models. This strategy is distinguished from other experimental approaches, where prevention has been utilized to examine the role of participants. Herein we take advantage of the potential restorative properties of PGE2.
MATERIALS AND METHODS
Animals, Cells, and Reagents
Female C57BL/6 mice were purchased from The Jackson Laboratory. All experiments were performed in compliance with federal laws and institutional guidelines. The animal protocol was approved by the Georgia Health Sciences University Institutional Animal Care and Use Committee (no. A3307-01).
Eight- to-ten-week-old mice (18–20 g) were used for all experiments. Established cloned immortalized mouse podocyte and glomerular endothelial cell clones were employed as described previously (1). For passage, the cells were grown under “growth permissive” conditions (33°C), whereas to acquire a differentiated and quiescent phenotype for use in experiments, the cells were grown under “restrictive conditions” at 37°C in 95% air-5% CO2. EP1–4 receptor antagonists (SC 19220, AH 6809, L-798106, and L-161982; Tocris Bioscience), annexin V apoptosis kit (Phoenix Flow Systems), DeadEnd Fluorometric TUNEL System (Promega), anti-synaptopodin antibodies (Santa Cruz Biotechnology), PGE2 (Sigma), and FITC-conjugated anti-rabbit antibodies were purchased (Abcam).
Nephrotoxic Nephritis
Sheep nephrotoxic sera was prepared and administered as described previously (1, 19). In brief, glomeruli were isolated from normal C57BL/6 mice by differential sieving and used to immunize sheep. The nephrotoxic sheep serum was heat inactivated and absorbed with an excess amount of murine blood cells before use. Nephrotoxic sera were aliquoted and frozen at −80°C. For disease induction, NTS was administered as a single dose intraperitoneally (13.5 μl of serum per g mouse weight). Mice were then followed with measurement of proteinuria (BCA; Pierce) and serum urea level using the urea nitrogen direct kit (Stanbio Laboratory). At days 2, 4, 7, 9, kidneys were removed and either fixed in 10% buffered formalin or frozen in OCT medium. Four-micrometer sections from paraffin-embedded samples were stained with hematoxylin and eosin. Pathological evaluation by light microscopy was done in a blinded manner by M. P. Madaio.
PGE2 (5 μg/g) was injected intraperitoneally on days 2 and 4 after NTS dosing. In other experiments, different doses of PGE2 (10, 5, and 1 μg/g) were given daily to mice the day after NTS treatment (day 2). EP1–4 receptor antagonists (5 and 25 μg/g of body wt) were injected in separate groups of mice 4 h in advance of PGE2 infusion. Proteinuria and blood urea nitrogen (BUN) levels were measured as described previously (9).
Histological Evaluation
Kidneys were removed and either fixed in 10% buffered formalin or frozen in OCT medium. Light microscopy and immunofluorescence analysis were performed and evaluated in a blinded manner for severity of nephritis (22). Indirect immunofluorescence was used to evaluate synaptopodin expression using anti-synaptopodin and FITC conjugated anti-rabbit Abs. F-actin filaments were visualized with CytoPainter F-actin stainig kit (Abcam) according to the manufacturer's instructions. Images were acquired using Zeiss 7 Live Duoscan Confocal Microscope. Mean fluorescence intensity of stained glomeruli was quantified using Zen 2009 Software for laser scanning microscopes. In brief, by using a two-dimensional grid, the fluorescence intensity was quantified from the same pixel sized area of specimen, which related to the fluorophores present at the corresponding area and therefore to the local concentration of studied protein. Mean fluorescent intensity was determined by average value of intensities calculated from three similar images.
Electron microscopy.
Cells/tissue were fixed in 2% glutaraldehyde in 0.1 M sodium cacodylate (NaCac) buffer, pH 7.4, postfixed in 2% osmium tetroxide in NaCac, stained with 2% uranyl acetate, dehydrated with a graded ethanol series, and embedded in Epon-Araldite resin. Thin sections were cut with a diamond knife on a Leica EM UC6 ultramicrotome (Leica Microsystems, Bannockburn, IL), collected on copper grids, and stained with uranyl acetate and lead citrate. Cells/tissue were observed in a JEM 1230 transmission electron microscope (JEOL, Peabody, MA) at 110 kV and imaged with an UltraScan 4000 CCD camera and First Light Digital Camera Controller (Gatan, Pleasanton, CA).
Apoptosis of Cultured Glomerular Endothelial Cells and Podocytes: Regeneration Treatment Conditions
Glomerular epithelial cells (GEC) and podocytes were detached by mild enzymatic treatment from tissue flasks, placed in 15-ml conical tubes, 1 × 106, incubated with NTS (6%) and NTS and PGE2 (0.01–1 μg/ml) for 5 h. Apoptotic cells were enumerated using annexin V assay (Phoenix Flow Sytems), and the DeadEnd Fluorometric Tunel System (Promega) according to manufacturer's instructions. For regeneration experiments, podocytes and GEC were placed in Petri dishes overnight, starved for 24 h, and then 4% NTS was added for 9 h, after which the NTS removed, fresh media with/without PGE2 were added, and the cells were cultured for and additional 12 h. The cells were gently detached, and absolute cell enumeration was carried out by flow cytometry. Glomerular cell enumeration in kidney sections was carried out as described previously (37).
Synaptopodin Detection in Cultured Podocytes
Podocytes in four-well chamber slides were pretreated with PGE2 (0.01–1 μg/ml) for 1 h, followed by addition of NTS (6%) for 5 h. Distribution of synaptopodin was evaluated by indirect immunofluorescence using anti-synaptopodin and FITC-conjugated anti-rabbit antibodies. F-actin filaments were visualized with CytoPainter F-actin stainig kit (Abcam) according to manufacturer's instructions.
Microarray Analysis
Following RNA isolation (Trizol extraction) from the kidneys of control, NTS-, and NTS/PGE2-treated mice, the RNAs were processed for microarray analysis at the Georgia Health Sciences University Genomic Core Facility. Affymetrix Gene Chip: Mouse Gene 1.0 ST Array was used for gene expression profiling. Further statistical analysis and biological processes were generated through the use of iReport (Ingenuity Systems).
Accession Number
The gene array data discussed in the publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series Accession No. GSE27126.
Statistical Analysis
All values are presented as means ± SD. A Student's t-test was used for significant differences between two data sets and ANOVA for multiple data sets (proteinurea and BUN).
RESULTS
PGE2 improves Clinical and Pathologic Parameters in Mice with Established NTN
Animals who received NTS without PGE2, uniformly developed severe nephritis with proteinuria, ascites, weight gain, and azotemia, whereas PGE2 limited these elevations (Fig. 1, A and B). Among several doses (1, 5, and 10 μg/g) of PGE2 tested, 5 μg/g of animal body wt was the most effective (data not shown). Particularly noteworthy, initiation of therapeutic administration of PGE2 on day 4, after nephritis was well established, resulted in normalization and significant decrease in urine protein and BUN levels on day 9 (Fig. 1C). At days 7 and 9, the kidneys of the NTS animals showed signs of severe nephritis with profound glomerular and tubulointerstital injury, necrosis, and heavy proteinuria. By contrast, NTN mice treated with PGE2 demonstrated recovery, with no or limited evidence of nephritis (Fig. 2, A and C). The NTN mice had a reduced life expectancy: on day 9 one mouse died and the other four appeared moribund with oliguia, which limited evaluation of proteinuria in these animals, whereas all mice in NTS/PGE2 group acted normally in their cages.
Fig. 1.

Prostaglandin E2 (PGE2) administration limits clinical evidence of nephrotoxic serum-induced nephritis (NTN). Time-dependent development of proteinuria and elevation in blood urea nitrogen (BUN; A and B) in NTN mice with inhibition after PGE2 administration [started either on day 2 (A and B) or day 4 (C) following nephrotoxic serum (NTS) administration] Initially, NTS (13.5 μl/g wt) was injected into C57/Bl6 mice, followed by daily PGE2 administrations (5 μg/g ip) starting on days 2 or 4, following the NTS injection. Urine protein levels could not be evaluated in the NTS, day 9 mice, due to oliguria or death. Even late administration of PGE2 started on day 4 resulted in decrease in BUN and proteinuria on day 9 (C). Results are shown as means ± SD; 5 mice were used in each group. *P ≤ 0.05, **P ≤ 0.01.
Fig. 2.

PGE2 treatment limits histologic evidence of nephritis. Representative microscopies (hematoxylin and eosin) from day 7 mice are shown (A). Kidneys of the NTS animals showed signs of severe nephritis with profound glomerular and tubulointerstital injury, necrosis, and heavy proteinuria. No or minimal evidence of nephritis was observed in NTS mice treated with PGE2. (top and bottom: ×100 magnification; middle: ×400). Morphometric evaluation of glomerular cells (B). Glomerular cell numbers were significantly higher in NTS/PGE2-treated mice. Cells were enumerated per glomeruli in 6 representative glomeruli. Representative transmission electron microscopy of glomeruli from an NTS-treated mouse after 24 h on day 7 and NTS/PGE2-treated mouse on day 7 and PBS-injected control mouse (C). After NTS administration, foot processes were variably shortened and widened on day 1 and completely effaced on day 7 (C). By contrast in NTS and PGE2-treated mice, foot processes were mostly intact on day 7. Results are shown as means ± SD. **P ≤ 0.01; ***P ≤ 0.001.
To determine which of four PGE2 receptors (EP1–4) were responsible for the observed beneficial effect, EP1–4 receptor antagonists (5 μg/g wt) were injected daily, beginning 4 h in advance of PGE2 administration to the NTN mice. Only the EP3 receptor antagonist fully inhibited the beneficial effects of PGE2 (Fig. 3). To further confirm that EP1, 2, and 4 receptors do not participate in PGE2-mediated recovery, higher concentrations (25 μg/g wt) of either EP1, EP2, and EP4 receptor antagonists were infused in separate groups of mice. By contrast with the EP3 receptor antagonist, they did not reverse the beneficial effects of PGE2.
Fig. 3.

EP3 receptor antagonist inhibits the beneficial effect of PGE2. EP1–4 receptor antagonists (SC 19220, AH 6809, L-798106, and L-161982, respectively) were injected in groups of mice (5 and 25 μg/g) 4 h in advance of PGE2 infusion starting on day 2 after NTS treatment. BUN levels were measured in mice on day 7. Results are shown as means ± SD. *P < 0.05.
PGE2 Limits Podocyte Injury in Established NTN
Since PGE2 treatment limited NTS-induced proteinuria, we reasoned that improvement in podocyte injury and restoration of actin associated proteins were involved. Therefore, we investigated modulation of expression of synaptopodin, which regulates the actin cytoskeleton and is expressed in foot processes as the Synpo-long isoform (25, 39). On initial analysis, there were differences in the pattern and intensity of synaptopodin staining, where normal animals displayed a “fine linear” staining pattern with intervals between lines, likely corresponding to intact foot processes in podocytes (Fig. 4A). By contrast, in the NTN mice, the lines and intervals disappeared, presumably due to foot process effacement. This normal pattern was restored in the NTS/PGE2 mice. Consistent with this conclusion, assessment of synaptopodin expression between the groups revealed quantitative differences: the mean fluorescence intensity of stained synaptopodin (evaluated from equal pixel sized area of specimens, see materials and methods) from the NTS-treated mice was reduced (487.4 ± 128.211), while the NTS/PGE2 mice had significantly higher values (760.05 ± 65.02; P < 0.05). Modulation in podocyte ultrastructure and foot processes was also observed by electron microscopy, where extensive loss of foot processes in nephritic mice was present (Fig. 2C). By contrast, foot process fusion had been largely restored after PGE2 administration.
Fig. 4.
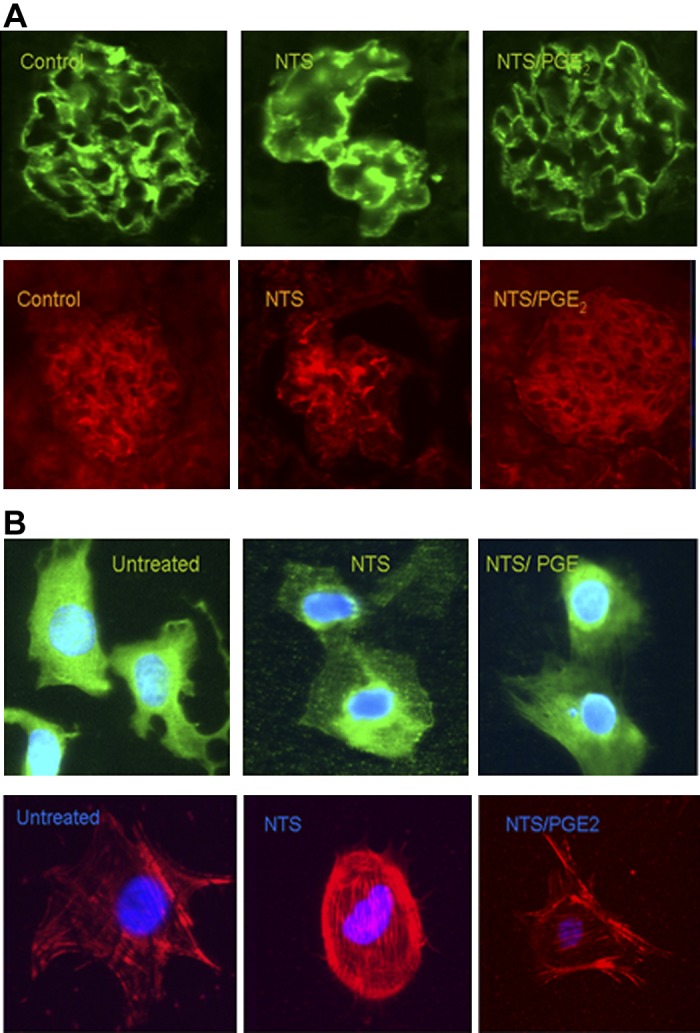
PGE2 treatment restores alterations in synaptopodin and F-actin staining in NTS-treated mice and cultured podocytes. A: in mice, the “fine linear” pattern of synaptopodin and F-actin staining in normal glomeruli was mostly eliminated in NTS mice, presumably due to foot process effacement. This normal pattern was restored in the NTS/PGE2 mice. B: in culture, podocytes were pretreated with PGE2 (0.01–1 μg/ml) 1 h, followed by addition of NTS (6%) for 5 h. Following NTS treatment of cultured podocytes, synaptopodin localization became less cytoplasmic and more perinuclear and PGE2 treatment limited synaptopodin distribution and F-actin filaments to nearly the untreated condition (C). Results are shown as means ± SD. *P < 0.05.
Alterations in cytoskeleton and associated proteins were evaluated in cultured podocytes by assessment of synaptopodin staining and F-actin arrangement after NTS/ ± PGE2 treatment conditions. Disarrangement of F-actin cytoskeleton, perinuclear dislocation, and loss of cytoplasmic staining of synapotopodin were observed after NTS treatment (Fig. 4B). PGE2 treatment maintained synaptopodin distribution and F-actin filaments nearly to the untreated condition (Fig. 4B).
PGE2 Upregulates Cell Cycle Progression Genes in the Kidney Following NTS Exposure
To begin to define genes/pathways responsible for PGE2-mediated recovery, microarray gene expression and pathway analysis (Ingenuity Systems) was carried out. Kidneys from NTS- and NTS/PGE2-exposed animals were analyzed. The most significant biological processes affected by prostanoid treatment are listed in Table 1. Proliferation of cells was ranked highest based on P value enrichment score and the number of differentially expressed genes, consistent with the in vivo results. Notably, genes specific for cell cycle/growth-promoting pathways were upregulated: cyclin-dependent kinase 1 (CDK1; 5.568-fold change), FOXM1 (3.882-fold), S100A6 (8.484-fold), CCNA2 (2.003-fold), CCNF (2.014-fold), and ANLN (8.698-fold) in PGE2-treated animals. These findings are consistent with the reduction in glomerular cell numbers observed in NTS-injected mice as evaluated by morphometric quantification on days 4 and 7. By contrast glomerular cell numbers were higher in the PGE2-treated mice (Fig. 2B) consistent with the conclusion that enhanced glomerular cell proliferation contributed to the improvement.
Table 1.
Ingenuity iReport reveals most significant biological processes affected by PGE2 treatment
| Biological Process | DEG | P Value |
|---|---|---|
| Proliferation of cells | 272 | 6.52879E-14 |
| Tissue development | 232 | 1.38971E-07 |
| Growth of cells | 195 | 6.83175E-10 |
| Cell movement | 183 | 9.61159e-09 |
| Development of organ | 146 | 0.000503125 |
| Immune response | 138 | 1.97659E-05 |
| Transport of molecules | 133 | 4.16644E-08 |
| Organismal death | 106 | 0.000186372 |
| Cell death of organs | 89 | 9.45585E-05 |
| Cell movement of leukocytes | 71 | 0.000250124 |
| Survival of organism | 67 | 1.92577E-07 |
| Cell movement of phagocytes | 52 | 0.000250844 |
| Kidney development | 26 | 0.000354856 |
PGE2, prostaglandin E2; DEG, differentially expressed genes.
To further address this issue, cellular recovery was investigated in cultured glomerular cells following NTS treatment. In brief, the cells were exposed to NTS, which time and dose-dependently reduced glomerular cell numbers. The NTS was then removed, and the cells were either treated with PGE2 or allowed to grow without it in normal media. Cellular numbers were then evaluated. After removal of NTS, PGE2 increased cell proliferation rates (Fig. 5, A and B), and more of the PGE2-treated cells regained a normal morphology (Fig. 5, C and D). Noteworthy, PGE2 had no effect on cells under normal conditions (data not shown); the growth-promoting effect was observed only after following NTS exposure.
Fig. 5.

PGE2 enhances mouse glomerular epithelial cell (GEC) proliferation after NTS-induced cell injury. Podocytes were treated with NTS for 9 h followed by media change to remove NTS; thereafter cultures were with or without PGE2 (0.01–1 μg/ml, overnight). Cells were detached and cell numbers assessed by FACS (A and B). Endothelial cells were incubated with NTS overnight, followed by changing media to remove NTS, with addition of fresh media with or without PGE2 0.1 μg/ml (C and D).
PGE2 Has Direct Effects on Cultured Mouse Glomerular Cells
To further evaluate the effects of PGE2 on glomerular cells, the capacity of PGE2 to limit NTS-induced cellular injury was evaluated. In culture NTS treatment reduced murine podocyte and endothelial cell numbers in a time- and dose-dependent manner, whereas normal sheep serum had no effect (data not shown). This effect was modulated by PGE2, as the number of viable cells after NTS exposure was increased. Furthermore, the cells appeared more confluent in the presence of PGE2, and their morphology and adherence appeared normal. To determine whether inhibition of cell death and/or apoptosis contributed to the benefit, podocytes were preincubated with PGE2 before NTS and the number of apoptotic cells was compared by FACS. NTS induced an increase in annexin V-positive cells, whereas, preincubation with PGE2 significantly reduced the NTS-mediated increase (Fig. 6A). Similar results were obtained with endothelial cells (data not shown). Apoptosis was also examined by terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL); PGE2 reduced TUNEL-positive staining in a dose-dependent manner (Fig. 6B).
Fig. 6.

PGE2 attenuates apoptotic actions of NTS on glomerular cells. Cultured podoctyes (POD) or GEC were pretreated with or without PGE2. Thereafter, NTS was added for 5 h. The number of apoptotic cells were determined using annexin V assay by FACS. Pretreatment with PGE2 suppressed NTS induced increase of phosphatidyl serine (A) and terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling-positive/apoptotic cells (B). Results are shown as means ± SD. *P < 0.05.
DISCUSSION
Our results confirm the hypothesis that PGE2 promotes recovery during the course of established nephritis in mice. The experimental approach involved treating animals after disease induction, with evaluation of the course of disease following PGE2 administration. PGE2 reversed the clinical and pathologic parameters of nephritis. This was not likely due to an effect on prevention of newly established disease (i.e., with ongoing NTS deposition), since nephritis was well established at 24 h and most of the nephritogenic antibodies are no longer circulating at this time (22). Thus the improvement was most likely due to a restorative effect of PGE2 on disease by promoting tissue repair after injury.
The effects are likely mediated through the EP3 receptor, as its selective antagonist, but not other selective EP receptor antagonists limited the PGE2 benefit. PGE2 signaling through EP3 receptors has been shown to mediate reorganization of damaged neuronal connections (16), attenuate myocardial injury during ischemia reperfusion (24), and regulate COX-2 expression in the kidney (38). This is a well-characterized G protein-coupled receptor (4), which exists in multiple isoforms, and each of them can be involved in variety of signaling pathways, including the prosurvival pathways of mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (11, 12), and in the modulation of Ca2+/calmodulin reaction and Rho kinases (16), which all play important role in maintaining/regulating podocyte ultrastructure (20).
Improvement in proteinuria by PGE2 is likely explained by a direct effect on podocytes, as restoration of synaptopodin and podocyte foot processes were observed. Synaptopodin is essential for the integrity of the podocyte actin cytoskeleton, and when phosphorylated, it is protected from degradation (25, 39). This particular benefit was likely due, at least in part, by a PGE2-mediated increase in cAMP; this pathway is known to activate cAMP dependent PKA, inhibit Ca immobilization, and reduce inositol 3-phosphate turnover (35). These effects diminish the activity of calcineurin, thereby protecting synaptopodin from degradation (31).
The cell culture experiments provide further insights. PGE2 decreased NTS-induced apoptosis of both podocytes and endothelial cells and enhanced GEC proliferation rates following NTS-mediated damage. Although we cannot eliminate the possibility that a protective effect on adhesion may have contributed, modulation of apoptosis/survival was definitely observed. These findings are consistent with the Ingeniuty gene expression profiling/results, where 272 differentially expressed genes were identified as responsive to PGE2 treatment and related to cell proliferation processes, Among them FOXM1 was upregulated (3.882-fold); its expression is known to be restricted to proliferative tissues and has been detected in regenerative tissues and various malignancies (30). A number of cyclins and cyclin dependent kinases were also significantly elevated, such as CDK1 (5,588-fold), cyclin A2 (CCN2, 2.003-fold), and cyclin F (2.014-fold), consistent with their role in cell cycle progression and activators of FOXM 1 gene. Particularly noteworthy, cyclins have been shown to interact with DNA repair proteins and contribute to repair processes (33, 18). Enhanced S100A6 gene expression, known to be preferentially expressed in proliferating cells (21), is also consistent with the beneficial effect of PGE2. Collectively, the results indicate benefit mediated at least in part, by eliciting growth-promoting genes regulating cell cycle progression and recovery processes.
In conclusion, regenerative and renoprotective effects of PGE2 during established NTN and after injury to glomerular cells in culture were observed. The results of these studies provide the rational to further define the cellular pathways involved in the process and to investigate the efficacy of PGE2 during the course of human glomerulonephritis. Ideally, regeneration-promoting therapeutics will be small molecule/s that can easily be produced in large amounts, facilitate regeneration, and have limited adverse effects in other tissues. PGE2 is an excellent candidate, given its physiologic and restorative effects.
GRANTS
The work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R21-DK-081140.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: N.K., M.B., and M.P.M. conception and design of research; N.K., M.M., and K.C. performed experiments; N.K., M.M., K.C., and M.P.M. analyzed data; N.K. and M.P.M. interpreted results of experiments; N.K. prepared figures; N.K. drafted manuscript; M.P.M. edited and revised manuscript; M.P.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We greatly acknowledge Robert Smith's skillful assistance in providing electron microphotographs and processing data. We thank Dr. Michael Duncan for helpful discussions and Dr. Irina Sazonova for providing additional mice.
Portions of this work have been presented at American Society of Nephrology Annual Meeting, 2010, 2011, and 2012, appeared in the abstract form (J Am Soc Nephrol 23: 140A, 2012, J Am Soc Nephrol 22: 630A, 2011, J Am Soc Nephrol 21: 273A, 2010). Also, portions of this work were also presented at the Experimental Biology 2011 meeting and appeared as a printed abstract of that meeting (FASEB J 25: lb341, 2011).
REFERENCES
- 1. Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int 65: 2223–2227, 2004 [DOI] [PubMed] [Google Scholar]
- 2. Aronoff DM, Bergine IL, Lewisb C, Goelf D, O'Brienf E, Peters-Goldend M, Mancusod P. E-prostanoid 2 receptor signaling suppresses lung innate immunity against Streptococcus pneumonia. Prostaglandins Other Lipid Mediat 98: 23–30, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadéa G, Bouquier N, Bibeau F, Lemmers B, Blanchard JM. A novel function of cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol 196: 147–162, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Audoly LP, Ma L, Feoktistov I, de Foe SK, Breyer MD, Breyer RM. Prostaglandin E-prostanoid-3 receptor activation of cyclic AMP response element-mediated gene transcription. J Pharmacol Exp Ther 289: 140–148, 1999 [PubMed] [Google Scholar]
- 5. Bondesen BA, Mills ST, Pavlath GK. The COX-2 pathway regulates growth of atrophied muscle via multiple mechanisms. Am J Physiol Cell Physiol 290: C1651–C1659, 2006 [DOI] [PubMed] [Google Scholar]
- 6. Bunce CM, Velica P. Prostaglandins in muscle regenaration. J Muscle Res Cell Motil 29:163–167, 2008 [DOI] [PubMed] [Google Scholar]
- 7. Cattell V, Smith J, Cook HT. Prostaglandin E1 suppresses macrophage infiltration and ameliorates injury in an experimental model of macrophage-dependent glomerulonephritis. Clin Exp Immunol 79: 260–265, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan & Moore ARMM. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J Immunol 184: 6418–6426, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Christensen M, Su AW, Snyder RW, Greco A, Lipschutz JH, Madaio MP. Simvastatin protection against acute immune-mediated glomerulonephritis in mice. Kidney Int 69: 457–463, 2006 [DOI] [PubMed] [Google Scholar]
- 10. Couser WG: Mechanisms of glomerular injury: an overview. Semin Nephrol 11: 254–258, 1991 [PubMed] [Google Scholar]
- 11. Feng C, Beller EM, Bagga S, Boyce JA. Human mast cells express multiple EP receptors for prostaglandin E2 that differentially modulate activation responses. Blood 107: 3243–3250, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65: 45–79, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294:1871–1875, 2001 [DOI] [PubMed] [Google Scholar]
- 14. Goessling W, Allen RS, Guan X, Jin P, Uchida N, Dovey M, Harris JM, Metzger ME, Bonifacino AC, Stroncek D, Stegner J, Armant M, Schlaeger T, Tisdale JF, Zon LI, Donahue RE, North TE. Prostaglandin E2 enhances human cord blood stem cell Xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell 8: 445–458, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 136: 1136–1147, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hatae N, Sugimoto Y, Ichikawa A. Prostaglandin receptors: advances in the study of EP3 receptor signaling. J Biochem 131: 781–784, 2002 [DOI] [PubMed] [Google Scholar]
- 17. Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood 113: 5444–5455, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, Correll M, Quackenbush J, Livingston DM, Gygi SP, Sicinski P. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 474: 230–234, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaneko Y, Nimmerjahn F, Madaio MP, Ravetch JV. Pathology and protection in nephrotoxic nephritis is determined by selective engagement of specific Fc receptors. J Exp Med 203: 789–797, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim EY, Suh JM, Chiu YH, Dryer SE. Regulation of podocyte BKCa channels by synaptopodin, Rho, and actin microfilaments. Am J Physiol Renal Physiol 299: F594–F604, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komatsu K, Kobune-Fujiwara Y, Andoh A, Ishiguro S, Hunai H, Suzuki N, Kameyama M, Murata K, Miyoshi J, Akedo H, Tatsuta M, Nakamura H. Increased expression of S100A6 at the invading fronts of the primary lesion and liver metastasis in patients with colorectal adenocarcinoma. Br J Cancer 83: 769–774, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Madaio MP, Salant DJ, Adler S, Couser WG. Effect of antibody charge and concentration on deposition of antibody to glomerular basement membrane. Kidney Int 26: 397–403, 1984 [DOI] [PubMed] [Google Scholar]
- 22a. Mathieson PW. Podocyte actin in health, disease and treatment. Nephrol Dial Transplant 25: 1772–1773, 2010 [DOI] [PubMed] [Google Scholar]
- 23. Mandapathil M, Szczepanski MJ, Szajnik M, Ren J, Jackson EK, Johnson JT, Gorelik E, Lang S, Whiteside TL. Adenosine and prostaglandin E2 cooperate in the suppression of immune responses mediated by adaptive regulatory T cells. J Biol Chem 285: 27571–275780, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin M, Meyer-Kirchrath J, Kaber G, Jacoby C, Flogel U, Schrader J, Ruther U, Schror K, Hohlfeld T. Cardiospecific overexpression of the prostaglandin EP3 receptor attenuates ischemia-induced myocardial injury. Circulation 112: 400–406, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Mathienson PW. Podocyte actin in health, disease and treatment. Nephol Dial Transplant 25: 1772–1773, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Mendrick DL, Rennke HG, Cotran RS, Springer TA, Abbas AK. Monoclonal antibodies against rat glomerular antigens: production and specificity. Lab Invest 49: 107–117, 1983 [PubMed] [Google Scholar]
- 27. Nakagawa N, Yuhki K, Kawabe J, Fujino T, Takahata O, Kabara M, Abe K, Kojima F, Kashiwagi H, Hasebe N, Kikuchi K, Sugimoto Y, Narumiya S, Ushikubi F. The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int 82: 158–171, 2012 [DOI] [PubMed] [Google Scholar]
- 28. Orikasa M, Matsui K, Oite T, Shimizu F. Massive proteinuria induced in rats by a single intravenous injection of a monoclonal antibody. J Immunol 141: 807–814, 1988 [PubMed] [Google Scholar]
- 29. Otsuka S, Aoyama T, Furu M, Ito K, Jin Y, Nasu A, Fukiage K, Kohno Y, Maruyama T, Kanaji T, Nishiura A, Sugihara H, Fujimura S, Otsuka T, Nakamura T, Toguchida J. PGE2 signal via EP2 receptors evoked by a selective agonist enhances regeneration of injured articular cartilage. Osteoarthritis Cartilage 17: e529–e538, 2009 [DOI] [PubMed] [Google Scholar]
- 30. Raychaudhury P, Park HJ. FoxM1: a master regulator of tumor metastasis. Cancer Res 71: 4329–4333, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rusnak F, Mertz P. Calcineurin: form, function. Physiol Rev 80: 1483–1521, 2000 [DOI] [PubMed] [Google Scholar]
- 32. Ruwart MJ, Rush BD, Friedle NM, Piper RC, Kolaja GJ. Protective effects of 16,16-dimethyl PGE2 on the liver and kidney. Prostaglandins 21, Suppl 97–102, 1981 [DOI] [PubMed] [Google Scholar]
- 33. Salant DJ, Belok S, Madaio MP, Couser WG. A new role for complement in experimental membranous nephropathy in rats. J Clin Invest 66: 1339–1350, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol 184: 7207–7218, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56: 387–343, 2004 [DOI] [PubMed] [Google Scholar]
- 36. Tarzi RM, Davies KA, Classens JW, Verbeek JS, Walport MJ, Cook HT. Both Fc gamma receptor I and Fc gamma receptor III mediate disease in accelerated nephrotoxic nephritis. Am J Pathol 162: 1677–1683, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vlahakos D, Foster MH, Ucci AA, Barrett KJ, Datta SK, Madaio MP. Murine monoclonal anti-DNA antibodies penetrate cells, bind to nuclei, and induce glomerular proliferation and proteinuria in vivo. Am Soc Nephol 2: 1345–1354, 1992 [DOI] [PubMed] [Google Scholar]
- 38. Vio CP, Quiroz-Munoz M, Cuevas CA, Cespedes C, Ferreri NR. Prostaglandin E2 EP3 receptor regulates cyclooxygenase-2 expression in the kidney. Am J Physiol Renal Physiol 303: F449–F457, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Welsh GI, Saleem MA. The podocyte cytoskeleton-key to a functioning glomerulus in health and disease. Nat Rev Nephrol 8: 14–21, 2012 [DOI] [PubMed] [Google Scholar]
- 40. Zhu Z, Fu C, Li X, Song Y, Li C, Zou M, Guan Y, Zhu Y. Prostaglandin E2 promotes endothelial differentiation from bone marrow-derived cells through AMPK activation. PLos One 6: e23554, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]