

Abstract
Ozone (O3) causes significant adverse health effects worldwide. Nasal epithelial cells (NECs) are among the first sites within the respiratory system to be exposed to inhaled air pollutants. They recruit, activate, and interact with immune cells via soluble mediators and direct cell-cell contacts. Based on our recent observation demonstrating the presence of natural killer (NK) cells in nasal lavages, the goal of this study was to establish a coculture model of NECs and NK cells and examine how exposure to O3 modifies this interaction. Flow cytometry analysis was used to assess immunophenotypes of NK cells cocultured with either air- or O3-exposed NECs. Our data show that coculturing NK cells with O3-exposed NECs decreased intracellular interferon-γ (IFN-γ), enhanced, albeit not statistically significant, IL-4, and increased CD16 expression on NK cells compared with air controls. Additionally, the cytotoxicity potential of NK cells was reduced after coculturing with O3-exposed NECs. To determine whether soluble mediators released by O3-exposed NECs caused this shift, apical and basolateral supernatants of air- and O3-exposed NECs were used to stimulate NK cells. While the conditioned media of O3-exposed NECs alone did not reduce intracellular IFN-γ, O3 enhanced the expression of NK cell ligands ULBP3 and MICA/B on NECs. Blocking ULBP3 and MICA/B reversed the effects of O3-exposed NECs on IFN-γ production in NK cells. Taken together, these data showed that interactions between NECs and NK cells in the context of O3 exposure changes NK cell activity via direct cell-cell interactions and is dependent on ULBP3/MICA/B expressed on NECs.
Keywords: air pollution, immune response, cell-cell interaction, conditioned media, natural killer cell ligands
air pollution and in particular ozone (O3) causes significant adverse health effects worldwide (14). Nasal epithelial cells (NECs) are among the first sites within the respiratory system to be exposed to inhaled air pollutants and respond to O3 exposure with the release of proinflammatory cytokines and chemokines (3, 11, 36, 38, 52, 70). In addition to epithelial cells (ECs), the respiratory mucosa also harbors resident immune cells, such as monocytes/macrophages, neutrophils, dendritic cells (DCs), and T cells (21, 34, 43, 53, 57), and exposure to O3 has been shown to modify their activity (4, 23, 30, 31). We have recently shown that natural killer (NK) cells are also important immune cells present in the nasal lavage (33), yet whether and how exposure to O3 affects NK cell activity is largely unknown.
ECs do not only provide a protective barrier to inhaled agents like O3 but also release soluble factors such as chemokines, cytokines, and lipid mediators (such as prostaglandins) that recruit and activate immune cells, including NK cells (26). For example, IL-8, monocyte chemotactic protein (MCP)-1, regulated on activation normal T cell expressed and secreted (RANTES), and eotaxin-3 are known to be produced by ECs after O3 (3, 11, 36). Through interaction with chemokine receptors, such as CXCR3 (73), these chemokines tightly regulate the recruitment of neutrophils, monocytes, NK cells, and other immune cells to the airways (50, 59, 78). In addition, it has been shown that ECs and NK cells not only communicate via soluble factors but also through direct receptor-ligand cell-cell interactions (69). Stressed ECs express myosin heavy chain class I polypeptide-related sequence (MIC) A and B and UL16-binding proteins (ULPBs) (10), which are ligands for the activating NK cell receptor NKG2D (15, 20, 54).
In the respiratory mucosa, NK cells have two important functions in the context of an immune response. First, they patrol in the epithelium and kill virus-infected and transformed tumor cells (74). NK cells express inhibitory receptors, such as KIR-L (73), recognizing self-antigens on host cells, and activating receptors, such as NKp46 or NKG2D (41, 54). Cytotoxicity and the ability of NK cells to kill target cells are mediated by the ability to release perforin and granzyme B. Second, NK cells are an important source of chemokines and cytokines (73), such as Th1 cytokines like interferon-γ (IFN-γ) and Th2 cytokines like IL-4 (2, 8), which are both crucial for the early immune response, especially in the context of viral infections (68, 73). NK cell-derived cytokine production can shift toward Th2-type cytokines, which has been demonstrated in subjects with allergic diseases (22, 80). However, whether the balance between NK cell-derived Th1 (e.g., IFN-γ) and Th2 (e.g., IL-4) cytokines can be modified in a nonallergic setting is not known.
Based on our previous observation that NK cells constitute an important immune cell type in the nasal lavage (33), we wanted to explore whether and how ECs can modify NK cell activity, how this interaction is altered in the context of O3 exposure, and determine the role of soluble mediators and ligand-receptor interaction in these responses. To do so, we developed a novel EC-NK cell coculture model using differentiated human NECs and peripheral blood NK cells. Our data show that, following exposure to O3, NK cells cocultured with NECs modify their activity and that NEC-derived soluble mediators do not solely mediate these changes.
MATERIALS AND METHODS
Nasal epithelial cells.
Human nasal biopsies were obtained from nonsmoking healthy adult volunteers [age = 27.9 ± 5.96 yr, body mass index (BMI) = 26.8 ± 7.00, female/male = 14/7, African American/white/Asian = 11/10/1, all were non-Hispanic] as described previously (37) using a protocol approved by the University of North Carolina School of Medicine Institutional Review Board for Biomedical Research. In addition, written informed consent was provided by each study participant. Briefly, nasal biopsies were obtained by gently stroking the interior surface of the turbinate several times with a Rhino-Probe cuvette (Arlington Scientific, Arlington, TX). For in vitro cell cultures, NECs were expanded to passage 2 in bronchial epithelial growth medium (Cambrex Bioscience Walkersville, Walkersville, MD) and plated on collagen-coated transwells with a 0.4-μM pore size (Trans-CLR; Costar, Cambridge, MA) and cultured in a 1:1 mixture of bronchial ECs basic medium and DMEM-H with SingleQuot supplements (Cambrex Bioscience), bovine pituitary extracts (13 mg/ml), BSA (1.5 mg/ml), and nystatin (20 units). Upon confluency, apical media was removed, and all-trans retinoic acid was added to the basolateral medium to establish air-liquid interface (ALI) culture conditions to promote differentiation. Mucociliary differentiation was achieved after 28 days post-ALI.
Natural killer cells.
Peripheral blood NK cells were isolated from peripheral blood mononuclear cells (PBMCs) obtained from healthy nonsmoking human volunteers (age = 27.0 ± 6.29 yr, BMI = 25.9 ± 5.03, female/male = 12/15, African American/white/Asian = 11/16/0) using a protocol approved by the University of North Carolina School of Medicine Institutional Review Board for Biomedical Research. In addition, written informed consent was provided by each study participant. PBMCs were isolated using a Lymphoprep (GIBCO) gradient as previously described (33). NK cells were isolated from PBMCs using Dynabeads NK isolation kits [Dynabeads Untouched Human NK Cells (Invitrogen); a negative selection, purity always >90%, around 95% in average] according to the supplier's instruction. After isolation, the NK cells were resuspended in NK cell media consisting of RMPI 1640 with l-glutamine, 10% heat-inactivated FBS, and 1% penecillin/streptomycin (GIBCO, Invitrogen, Grand Island, NY).
Coculture model of NEC and NK cells.
NK cells (2.5 × 105) were added in 100 μl of media to the apical side of NECs and allowed to incubate for 24 h after which NECs and NK cells were harvested for analysis of coculture effects. Basolateral supernatants and apical washes were collected and stored at −80°C until analysis was conducted as described by us before (32). As controls, NECs alone or NK cells alone were analyzed in comparison.
Microscopic analysis.
For histological analysis of fresh tissue, biopsies were fixed with ethanol and embedded in paraffin, and 4-μm sections were stained with hematoxylin and eosin. Cocultures were fixed with ethanol and stained with cyto blue (Innovex Bioscience, Richmond, CA). Both were visualized using light microscopy.
For confocal laser-scanning microscopy analysis, NK cells were labeled with a fluorescein-emitting dye (Vybrant Mulitcolor Cell-Labeling Kit; Invitrogen) before addition to the NECs. The cocultures were fixed with 4% paraformaldehyde (PFA) and stained with phalloidin-rhodamine for F-actin cytoskeleton of the NECs. The cell nuclei were stained with DAPI. Samples were visualized using a Nikon C1Si laser-scanning confocal microscope, and images were processed using the EZ-C1 FreeViewer software (Nikon Instruments, Melville, NY).
O3 exposure.
Differentiated NECs were exposed to 0.4 parts/million (ppm) O3 or filtered air for 4 h under ALI conditions using exposure chambers (80% relative humidity, 5% CO2) operated by the United States Environmental Protection Agency, Environmental Public Health Division. At 2 h postexposure, NK cells were added to the apical side of NECs to establish the cocultures. NECs and NK cells were harvested 24 h after establishing the coculture for analysis, and samples were collected and stored as described above.
Flow cytometry.
For flow cytometric analysis of fresh nasal superficial scrape biopsies, the tissue was incubated for 30 min in RPMI media with 15 μg/ml DNase I (catalog no. DN25-100MG; Sigma) and 5 μg/ml Pronase E (catalog no. P6911; Sigma) and consecutively stained for analysis (antibody cocktails see Table 1).
Table 1.
Flow cytometry antibody cocktails used for the different endpoints
| Tube | FITC | PE (PacBlue for NK surface and biopsy) | PE-TR | APC | PerCP-Cy5.5 | APC-Cy7 |
|---|---|---|---|---|---|---|
| Biopsy | CD31 | Live/dead3 | CD563 | NKG2D4 | CD161 | CD451 |
| Biopsy isotype control | IgG11 | Live/dead3 | IgG23 | IgG11 | IgG14 | CD451 |
| EC | MICA/B2 | ULBP36 | CD563 | CD31 | CXCR34 | CD451 |
| EC isotype control | IgG22 | IgG26 | IgG23 | IgG11 | IgG14 | CD451 |
| NK cell surface marker | CD165 | NKp464 | CD563 | NKG2D4 | CXCR34 | CD451 |
| NK surface isotype control | IgG11 | IgG11 | IgG23 | IgG11 | IgG14 | CD451 |
| NK cell intracellular | Granzyme B*1 | IL-4*2 | CD563 | IFN-γ*2 | CD161 | CD451 |
| NK intracellular isotype control | IgG1*1 | IgG1*2 | IgG23 | IgG1*1 | IgG14 | CD451 |
CD45 was included in all cocktails (also in isotype control tubes) for differentiation between epithelial cells (ECs) and natural killer (NK) cells. PE, phytoerythrin; PacBlue, Pacific Blue; PE-TR, phytoerythrin-Texas red; APC, allophycocyanin; PerCP-Cy5.5, peridinin chlorophyll protein-cyanine 5.5; APC-Cy7, allophycocyanin-cyanine 7. The antibodies were purchased by the following companies: 1BD Biosciences, 2eBioscience, 3Invitrogen, 4Biolegend, 5Beckman Coulter/IO Test, and 6R&D.
These antibodies were added during the second staining step after fixation and permeabilization.
Cocultures were digested for 30 min at 37°C in 1 ml RPMI, 15 μg/ml DNase I (Sigma, St. Louis, MO), and 5 μg/ml Pronase E (Sigma). The digestion activity was stopped by adding 100 μl FBS, the cell suspension was centrifuged (500 g, 10 min, 4°C), and the cell pellet was resuspended in 1 ml flow-staining buffer [PBS (without Ca2+ and Mg2+; GIBCO), 1% heat-inactivated FBS, and 0.09% sodium azide (Sigma)]. Cells were divided in various tubes for different antibody panels (see Table 1 for details) and incubated 20 min at room temperature in the dark. For analysis of surface markers only (NEC focus and NK cell surface focus), cells were resuspended in 300 μl 0.5% PFA. For analysis of intracellular cytokines, cells, which were treated with a Golgi block (Brefeldin A; eBioscience) for 4 h before cell harvesting, were stained using the BD Cytofix/Cytoperm staining kit following the supplier's instructions. All samples were analyzed within 24 h on a BD LSRII flow cytometer (BD Biosciences, San Jose, CA).
Chemokine and cytokine.
Apical washes and basolateral supernatants were analyzed for eotaxin, eotaxin-3, MCP-1, MCP-4, IL-8, macrophage-derived chemokine (MDC), interferon-γ-induced protein (IP)-10, macrophage inflammatory protein (MIP)-1β, and thymus- and activation-regulated chemokine (TARC) (MS6000 Human Chemokine-9 Plex Tissue Culture Kit; Meso Scale Discovery) according to the supplier's instruction.
Blocking of ULBP3 and MICA/B and stimulation with recombinant ULBP3.
NECs were treated with antibodies against NK ligands ULBP3 and MICA/B [human ULBP-3 antibody, clone 166510, no. FAB1517, 20 μl (R&D Systems) and anti-human MICA/B, clone 6D4, no. 53-5788, 10 μl (eBioscience)] or the matching isotype controls (20 μl for ULBP3 and 10 μl of 1:10 diluted for MICA/B). The target antibodies or the isotype controls were added 1 h post-O3 exposure simultaneously with adding the NK cells to the NECs. In separate experiments, NK cells were stimulated with 10 ng/ml of human recombinant ULBP3 (R&D System, Minneapolis, MN) for 4 h and analyzed for changes in intracellular cytokines as described above.
Cell-mediated cytotoxicity assay for NK cells.
The cell-mediated cytotoxicity assay (7-AAD/CFSE Cell-Mediated Cytotoxicity Assay Kit; Cayman Chemical, Ann Arbor, MI) was performed to quantify the potential of NK cells to kill target cells. The human erythromyeloblastoid leukemia K562 cell line (ATCC, Manassas, VA) was used as target cells. NK cells were added to the air- or O3-exposed NECs as described earlier. After 24 h, the 5-(6)-carboxyfluorescein diacetate succinimidyl ester-labeled K562 cells were added to the cocultures for 4 h at a ratio of 1:5 (target-NK cells). After 4 h incubation, the target-NK cell mixtures were collected, stained for dead cells with 7-amino-actinomycin D, and acquired immediately with a BD LSRII flow cytometer (BD Biosciences).
Statistical analysis.
Data for the coculture effects are presented as means ± SE and were tested using the Wilcoxon matched-pairs test to compare monoculture with coculture data. Data obtained from mono- and cocultures using NECs from the same donor were considered matched pairs. Results obtained from cells exposed to O3 were normalized to matched air-exposed samples using cells from the same subjects and presented as fold induction of O3 over the respective air-exposed controls. Data were analyzed using the Wilcoxon signed-rank test to test the fold induction to a theoretical value of 1. A value of P < 0.05 was considered significant; n = 3–9 for each endpoint.
RESULTS
NEC and NK cell interactions in vivo and in coculture.
Based on our previous study demonstrating NK cells in nasal lavages of human volunteers (33), we examined whether NK cells are closely associated with or embedded in the nasal mucosa. We conducted flow cytometry analysis of fresh superficial nasal scrapes to determine the presence of NK cells (CD45+CD56+CD3− cells) (Fig. 1A), which demonstrated a robust population of NK cells in superficial nasal scrapes. To visualize NK cells in the nasal mucosa, we used histological analysis, providing further evidence for the location of NK cells near the apical surface of NECs (Fig. 1B).
Fig. 1.

Nasal epithelial cells (NECs) and natural killer (NK) cell interactions in vivo are mimicked by NEC-NK cocultures in vitro. A: flow cytometric analysis of superficial nasal scrape biopsies. The biopsies were digested to obtain a single cell suspension and analyzed for the presence of NK cells (CD45+CD56+CD3−). Representative scatterplot is shown. B: hematoxylin- and eosin-stained paraffin-embedded sections of superficial nasal scrape biopsies. The image shows ciliated NECs above multiple layers of basal ECs and an apically located lymphocytic cell (white arrowhead). C: in vitro coculture model of differentiated human NECs and human peripheral blood NK cells (arrow) added on the apical side. NK cells are located on the apical side of NECs and are in contact with the luminal border of the NECs. D: laser-scanning microscopy image of a NEC-NK cell coculture. NK cells (green) were labeled with a fluorescein-emitting dye before addition to the NECs. The F-actin cytoskeleton of the NECs (red) was stained with phalloidin-rhodamine and the cell nuclei of both cell types (blue) with DAPI. En face view demonstrates the presence of several NK cells located apically on the NEC monolayer. Cross-sectional view of the cocultures shows NK cells in direct contact with NECs and partially enclosed in the NEC monolayer (white arrowhead).
To further investigate potential cell-cell communication between NECs and NK cells, we developed a coculture model. Peripheral blood NK cells were added to the apical side of differentiated NECs and analyzed 24 h later. Figure 1C shows histological examination using light microscopy, demonstrating the presence of NK cells located on the apical side of differentiated NECs (Fig. 1C), similar to our observations made in nasal scrapes in vivo (Fig. 1B). To further characterize NEC-NK cell interaction, we used confocal microscopy. Figure 1D shows that some NK cells have direct cell-to-cell contact with NECs and appear to be embedded in the NEC monolayer, indicating direct interactions between NECs and NK cells.
Effects of coculturing NECs and NK cells on surface markers, cytokine production, and chemokine release.
To investigate the effect of coculturing peripheral blood NK cells with NECs, we analyzed the expression of NEC surface markers, NK surface, and intracellular markers with flow cytometry and the release of chemokines 24 h after establishing the coculture. No significant changes in lactate dehydrogenase levels as a marker for cytotoxicity were detected (data not shown). Flow cytometry analysis showed no changes in the expression of the potential NK cell ligands MICA/B and ULBP3 but a statistically significant enhancement of the chemokine receptor CXCR3 expression on the NECs (Table 2). NK cells cocultured with NECs had lower expression of CXCR3, NKG2D, and NKp46 (P = 0.06) (Table 2). Similarly, intracellular granzyme B levels were lower in NK cells cocultured with NECs, albeit not statistically significant (Table 2). To determine whether coculture altered baseline mediator expression, we analyzed the levels of nine chemokines (eotaxin, eotaxin-3, IP-10, MDC, MCP-1, MCP-4, MIP-1β, TARC, and IL-8) in the apical washes and basolateral supernatants of NECs alone and NECs cocultured with NK cells. No differences were observed for any of the measured chemokines (Table 3). Taken together, these data suggest that coculturing NK cells with NECs reduced markers of activation in NK cells but had no effect on chemokine release or NK cell ligand expression on NECs.
Table 2.
Flow cytometry analysis of the effect of coculturing on NEC and NK cell surface markers and cytokine expression
| Epithelial Cell Markers | |||
|---|---|---|---|
| Marker | Epithelial cells only | Cocultures | P Value |
| MICA/B | 5,429 ± 485 | 5,669 ± 552 | 0.25 |
| ULBP3 | 3,268 ± 331 | 3,349 ± 324 | 0.38 |
| CXCR3 | 16,785 ± 3312 | 27,200 ± 6,572 | 0.02* |
| Natural Killer Cell Markers | |||
|---|---|---|---|
| Marker | NK Cells Only | Cocultures | P Value |
| NKG2D | 818 ± 89 | 558 ± 78 | 0.03* |
| NKp46 | 586 ± 153 | 240 ± 37 | 0.06 |
| CD16 | 2,102 ± 898 | 1,941 ± 772 | 0.69 |
| CXCR3 | 2,979 ± 732 | 1,509 ± 732 | 0.02* |
| IFN-γ | 466 ± 74 | 430 ± 67 | 0.16 |
| IL-4 | 1,741 ± 1,201 | 2,405 ± 2,020 | 0.44 |
| Granzyme B | 2,007 ± 960 | 1,860 ± 973 | 0.09 |
Data show mean fluorescence intensity ± SE; n = 6–8 experiments. NEC, nasal epithelial cells.
P < 0.05.
Table 3.
Effect of coculturing on chemokine release
| Apical |
Basolateral |
|||||
|---|---|---|---|---|---|---|
| Chemokine, pg/ml | NECs only | Cocultures | P value | NECs only | Cocultures | P value |
| Eotaxin | ND | ND | ND | ND | ||
| Eotaxin 3 | 311 ± 118 | 168 ± 32 | 0.44 | 142 ± 36 | 194 ± 38 | 0.44 |
| IP-10 | 5,209 ± 2,830 | 4,973 ± 3,712 | 0.56 | 2,305 ± 802 | 1,791 ± 607 | 0.17 |
| MCP-1 | 232 ± 50 | 186 ± 67 | 0.81 | 246 ± 75 | 359 ± 147 | 0.81 |
| MCP-4 | 156 ± 54 | 131 ± 40 | 0.44 | 110 ± 70 | 114 ± 74 | 0.63 |
| MIP-1β | 11.7 ± 6.6 | 21.9 ± 7.9 | 0.25 | 15.1 ± 3.5 | 16.4 ± 2.8 | 0.63 |
| IL-8 | ND | ND | ND | ND | ||
| MDC | 381 ± 187 | 490 ± 277 | 0.56 | 80.8 ± 30.3 | 83.2 ± 48 | 1.0 |
| TARC | 43.3 ± 15.4 | 56.5 ± 30.2 | 1.1 | 32.8 ± 19.4 | 24.2 ± 14.2 | 0.13 |
Data show mean concentration (pg/ml) ± SE; n = 3–6. IP-10, interferon-γ-induced protein-10; MCP, monocyte chemotactic protein; MIP-1β, macrophage inflammatory protein-1β; MDC, macrophage-derived chemokine; TARC, thymus- and activation-regulated chemokine. ND, not determined.
O3 alters the chemokine release and changes NK cell imunophenotype in the coculture model.
Exposure to O3 has been shown to increase the release of chemokines involved in the recruitment and activation of immune cells (36, 52, 59, 70, 78). To determine whether coculturing NK cells with O3-exposed NECs alters the release of chemokines, we analyzed apical washes and basolateral supernatants for nine chemokines and normalized to levels found in air-exposed cells (Table 4). Compared with air, exposure of NECs to O3 and subsequently cultured either alone or in the presence of NK cells had no effect on the release of eotaxin, eotaxin-3, MDC, MCP-4, and TARC. Basolateral levels of IP-10 and apical and basolateral levels of MCP-1 were enhanced in NECs exposed to O3 and cultured alone, an effect that was abolished by coculturing with NK cells. MIP-1β levels were enhanced in the apical compartment of NECs exposed to O3 and cultured alone and in the basolateral compartment of NECs exposed to O3 and cultured with NK cells. Basolateral levels of IL-8 were significantly increased in cocultures after O3 exposure.
Table 4.
Chemokine release of NECs alone or cocultured with NK cells after exposure to air or O3
| Apical |
Basolateral |
|||
|---|---|---|---|---|
| Fold induction O3/air of chemokines, pg/ml | NECs only | Cocultures | NECs only | Cocultures |
| Eotaxin | 1.08 ± 0.47 | 1.13 ± 0.32 | ND | ND |
| Eotaxin 3 | 1.20 ± 0.24 | 0.88 ± 0.16 | 1.32 ± 0.26 | 0.88 ± 0.12 |
| IP-10 | 1.73 ± 0.76 | 0.77 ± 0.27 | 2.10 ± 0.50* | 1.38 ± 0.45 |
| MCP-1 | 4.54 ± 3.12+ | 1.33 ± 0.66 | 3.02 ± 1.64+ | 1.23 ± 0.38 |
| MCP-4 | 1.08 ± 0.21 | 1.02 ± 0.24 | 1.04 ± 0.11 | 1.23 ± 0.38 |
| MIP-1β | 1.76 ± 0.44+ | 2.27 ± 1.4 | 1.78 ± 0.12 | 1.55 ± 0.15+ |
| IL-8 | 1.04 ± 0.03 | 1.03 ± 0.04 | 1.31 ± 0.31 | 1.35 ± 0.15* |
| MDC | 0.95 ± 0.19 | 1.02 ± 0.16 | 1.38 ± 0.22 | 1.11 ± 0.11 |
| TARC | 0.97 ± 0.33 | 0.66 ± 0.16 | 0.87 ± 0.15 | 1.42 ± 0.36 |
Data are presented as fold induction of O3 over air ± SE; n = 3–6.
P < 0.05 and +P = 0.06.
To determine whether exposure of NECs to O3 modifies NEC-NK cell interactions, we examined markers of NK cell immunophenotype by flow cytometry. Specifically, we examined markers of NK cell activation and maturation (CD16, NKG2D, and NKp46) and recruitment (CXCR3) as well as intracellular mediator production (IL-4, IFN-γ, and granzyme B). Figure 2A shows that NKG2D, NKp46, and CXCR3 expression was unchanged in NK cells cocultured with O3-exposed NECs, whereas the expression of CD16 was enhanced. However, the ratio of CD16bright/CD16dim NK cells, which is often used as a marker of cytotoxic potential of NK cells (19), was not affected by coculturing with O3-exposed NECs (data not shown).
Fig. 2.
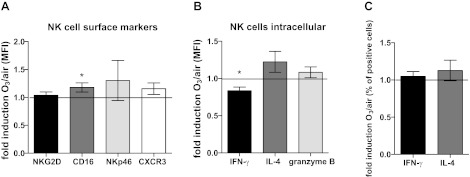
Flow cytometry analysis of NK cells cocultured with air- or O3-exposed NECs. A: NK cell surface markers. B: intracellular mediator expression as mean fluorescence intensity fold induction of O3 over air of the entire population. C: percent interferon-γ (IFN-γ)- and IL-4-positive NK cells. Data are presented as fold induction of the mean fluorescence intensity (MFI) (A and B) or of percentage of positive cells (C) ± SE of cells exposed to O3 over cells exposed to air (n = 5–6), *P < 0.05.
Interestingly, coculturing NK cells with O3-exposed NECs decreased the expression of IFN-γ and enhanced, albeit not statistically significant, the expression of IL-4 in NK cells (Fig. 2B), without affecting the percentage of cells staining positive for these cytokines (Fig. 2C). This indicates that the entire NK cell population was stimulated to produce less IFN-γ rather than shifting toward separate cell populations. Granzyme B expression was not affected by coculturing NK cells with O3-exposed NECs. These data indicate a suppression of the IFN-γ production in NK cells after coculturing them with O3-exposed NECs.
Soluble factors are not sufficient to induce the change in NK cell activity.
To investigate potential mechanisms mediating the suppression of IFN-γ production, we examined whether soluble mediators released by O3-exposed NECs could induce this effect. Apical washes and basolateral supernatants of NECs exposed to O3 and air were removed 24 h postexposure, and NK cells were incubated with these conditioned media for 4 h upon which NK cells were analyzed for surface markers and intracellular mediator expression by flow cytometry. As shown in Fig. 3, A and B, neither surface markers of NK cell activation nor intracellular mediators were affected by stimulating NK cells with conditioned media from O3-exposed NECs. These data suggest that NEC-derived soluble factors alone are not sufficient to suppress the IFN-γ production by the NK cells seen in Fig. 2B.
Fig. 3.
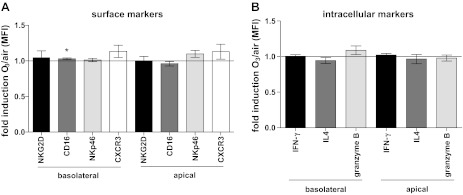
Flow cytometry data of NK cells incubated with conditioned media from air- and O3-exposed NECs. A: surface markers. B: intracellular markers. Data are presented as fold induction of the mean fluorescence intensity ± SE of cells exposed to O3 over air (n = 8), *P < 0.05.
O3 enhances the expression of EC markers MICA/B and ULBP3.
After recognizing that NEC-derived soluble mediators alone are not sufficient to induce the suppression of IFN-γ production by NK cells, we determined whether exposure to O3 modified NK cell ligand expression on NECs. NECs were exposed to air or O3 for 4 h, cultured with NK cells or vehicle, and analyzed by flow cytometry 24 h later. We focused our analysis on the NK cell ligands (MICA/B and ULBP3) as well as the chemokine receptor CXCR3, which regulates cell proliferation in ECs (1). Exposure of NECs to O3, regardless of whether NK cells were added postexposure or not, enhanced the expression of ULBP3 on NECs (Fig. 4, A and B). The expression of MICA/B was only enhanced in NECs cocultured with NK cells (Fig. 4B), and the expression of CXCR3 was unchanged after O3 exposure. Taken together, these data suggest O3 exposure alters the expression of NK cell ligands on NECs.
Fig. 4.

Flow cytometry analysis of NEC surface makers. Analysis of MICA/B, ULBP3, and CXCR3 in NECs alone (A) or NECs cocultured with NK cells (B). Data are presented as fold induction of mean fluorescence intensity ± SE of cells exposed to O3 over air (n = 9), *P < 0.05 and **P < 0.01.
O3-induced changes in NECs reduce the cytotoxicity potential of cocultured NK cells.
To determine whether the changes in cytokine production affect NK cell function, we assessed cytotoxic potential of NK cells cocultured with NECs. Whereas NK cells cocultured with air-exposed NECs killed on average 25.88 ± 7.28% cells, samples of NK cells cocultured with O3-exposed NECs were only able to kill 22.32 ± 5.71% of the target cells (Fig. 5), indicating a reduction in NK cell cytotoxic function.
Fig. 5.

Cytotoxic potential of NK cells was reduced after coculturing with O3-exposed NECs. The data represent the percentage of lysed target cells as a measure of NK cell cytotoxic potential. *P < 0.05.
O3-induced upregulation of MICA/B and ULBP3 is responsible for the reduction in IFN-γ production in cocultured NK cells.
To test whether the O3-induced upregulation of the NK cell ligands ULBP3 and MICA/B is responsible for the reduction of the IFN-γ production by NK cells, we performed two types of experiments. First, we incubated NK cells with recombinant ULBP3, and, second, we blocked ULBP3 and MICA/B after the O3 exposure.
Recombinant ULBP3 at a concentration of 10 ng/ml reduced the intracellular IFN-γ and enhanced, albeit not statistically significant, the intracellular IL-4 (Fig. 6A), suggesting that ULBP3 plays a role in suppressing IFN-γ production. Blocking of ULPB3 and MICA/B after exposing NECs to O3 reversed the reduction of IFN-γ, whereas IL-4 was not changed (Fig. 6, B and C). These data show that the O3-induced upregulation of ULBP3 and MICA/B on NECs mediates the suppression of IFN-γ.
Fig. 6.
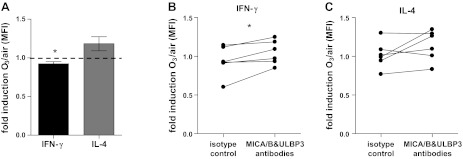
Analysis of the role of ULBP3 and MICA/B in the effects on NK cells. A: effect of stimulation with recombinant ULBP3 (10 ng for 4 h) on intracellular IFN-γ and IL-4 of NK cells. B and C: effect of blocking ULBP3 and MICA/B after exposing NECs to air or O3 on IFN-γ (B) and IL-4 (C) in NK cells cocultured with NECs. Data are presented as fold induction of mean fluorescence intensity (MFI) of cells exposed to O3 over air (A, n = 6). *P < 0.05.
DISCUSSION
We have recently demonstrated that NK cells comprise a significant portion of leukocytes recoverable by nasal lavage (33). NK cells receive signals leading to maturation and activation from their microenvironment (69), including structural cells like ECs. The communication between NK cells and ECs occurs via the release of chemokines and cytokines (59, 66, 78) and through direct cell-cell contacts (8, 15, 20, 54). To investigate how ECs communicate with and modify NK cells and whether these interactions are changed by exposure to oxidant air pollution, we established a coculture model of NECs and NK cells, mimicking the human nasal mucosa (Fig. 1). In this study, we showed that O3-exposed NECs modify the activity of NK cells, resulting in suppressed IFN-γ production (Fig. 2) and reduced cytotoxic potential to kill cancer cells (Fig. 5). Our data also show that these effects were not induced by soluble mediators released from NECs (Fig. 3) alone but are mediated via change in the expression of the NK ligands ULBP3 and MICA/B on the NEC surface after O3 exposure (Figs. 4 and 6).
NK cells are known for their important role in patrolling the epithelium to kill (virus-) infected cells and transformed tumor cells (74) and more recently as a source for cytokines (73). Specifically, NK cells can be classified in cytotoxic (CD56dimCD16bright) and secretory (CD56brightCD16dim/negative) NK cells (19). NK cells' cytokine-producing potential can change in the context of allergic diseases (22, 80) and upon stimulation with soluble mediators (12, 77), but it is not known whether exposure to common air pollutants can modify the immunophenotype of NK cells. Our data demonstrate that an acute exposure (4 h) of the NECs to the oxidant air pollutant O3 (0.4 ppm) and subsequent coculturing with NK cells reduced intracellular IFN-γ levels in NK cells (Fig. 2). It has been shown that NK cells are an important source of chemokines and cytokines (73), including IFN-γ and IL-4 (2, 8), which are both crucial for the early immune response, especially in the context of viral infections (68, 73). NK cells are important during antiviral defense responses and rely on their ability to produce cytolytic agents like granzyme B and cytokines like IFN-γ to fulfill that function (7, 19, 24). In addition to reduced IFN-γ levels, coculturing NK cells with O3-exposed NECs also reduced cytolytic function of NK cells (Fig. 5). The ability of O3 to reduce the cytotoxic potential of pulmonary NK cells has previously been shown in rats (16, 72). With the work presented here, we not only confirm these previous observations using human NK cells but also show that the effect is at least partially mediated by ECs. O3-induced decreases in the ability of NK cells to produce IFN-γ and to kill target cells could contribute to the attenuated immune response and higher susceptibility to infection with viruses like influenza, which has been shown in epidemiological studies (83).
NK cells receive signals inducing maturation and activation from their microenvironment (69) via soluble factors released into the milieu (59, 66, 78) and through direct receptor-ligand interaction with other cells (8, 15, 20, 54). We found slight changes in chemokine release by NECs after O3 exposure (Table 1). The factors measured here as well as others that can be produced by ECs, such as IL-4, IL-12, IL-15, or IL-18 or RANTES (17, 44, 58, 64), can activate NK cells and also induce changes in NK cell immunophenotypes (73). Therefore, we tested whether soluble factors released by NECs can change NK cell activity. Neither apical washes nor basolateral supernatants of air- and O3-exposed NECs induced statistically significant changes in NK cell activity (Fig. 3). Various studies have shown the activating potential of different cytokines, such as IL-2, IL-4, IL-12, IL-15, IL-18, IL-21, and type I IFNs (12, 13, 19, 74, 77), on NK cells, especially to induce IFN-γ production. IL-18 and type I IFNs are potentially produced by ECs (17, 18, 39, 66), and the IL-18 expression could be modified by exposure to O3 (46). However, the inability of conditioned media obtained from O3-exposed ECs to affect NK cell immunophenotypes in the absence of cell-cell interactions indicates that soluble mediators alone are not sufficient to induce changes in NK cells in our coculture model.
Because soluble factors released by O3-exposed NECs alone did not account for the change in NK cell activity observed in the coculture model, we examined whether contact-dependent mechanisms could mediate these effects. Specifically, we analyzed whether and how exposure to O3 affects potential NK cell ligands expressed on NECs. Our data show that O3 exposure increased the expression of the NK ligands ULBP3 (in NECs only and in cocultures) and MICA/B (in cocultures only) (Fig. 4) on NECs. These ligands are known to be expressed on stressed ECs following viral infections (27, 81) or stimulation with hydrogen peroxide (10). MICA/B and ULBP3 are ligands for the activating NK cell receptor NKG2D (54). Binding of ligands to the NKG2D receptor activates NK cells by triggering cytotoxic responses and/or cytokine secretion (54, 79). However, the output of NK cell activation depends on costimulation with other activating and inhibitory receptors (15, 79). In our study we found enhanced cytotoxic markers (CD16, trend for NKG2D, NKp46, and granzyme B), and the NK cells appear to be generally activated, most likely through the NKG2D-ligand pathway, when cocultured with O3-exposed NECs. However, we also found decreased intracellular IFN-γ and reduced cytotoxic potential. The reduction of IFN-γ was driven by the upregulation of ULBP3 and MICA/B on O3-epoxsed NECs, indicating that direct cell-cell interaction between NECs and NK cells modifies NK cell activity. However, as indicated above, soluble factors, such as IL-12, IL-15, and IL-18, are known to alter the maturation and activation and enhance the IFN-γ production as well as the cytotoxicity potential of NK cells (12). Thus, in combination with effects on ligand-receptor interactions, O3-induced changes in the release of soluble mediators may contribute to the change in NK cell activity seen here.
In general, direct cell-cell interactions are important during immune cell activation in the respiratory mucosa, and coculture models can be important tools to study those interactions (60, 65, 71). Therefore, one of the goals of this study was to establish a coculture model of human NECs and peripheral NK cells mimicking the nasal mucosa. Although there are several studies, including our own, that have used coculture models of either ECs with immune cell types, such as DCs or macrophages (9, 32, 49), or of NK cells and DCs (28, 84), this is, to our knowledge, the first published coculture model of ECs and NK cells. This model can be a helpful tool to study the effects of ECs not only in the context of pollutant exposure as shown in this study but also in the context of viral infections or diseases, including cancer, as well as to explore therapeutic strategies. For example, a recently published study (45) showed increased antibody-dependent, cell-mediated cytotoxicity of NK cells against tumor cells after treatment with IL-12, a cytokine that can also be produced by ECs (64). This model could be used to identify novel treatments with NECs as targets to specifically enhance IL-12 production by ECs, which would act on NK cells and increase the cytotoxicity against cancer cells. Because of the importance of NK cells in fighting viral infections, another possibility for novel therapeutic strategies is the enhancement of the NK cell potential to kill virus-infected cells and to produce IFN-γ. Agents known to increase NEC-dependent production of factors that could enhance NK cell-dependent antiviral activities, such as cytokines (IL-12, IL-15, IL-18, or type I IFNs) (76), could be explored as antiviral strategies. Furthermore, the model could be used to improve mechanistic understanding of EC-NK cell interactions by blocking or upregulating specific receptors, other ligands, or soluble mediators.
Development of asthma (6) and asthma exacerbations (25, 55, 61–63, 82) are known to be associated with O3 exposure, yet the mechanisms mediating these responses are unknown. The shift from the production of Th1 (mainly IFN-γ) to the classical Th2 cytokines IL-4, IL-5, and IL-13 is often associated with allergy and asthma (51). Various other studies have investigated the effect of O3 on immune cells with different results. Becker et al. found no alteration of either IL-4 or IFN-γ in lymphocytes exposed to O3 in vitro (5). In vitro O3 exposure of PBMCs stimulated IFN-γ and suppressed IL-4 production (42), and, in bronchoalveolar lavages of O3-exposed mice, IL-4 and IFN-γ concentrations were enhanced after allergic challenges compared with controls without O3 exposure (35). However, because NK cells play crucial roles in linking the innate and the adaptive immune response (73) and are of special importance in allergic diseases (48, 67, 74, 75, 80), the effect of O3 on NK cells rather than on all lymphocytes could be of particular interest. Pichavant and colleagues (56) showed that O3 shifts NKT cell cytokine profile in mice from IFN-γ to IL-4, similarly to our results, and that repeated O3 exposure leads to an asthmatic phenotype. In general, a shift of NK cells toward production of IFN-γ can suppress airway hyperresponsiveness and eosinophilia (47). In addition, depletion of NK cells during allergic sensitization significantly reduces eosinophilia and allergic inflammation in mice (40), indicating that NK cells are important during development of allergy. Last, NK cells have been shown to be important during resolution of allergic inflammation (29), suggesting modified function of these cells could affect clearing of eosinophils and T cells after exacerbation of allergic inflammation. Thus, the shift of NK cells cocultured with O3-exposed NECs could have consequences for allergic inflammation, via reduced expression of IFN-γ.
In conclusion, our data showed that exposure of NECs to O3 changes activation of resident NK cells. Cytotoxic potential and IFN-γ production was suppressed via an upregulation of ULBP3 and MICA/B on the NECs. These O3-induced effects at the level of the epithelium could play important roles in the development, exacerbation, and resolution of allergic inflammation and clearance of viral infections. Understanding of the mechanisms by which EC-NK cell interactions following exposure to oxidant pollutants affects allergic airway diseases or respiratory virus infections could uncover new therapeutic strategies using EC as a target to modulate NK cell function.
GRANTS
L. Müller is supported by the Swiss National Science Foundation with a personal grant and declares no conflict of interest. I. Jaspers is supported by grants from the National Institutes of Health (ES-013611 and HL-095163), the Flight Attendant Medical Research Institute, and the Environmental Protection Agency (CR83346301) and declares no conflict of interest.
DISCLOSURES
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Although the research described in this article has been funded in part by the U.S. Environmental Protection Agency through cooperative agreement CR83346301 with the Center for Environmental Medicine, Asthma and Lung Biology at the University of North Carolina-Chapel Hill, it has not been subjected to the agency's required peer and policy review and therefore does not necessarily reflect the views of the agency, and no official endorsement should be inferred. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: L.M. and I.J. conception and design of research; L.M. and L.E.B. performed experiments; L.M., L.E.B., and I.J. analyzed data; L.M. and I.J. interpreted results of experiments; L.M. prepared figures; L.M. and I.J. drafted manuscript; L.M. and I.J. edited and revised manuscript; L.M., L.E.B., and I.J. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Wenli Zhang for the mesoscale analysis and Kimberlie Burns (Cystic Fibrosis/Pulmonary Research & Treatment Center, UNC at Chapel Hill) for microscopy preparations.
REFERENCES
- 1.Aksoy MO, Yang Y, Ji R, Reddy PJ, Shahabuddin S, Litvin J, Rogers TJ, Kelsen SG. CXCR3 surface expression in human airway epithelial cells: cell cycle dependence and effect on cell proliferation. Am J Physiol Lung Cell Mol Physiol 290: L909–L918, 2006 [DOI] [PubMed] [Google Scholar]
- 2.Aktas E, Akdis M, Bilgic S, Disch R, Falk CS, Blaser K, Akdis C, Deniz G. Different natural killer (NK) receptor expression and immunoglobulin E (IgE) regulation by NK1 and NK2 cells. Clin Exp Immunol 140: 301–309, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayram H, Sapsford RJ, Abdelaziz MM, Khair OA. Effect of ozone and nitrogen dioxide on the release of proinflammatory mediators from bronchial epithelial cells of nonatopic nonasthmatic subjects and atopic asthmatic patients in vitro. J Allergy Clin Immunol 107: 287–294, 2001 [DOI] [PubMed] [Google Scholar]
- 4.Becker S, Madden MC, Newman SL, Devlin RB, Koren HS. Modulation of human alveolar macrophage properties by ozone exposure in vitro. Toxicol Appl Pharmacol 110: 403–415, 1991 [DOI] [PubMed] [Google Scholar]
- 5.Becker S, Quay J, Soukup J. Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. J Immunol 147: 4307–4312, 1991 [PubMed] [Google Scholar]
- 6.Bernstein JA, Alexis N, Barnes C, Bernstein IL, Nel A, Peden D, Diaz-Sanchez D, Tarlo SM, Williams PB. Health effects of air pollution. J Allergy Clin Immunol 114: 1116–1123, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Biron CA, Brossay L. NK cells and NKT cells in innate defense against viral infections. Curr Opin Immunol 13: 458–464, 2001 [DOI] [PubMed] [Google Scholar]
- 8.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol 17: 189–220, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Bleck B, Tse DB, Jaspers I, Curotto de Lafaille MA, Reibman J. Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation. J Immunol 176: 7431–7437, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 291: L222–L231, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Bosson J, Stenfors N, Bucht A, Helleday R, Pourazar J, Holgate ST, Kelly FJ, Sandstrom T, Wilson S, Frew AJ, Blomberg A. Ozone-induced bronchial epithelial cytokine expression differs between healthy and asthmatic subjects. Clin Exp Allergy 33: 777–782, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Brady J, Carotta S, Thong RP, Chan CJ, Hayakawa Y, Smyth MJ, Nutt SL. The interactions of multiple cytokines control NK cell maturation. J Immunol 185: 6679–6688, 2010 [DOI] [PubMed] [Google Scholar]
- 13.Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol 172: 2048–2058, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Brunekreef B, Holgate ST. Air pollution and health. Lancet 360: 1233–1242, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Bryceson YT, March ME, Ljunggren HG, Long EO. Activation, coactivation, and costimulation of resting human natural killer cells. Immunol Rev 214: 73–91, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burleson GR, Keyes LL, Stutzman JD. Immunosuppression of pulmonary natural killer activity by exposure to ozone. Immunopharmacol Immunotoxicol 11: 715–735, 1989 [DOI] [PubMed] [Google Scholar]
- 17.Cameron LA, Taha RA, Tsicopoulos A, Kurimoto M, Olivenstein R, Wallaert B, Minshall EM, Hamid QA. Airway epithelium expresses interleukin-18. Eur Respir J 14: 553–559, 1999 [DOI] [PubMed] [Google Scholar]
- 18.Ciencewicki JM, Brighton LE, Jaspers I. Localization of type I interferon receptor limits interferon-induced TLR3 in epithelial cells. J Interferon Cytokine Res 29: 289–297, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol 22: 633–640, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Cosman D, Mullberg J, Sutherland CL, Chin W, Armitage R, Fanslow W, Kubin M, Chalupny NJ. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14: 123–133, 2001 [DOI] [PubMed] [Google Scholar]
- 21.Demange V, Wild P, Zmirou-Navier D, Tossa P, Bohadana A, Barbaud A, Paris C. Associations of airway inflammation and responsiveness markers in non asthmatic subjects at start of apprenticeship (Abstract). BMC Pulm Med 10: 37, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deniz G, Akdis M, Aktas E, Blaser K, Akdis CA. Human NK1 and NK2 subsets determined by purification of IFN-gamma-secreting and IFN-gamma-nonsecreting NK cells. Eur J Immunol 32: 879–884, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Devlin RB, McKinnon KP, Noah T, Becker S, Koren HS. Ozone-induced release of cytokines and fibronectin by alveolar macrophages and airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 266: L612–L619, 1994 [DOI] [PubMed] [Google Scholar]
- 24.Fehniger TA, Cai SF, Cao X, Bredemeyer AJ, Presti RM, French AR, Ley TJ. Acquisition of murine NK cell cytotoxicity requires the translation of a pre-existing pool of granzyme B and perforin mRNAs. Immunity 26: 798–811, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Gent JF, Triche EW, Holford TR, Belanger K, Bracken MB, Beckett WS, Leaderer BP. Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. J Am Med Assoc 290: 1859–1867, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Goldie RG, Fernandes LB, Farmer SG, Hay DW. Airway epithelium-derived inhibitory factor. Trends Pharmacol Sci 11: 67–70, 1990 [DOI] [PubMed] [Google Scholar]
- 27.Groh V, Rhinehart R, Randolph-Habecker J, Topp MS, Riddell SR, Spies T. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat Immunol 2: 255–260, 2001 [DOI] [PubMed] [Google Scholar]
- 28.Gros F, Cabillic F, Toutirais O, Maux AL, Sebti Y, Amiot L. Soluble HLA-G molecules impair natural killer/dendritic cell crosstalk via inhibition of dendritic cells. Eur J Immunol 38: 742–749, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Haworth O, Cernadas M, Levy BD. NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol 186: 6129–6135, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hollingsworth JW, Free ME, Li Z, Andrews LN, Nakano H, Cook DN. Ozone activates pulmonary dendritic cells and promotes allergic sensitization through a Toll-like receptor 4-dependent mechanism. J Allergy Clin Immunol 125: 1167–1170, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hollingsworth JW, Kleeberger SR, Foster WM. Ozone and pulmonary innate immunity. Proc Am Thorac Soc 4: 240–246, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvath KM, Brighton LE, Zhang W, Carson JL, Jaspers I. Epithelial cells from smokers modify dendritic cell responses in the context of influenza infection 1. Am J Respir Cell Mol Biol 45: 237–245, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horvath KM, Herbst M, Zhou H, Zhang H, Noah TL, Jaspers I. Nasal lavage natural killer cell function is suppressed in smokers after live attenuated influenza virus. Respir Res 12: 102, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jahnsen FL, Gran E, Haye R, Brandtzaeg P. Human nasal mucosa contains antigen-presenting cells of strikingly different functional phenotypes. Am J Respir Cell Mol Biol 30: 31–37, 2004 [DOI] [PubMed] [Google Scholar]
- 35.Jang AS, Choi IS, Takizawa H, Rhim T, Lee JH, Park SW, Park CS. Additive effect of diesel exhaust particulates and ozone on airway hyperresponsiveness and inflammation in a mouse model of asthma. J Korean Med Sci 20: 759–763, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaspers I, Flescher E, Chen LC. Ozone-induced IL-8 expression and transcription factor binding in respiratory epithelial cells. Am J Physiol Lung Cell Mol Physiol 272: L504–L511, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Jaspers I, Horvath KM, Zhang W, Brighton LE, Carson JL, Noah TL. Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am J Respir Cell Mol Biol 43: 368–375, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kesic MJ, Meyer M, Bauer R, IJ Exposure to ozone modulates human airway protease/antiprotease balance contributing to increased influenza a infection. PLoS ONE 7: e35108, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kesic MJ, Simmons SO, Bauer R, Jaspers I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic Biol Med 51: 444–453, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korsgren M, Persson CG, Sundler F, Bjerke T, Hansson T, Chambers BJ, Hong S, Van Kaer L, Ljunggren HG, Korsgren O. Natural killer cells determine development of allergen-induced eosinophilic airway inflammation in mice. J Exp Med 189: 553–562, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9: 495–502, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larini A, Bocci V. Effects of ozone on isolated peripheral blood mononuclear cells. Toxicol In Vitro 19: 55–61, 2005 [DOI] [PubMed] [Google Scholar]
- 43.Larsson BM, Grunewald J, Skold CM, Lundin A, Sandstrom T, Eklund A, Svartengren M. Limited airway effects in mild asthmatics after exposure to air pollution in a road tunnel. Respir Med 104: 1912–1918, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Leikauf GD, Simpson LG, Santrock J, Zhao Q, Abbinante-Nissen J, Zhou S, Driscoll KE. Airway epithelial cell responses to ozone injury. Environ Health Perspect 103, Suppl 2: 91–95, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luedke E, Jaime-Ramirez AC, Bhave N, Roda J, Carson WE., 3rd Cetuximab therapy in head and neck cancer: immune modulation with interleukin-12 and other natural killer cell-activating cytokines. Surgery 152: 431–440, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manzer R, Dinarello CA, McConville G, Mason RJ. Ozone exposure of macrophages induces an alveolar epithelial chemokine response through IL-1alpha. Am J Respir Cell Mol Biol 38: 318–323, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsubara S, Takeda K, Kodama T, Joetham A, Miyahara N, Koya T, Swasey CH, Okamoto M, Dakhama A, Gelfand EW. IL-2 and IL-18 attenuation of airway hyperresponsiveness requires STAT4, IFN-gamma, and natural killer cells. Am J Respir Cell Mol Biol 36: 324–332, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mesdaghi M, Vodjgani M, Salehi E, Hadjati J, Sarrafnejad A, Bidad K, Berjisian F. Natural killer cells in allergic rhinitis patients and nonatopic controls. Int Arch Allergy Immunol 153: 234–238, 2010 [DOI] [PubMed] [Google Scholar]
- 49.Muller L, Comte P, Czerwinski J, Kasper M, Mayer AC, Gehr P, Burtscher H, Morin JP, Konstandopoulos A, Rothen-Rutishauser B. New exposure system to evaluate the toxicity of (scooter) exhaust emissions in lung cells in vitro. Environ Sci Technol 44: 2632–2638, 2010 [DOI] [PubMed] [Google Scholar]
- 50.Nakayama T, Watanabe Y, Oiso N, Higuchi T, Shigeta A, Mizuguchi N, Katou F, Hashimoto K, Kawada A, Yoshie O. Eotaxin-3/CC chemokine ligand 26 is a functional ligand for CX3CR1. J Immunol 185: 6472–6479, 2010 [DOI] [PubMed] [Google Scholar]
- 51.Ngoc LP, Gold DR, Tzianabos AO, Weiss ST, Celedon Cytokines JC. Allergy, asthma. Curr Opin Allergy Cl 5: 161–166, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Nichols BG, Woods JS, Luchtel DL, Corral J, Koenig JQ. Effects of ozone exposure on nuclear factor-kappaB activation and tumor necrosis factor-alpha expression in human nasal epithelial cells. Toxicol Sci 60: 356–362, 2001 [DOI] [PubMed] [Google Scholar]
- 53.Noah TL, Zhou H, Monaco J, Horvath K, Herbst M, Jaspers I. Tobacco smoke exposure and altered nasal responses to live attenuated influenza virus. Environ Health Perspect 119: 78–83, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Obeidy P, Sharland AF. NKG2D and its ligands. Int J Biochem Cell Biol 41: 2364–2367, 2009 [DOI] [PubMed] [Google Scholar]
- 55.Peden D. Indoor and outdoor air pollution. In: Allergy: Principles and Practice, edited by Adkinson NFBW, Bochner B, Holgate S, Simons FES, Lemanske RF. Philadelphia, PA: Elsevier, 2009 [Google Scholar]
- 56.Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, Zhu M, Iwakura Y, Savage PB, DeKruyff RH, Shore SA, Umetsu DT. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med 205: 385–393, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Piotrowska VM, Piotrowski WJ, Kurmanowska Z, Marczak J, Gorski P, Antczak A. Rhinosinusitis in COPD: symptoms, mucosal changes, nasal lavage cells and eicosanoids. Int J Chron Obstruct Pulmon Dis 5: 107–117, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Regamey N, Obregon C, Ferrari-Lacraz S, van Leer C, Chanson M, Nicod LP, Geiser T. Airway epithelial IL-15 transforms monocytes into dendritic cells. Am J Respir Cell Mol Biol 37: 75–84, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol 71: 173–183, 2002 [PubMed] [Google Scholar]
- 60.Roggen EL, Soni NK, Verheyen GR. Respiratory immunotoxicity: an in vitro assessment. Toxicol In Vitro 20: 1249–1264, 2006 [DOI] [PubMed] [Google Scholar]
- 61.Romieu I, Meneses F, Ruiz S, Huerta J, Sienra JJ, White M, Etzel R, Hernandez M. Effects of intermittent ozone exposure on peak expiratory flow and respiratory symptoms among asthmatic children in Mexico City. Arch Environ Health 52: 368–376, 1997 [DOI] [PubMed] [Google Scholar]
- 62.Romieu I, Meneses F, Ruiz S, Sienra JJ, Huerta J, White MC, Etzel RA. Effects of air pollution on the respiratory health of asthmatic children living in Mexico City. Am J Respir Crit Care Med 154: 300–307, 1996 [DOI] [PubMed] [Google Scholar]
- 63.Romieu I, Meneses F, Sienra-Monge JJ, Huerta J, Ruiz Velasco S, White MC, Etzel RA, Hernandez-Avila M. Effects of urban air pollutants on emergency visits for childhood asthma in Mexico City. Am J Epidemiol 141: 546–553, 1995 [DOI] [PubMed] [Google Scholar]
- 64.Roschmann KI, Luiten S, Jonker MJ, Breit TM, Fokkens WJ, Petersen A, van Drunen CM. Timothy grass pollen extract-induced gene expression and signalling pathways in airway epithelial cells. Clin Exp Allergy 41: 830–841, 2011 [DOI] [PubMed] [Google Scholar]
- 65.Rothen-Rutishauser B, Blank F, Muhlfeld C, Gehr P. In vitro models of the human epithelial airway barrier to study the toxic potential of particulate matter. Expert Opin Drug Metab Toxicol 4: 1075–1089, 2008 [DOI] [PubMed] [Google Scholar]
- 66.Sanders CJ, Doherty PC, Thomas PG. Respiratory epithelial cells in innate immunity to influenza virus infection. Cell Tissue Res 343: 13–21, 2011 [DOI] [PubMed] [Google Scholar]
- 67.Scordamaglia F, Balsamo M, Scordamaglia A, Moretta A, Mingari MC, Canonica GW, Moretta L, Vitale M. Perturbations of natural killer cell regulatory functions in respiratory allergic diseases. J Allergy Clin Immunol 121: 479–485, 2008 [DOI] [PubMed] [Google Scholar]
- 68.See H, Wark P. Innate immune response to viral infection of the lungs. Paediatr Respir Rev 9: 243–250, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Siren J, Sareneva T, Pirhonen J, Strengell M, Veckman V, Julkunen I, Matikainen S. Cytokine and contact-dependent activation of natural killer cells by influenza A or Sendai virus-infected macrophages. J Gen Virol 85: 2357–2364, 2004 [DOI] [PubMed] [Google Scholar]
- 70.Song H, Tan W, Zhang X. Ozone induces inflammation in bronchial epithelial cells. J Asthma 48: 79–83, 2011 [DOI] [PubMed] [Google Scholar]
- 71.Steimer A, Haltner E, Lehr CM. Cell culture models of the respiratory tract relevant to pulmonary drug delivery. J Aerosol Med 18: 137–182, 2005 [DOI] [PubMed] [Google Scholar]
- 72.Van Loveren H, Krajnc EI, Rombout PJ, Blommaert FA, Vos JG. Effects of ozone, hexachlorobenzene, and bis(tri-n-butyltin)oxide on natural killer activity in the rat lung. Toxicol Appl Pharmacol 102: 21–33, 1990 [DOI] [PubMed] [Google Scholar]
- 73.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science 331: 44–49, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol 9: 503–510, 2008 [DOI] [PubMed] [Google Scholar]
- 75.von Bubnoff D, Andres E, Hentges F, Bieber T, Michel T, Zimmer J. Natural killer cells in atopic and autoimmune diseases of the skin. J Allergy Clin Immunol 125: 60–68, 2010 [DOI] [PubMed] [Google Scholar]
- 76.Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l'union fait la force.” Blood 106: 2252–2258, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Walzer T, Dalod M, Vivier E, Zitvogel L. Natural killer cell-dendritic cell crosstalk in the initiation of immune responses. Expert Opin Biol Ther 5, Suppl 1: S49–S59, 2005 [DOI] [PubMed] [Google Scholar]
- 78.Walzer T, Vivier E. G-protein-coupled receptors in control of natural killer cell migration. Trends Immunol 32: 486–492, 2011 [DOI] [PubMed] [Google Scholar]
- 79.Watzl C. The NKG2D receptor and its ligands-recognition beyond the “missing self”? Microbes Infect 5: 31–37, 2003 [DOI] [PubMed] [Google Scholar]
- 80.Wei H, Zhang J, Xiao W, Feng J, Sun R, Tian Z. Involvement of human natural killer cells in asthma pathogenesis: natural killer 2 cells in type 2 cytokine predominance. J Allergy Clin Immunol 115: 841–847, 2005 [DOI] [PubMed] [Google Scholar]
- 81.Welte SA, Sinzger C, Lutz SZ, Singh-Jasuja H, Sampaio KL, Eknigk U, Rammensee HG, Steinle A. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol 33: 194–203, 2003 [DOI] [PubMed] [Google Scholar]
- 82.White MC, Etzel RA, Wilcox WD, Lloyd C. Exacerbations of childhood asthma and ozone pollution in Atlanta. Environ Res 65: 56–68, 1994 [DOI] [PubMed] [Google Scholar]
- 83.Wong TW, Lau TS, Yu TS, Neller A, Wong SL, Tam W, Pang SW. Air pollution and hospital admissions for respiratory and cardiovascular diseases in Hong Kong. Occup Environ Med 56: 679–683, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang L, Du C, Chen T, Li S, Nie W, Zhu W, Fan F, Zhu J, Yan H. Distinct MAPK pathways are involved in IL-23 production in dendritic cells cocultured with NK cells in the absence or presence of angiotensin II. Mol Immunol 51: 51–56, 2012 [DOI] [PubMed] [Google Scholar]