

Abstract
Pulmonary endothelial cell (EC) apoptosis has been implicated in the pathogenesis of emphysema. Cigarette smoke (CS) causes lung EC apoptosis and emphysema. In this study, we show that CS exposure increased lung tissue adenosine levels in mice, an effect associated with increased lung EC apoptosis and the development of emphysema. Adenosine has a protective effect against apoptosis via adenosine receptor-mediated signaling. However, sustained elevated adenosine increases alveolar cell apoptosis in adenosine deaminase-deficient mice. We established an in vitro model of sustained adenosine exposure by incubating lung EC with adenosine in the presence of an adenosine deaminase inhibitor, deoxycoformicin. We demonstrated that sustained adenosine exposure caused lung EC apoptosis via nucleoside transporter-facilitated intracellular adenosine uptake, subsequent activation of p38 and JNK in mitochondria, and ultimately mitochondrial defects and activation of the mitochondria-mediated intrinsic pathway of apoptosis. Our results suggest that sustained elevated adenosine may contribute to CS-induced lung EC apoptosis and emphysema. Our data also reconcile the paradoxical effects of adenosine on apoptosis, demonstrating that prolonged exposure causes apoptosis via nucleoside transporter-mediated intracellular adenosine signaling, whereas acute exposure protects against apoptosis via activation of adenosine receptors. Inhibition of adenosine uptake may become a new therapeutic target in treatment of CS-induced lung diseases.
Keywords: mitochondria, nucleoside transporters, oxidative stress, adenosine, cigarette smoke
increased endothelial cell (EC) apoptosis has been observed in humans and animals with emphysema (31, 32), acute lung injury (ALI) (1, 25), idiopathic pulmonary fibrosis (IPF) (13, 34, 64), and ischemia-reperfusion (I/R) lung injury (48, 51). Inhibition of alveolar cell apoptosis attenuates the emphysematous changes (32, 50, 61), increases survival rate in ALI (33), and enhances the function of transplanted lungs (51) in animal models. These results indicate that excessive lung EC apoptosis plays a role in pathogenesis of some lung diseases (26, 40).
The purine nucleoside adenosine (Ado) is a potent signaling molecule. Extracellular Ado exists in low concentrations (40–600 nM) under homeostatic conditions and is elevated by platelet degranulation and cell necrosis. In response to inflammation and tissue injury, extracellular Ado levels are also increased due to increased expression and/or activation of ectonucleotidases (CD39 and CD73) and/or decreased expression and/or activation of Ado deaminase (ADA) (21, 22, 63, 66). It has been reported that Ado levels are significantly elevated in plasma of humans with sepsis-induced ALI (43, 60) and tissue ischemia (59). Ado levels are also increased in bronchoalveolar lavage (BAL) fluid of human smokers (20). However, it is unknown whether lung tissue Ado levels are altered in smokers or patients with cigarette smoke (CS)-associated lung diseases. It is well documented that CS is a major risk factor for emphysema. CS also increases susceptibility to ALI (10). Therefore, understanding the effect of CS on lung Ado levels and the role of sustained elevated Ado on lung EC apoptosis may provide novel insights into the mechanisms by which CS predisposes to a variety of lung diseases.
Ado achieves its physiological and pathological roles via either activation of G protein-coupled Ado receptors (AR), including A1R, A2AR, A2BR, and A3R, or nucleoside transporter (NT)-facilitated intracellular Ado uptake and metabolism. Activation of AR-mediated Ado signaling has been shown to be protective against apoptosis and tissue injury in multiple organs. For example, activation of A2AR (54) and A3R (14, 44, 55) reduces I/R brain and lung injury and apoptosis. CD39 transgenic mice are protected against renal I/R vascular injury and apoptosis, and these beneficial effects are attributed to increased extracellular Ado and subsequent activation of A2AR (17). Activation of A1R also blunts staurosporine-induced apoptosis of astrocytes (18). Additionally, activation of A2BR protects against apoptosis of remote cardiac myocytes in an infarcted heart (57).
In contrast to the protective effect of AR-mediated signaling, the stable Ado analog, 2-chloroadenosine, causes apoptosis and increases oxidative stress in prostate cancer cells (46). Studies of ADA-deficient mice indicate that sustained elevation of Ado caused chronic lung injury and alveolar cell apoptosis, effects that were exacerbated by deletion of A2BR (69). The mechanism by which sustained exposure to elevated Ado causes lung EC apoptosis is not known.
Since Ado is rapidly metabolized by ADA, we developed a model of sustained Ado exposure by incubating lung EC with Ado in the presence of an ADA inhibitor, deoxycoformicin (DCF) (38). We found that sustained Ado exposure caused lung EC apoptosis via NT-mediated intracellular events. We further showed that sustained elevated Ado after uptake caused activation of p38 and JNK, subsequent mitochondrial defects, and ultimately activation of the mitochondria-mediated intrinsic pathway of apoptosis. We also documented that lung tissue Ado levels were elevated in AKR mice exposed to CS for 3 wk, an effect associated with increased lung EC apoptosis and the development of emphysema. These results suggest that sustained elevated Ado in the lungs may contribute to CS-induced lung EC apoptosis and tissue injury.
MATERIALS AND METHODS
Cells and reagents.
Bovine pulmonary artery ECs were purchased from Vec Technologies (Rensselaer, NY) and were used between passages 3 and 9. The pulmonary artery ECs were propagated in MEM medium containing 10% fetal bovine serum and sodium pyruvate. Ado, dipyridamole (DPM), nitrobenzylthioinosine (NBTI), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), 1,3-dipropyl-7-methylxanthine (DPMX), 8-(4{[(4-cyano)phenylcarbamoylmethyl]oxy}epheyl)-1,3-di-(n-propyl)xanthine (MRS1754), and 3-ethyl 5 benzyl 2-methyl-6-phenylethynyl-1,4-(±)-dihydro-pyridine-3,5-dicarboxylate (MRS1191), N-acetyl-cysteine (NAC), apocynin, SB203580, and allopurinol were purchased from Sigma Aldrich (St. Louis, MO). Deoxycoformycin and SP600125 were from Tocris (Minneapolis, MN). Antibodies directed against caspase-3, Bax, and caspase-8 were obtained from Cell Signaling Technology (Danvers, MA). Antibodies directed against caspase-9 and Bad were purchased from Stressgen (San Diego, CA). Antibodies directed against Bcl-2 were purchased from BD Biosciences (San Jose, CA). Antibodies directed against actin and GAPDH were from Santa Cruz (Santa Cruz, CA). Antibody against von Willebrand factor (vWF) was from DAKO (Carpinteria, CA). JC-1 was from Cayman Chemical (Ann Arbor, MI). 4,6-Diamidino-2-phenylindole (DAPI) (Prolong Gold antifade reagent with DAPI) and MitoTracker Red CMXRos were from Invitrogen (Grand Island, NY). ApopTag peroxidase In Situ Apoptosis Detection Kit [terminal transferase-mediated dUTP nick end-labeling (TUNEL) kit] was purchased from EMD Millipore (Billerica, MA). DAB and VIP peroxidase substrates kits were from Vector Laboratories (Burlingame, CA).
CS exposure of mice.
Animal protocols were approved by the Providence Veterans Affairs Medical Center Institutional Animal Care and Use Committee and complied with the Health Research Extension Act and the Public Health Service policy. C57BL/6J and AKR mice were exposed to CS for 6 h/day, 4 days/wk, for 3 wk using a TE-10 mouse smoking machine (Teague Enterprises, Woodland, CA) and 3R4F reference cigarettes (University of Kentucky, Tobacco Research Institute, Lexington, KY), as described (52). Each cigarette was puffed for 2 s, once every minute for a total of eight puffs, at a flow rate of 1.05 l/min, to provide a standard puff of 35 cm3. The smoke machine was adjusted to produce a mixture of sidestream (89%) and mainstream (11%) smoke by burning three cigarettes at a time. The smoking chamber atmosphere was monitored for total suspended particles at concentration of 120 mg/m3. Fraction of inspired O2 and carbon monoxide were ∼20% and 100 ppm, respectively, in the smoking chamber.
Measurement of lung static compliance.
Lung mechanics were assessed using the FlexiVent system (SCIREQ, Montreal, Quebec, Canada). Mice were deeply anesthetized using 90 mg/kg pentobarbital via intraperitoneal injection. After tracheotomy with an 18-gauge blunt needle, the mice were placed on a computer-controlled small-animal ventilator at a constant tidal volume of 6 ml/kg, peak expiratory end pressure of 3 cmH2O, and respiratory rate of 150 breaths/min. Measurements of airway pressure were made at a side port of the tracheal cannula via a differential pressure transducer. Pressure-volume loops were obtained following manufacturer's recommendations. Specifically, volume history was standardized by three consecutive inflations at 30 cmH2O for 3 s. Pressure-volume curves were generated by stepwise inflation, with changes in pressure by 5 cmH2O until the maximal pressure of 30 cmH2O was reached, followed by stepwise deflation of the lungs. Quasi-static compliance was calculated at 5 cmH2O.
Quantification of Ado levels in lung tissue.
Ado can be rapidly metabolized by ADA. ATP, ADP, and AMP can be rapidly hydrolyzed to Ado by ectonucleotidases. To avoid changes in Ado levels during the process of tissue collection, after euthanasia of mice, lung tissue was immediately harvested into a preservative solution with inhibitors for ADA and ectonucleotidases and frozen in liquid nitrogen. Nucleosides were extracted from frozen lung tissue using 0.4 N perchloric acid, and Ado was separated and quantified using reverse-phase HPLC, as previously described (7, 37).
Assessment of lung endothelial apoptosis in lung tissue.
Apoptosis of lung EC was assessed by immunohistochemical analysis of TUNEL staining of apoptotic cells, costained with the EC marker, vWF. The total numbers of apoptotic EC, as indicated by dual positive staining of TUNEL and vWF in the entire lung slide, were counted and quantitated by an observer blinded to the source of the lungs using Aperio ScanScope (Vista, CA), which is a bright-field whole-slide scanner. The data are presented as number of apoptotic EC per millimeter square lung section area (TUNEL+/vWF+ cells/mm2).
Assessment of apoptosis in cultured EC.
EC apoptosis was assessed by assaying caspase-3 activation via immunoblot analysis of the levels of the cleaved (active) form of caspase-3, by nuclear morphological visualization of apoptotic nuclei using fluorescence microscopy after DAPI staining, as our laboratory previously described (39), and by TUNEL staining of apoptotic cells.
Mitochondrial morphological assay.
As our laboratory previously described (36), EC grown on slides were fixed with 4% paraformaldehyde and rendered permeable with 0.1% Triton X-100. Fixed cells were stained with MitoTracker Red CMXRos. Images were visualized by fluorescence microscopy at ×100 magnification and recorded.
Mitochondrial membrane potential assay.
Cells were treated as described in Fig. 4 legend. The treatments were removed, and cells were then incubated with JC-1 at a final dilution of 1:150 in serum-free MEM media for 20 min at 37°C. JC-1 was then removed from cells. After washing with warm serum-free MEM, coverslips were mounted onto microscope slides with a drop of warm serum-free MEM and sealed with nail polish. Images were captured by fluorescence microscopy in green channel (FITC) and red channel (Texas Red) and merged.
Fig. 1.
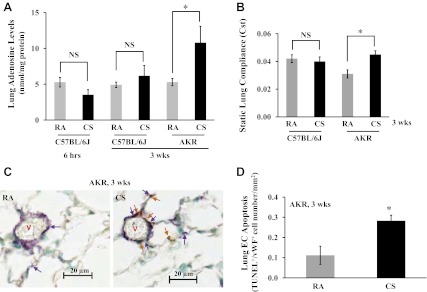
Subacute (3 wk) exposure to cigarette smoke (CS) increased lung tissue adenosine levels, static lung compliance (Cst), and lung endothelial apoptosis. 6-wk old male AKR mice and C57BL/6J mice were exposed to room air (RA) or CS for 6 h or 3 wk at 6 h/day, 4 days/wk, using a TE-10 mouse smoking machine and 3R4F reference cigarettes. Smoking chamber atmosphere was monitored for total suspended particles at 120 mg/m3, which was a mixture of 89% sidestream smoke and 11% mainstream smoke. Fraction of inspired O2 is 20%, and carbon monoxide is ∼100 ppm. A: by the end of CS exposure, lung tissue was collected for assessment of adenosine levels using HPLC. Four mice per group per time point were used. NS, not significant. B: Cst was assessed using FlexiVent system. Six mice per group were used. The lungs were then fixed at 25-cmH2O pressure, H&E staining was performed, and lung mean linear intercept was measured and quantitiated using Aperio ScanScope system (data not shown). C and D: lung endothelial cell (EC) apoptosis in mice exposed to RA or CS for 3 wk was assessed by terminal transferase-mediated dUTP nick end-labeling (TUNEL) costained with EC marker, von Wildebrand factor (vWF). C: arrows in brown indicate apoptotic cells, and arrows in purple indicate EC. V, vessel. Scale bars represent 20 μm. The total number of apoptotic EC (both brown and purple staining) were counted by a blinded observer and quantitated using Aperio ScanScope system. D: the data are presented as numbers of apoptotic EC (dual-positive staining for TUNEL and vWF) per mm2 lung tissue section area (TUNEL+/vWF+ cells/mm2). Three mice per group were used for C and D. Values are means ± SE. *P < 0.05 vs. RA exposure.
Gel electrophoresis and immunoblot analysis.
Lysates were solubilized in Laemmli buffer, and proteins were resolved using SDS-PAGE, and then transferred to PVDF membranes and immunoblotted with specific antibodies, as our laboratory previously described (36).
Data analysis.
For in vitro studies, all experiments were performed at least in triplicate. Means and SE were calculated based on the values of individual treatments and the numbers of the experiments performed in minimum of three individual experiments. For animal studies, three to six mice per group were used. Means and SE were calculated based on the values of each animal in each group and the numbers of animals used in that group. Data are presented as means ± SE. The difference between two means was assessed using Student's t-test, and the differences among three or more means were assessed using ANOVA followed by Tukey-Kramer post hoc test. Differences among means are considered significant when P < 0.05.
RESULTS
CS-induced increase in lung Ado levels was associated with lung EC apoptosis and early onset of emphysema.
CS exposure causes lung EC apoptosis and emphysema in humans (31). Ado levels are increased in BAL fluid of human smokers (20). Whether CS increases lung tissue Ado is unknown. We examined the effects of CS on lung tissue Ado in C57BL/6J mice, which are mildly susceptible to CS-induced emphysema, and in AKR mice, which are highly susceptible to CS-induced emphysema (27). We found that lung tissue Ado levels were significantly elevated in AKR mice exposed to CS for 3 wk, but not in C57BL/6J mice exposed to CS for either 6 h or 3 wk (Fig. 1A). As expected, after 3 wk of CS exposure, C57BL/6J mice did not show any sign of emphysema, with no change in either static lung compliance (Fig. 1B) or mean linear intercept (data not shown). In contrast, AKR mice displayed a trend of increased mean linear intercept (data not shown) and a significant increase in static lung compliance (Fig. 1B), indicating the onset of early emphysema. Temporally correlating with increased lung Ado and static lung compliance, AKR mice also exhibited increased lung EC apoptosis after 3 wk of CS exposure (Fig. 1, C and D). However, increased lung EC apoptosis was not seen in C57BL/6J mice exposed to CS for 3 wk (data not shown). Thus our data demonstrate a strong association among lung tissue Ado levels, lung endothelial apoptosis, and emphysema upon 3 wk of CS exposure. Similar to a previous report by others (28), we also noticed that the baseline levels of static lung compliance of AKR mice were significantly lower than that of C57BL/6J mice (Fig. 1B).
Sustained Ado exposure caused lung EC apoptosis.
Sustained elevated Ado in ADA-deficient mice causes alveolar cell apoptosis (69). To determine whether sustained exposure to elevated Ado causes lung EC apoptosis, we used our previously established in vitro model of sustained Ado exposure (38) by incubating lung EC with Ado plus ADA inhibitor, DCF, for up to 48 h. Because Ado can be rapidly metabolized by ADA, EC exposed to Ado plus DCF maintained high concentrations of intracellular Ado for up to 48 h, whereas EC exposed to vehicle, Ado, or DCF alone had very low levels of Ado (38). Similar to the in vivo effect of sustained Ado on alveolar cell apoptosis (69), lung EC exposed to elevated Ado for 24 and 48 h, but not 5 h, had an increased cleaved (active) caspase-3 (Fig. 2A). The effect of sustained elevated Ado on lung EC apoptosis was also demonstrated by increased chromatin condensation in apoptotic nuclei by DAPI staining (Fig. 2B) and by elevated TUNEL-positive cells (Fig. 3C) in cultures exposed to Ado plus DCF for 24 h.
Fig. 2.

Effect of sustained adenosine exposure on cultured lung EC apoptosis. A: bovine pulmonary artery ECs (PAEC) were incubated with vehicle (V) or 50 μM adenosine (A) in the absence or presence of 50 μM deoxycoformicin (D) for indicated times. Apoptosis was assessed by the proapoptotic, cleaved form of caspase-3. The same immunoblots were stripped and reprobed for actin to control for protein loading. Data represent three independent experiments. B: bovine PAEC were incubated with V or 50 μM A plus 50 μM D for 24 h, and apoptosis was assessed by 4,6-diamidino-2-phenylindole (DAPI) staining of apoptotic nuclei. Representative images from five independent experiments are shown. Arrows indicate apoptotic nuclei. Data are means ± SE, expressed as the ratio of apoptotic cells to the total counted cells × 100 (%). About 600 cells in three high-power fields for each group in each independent experiment were analyzed by a blinded observer. *P < 0.05 vs. V-treated EC.
Fig. 3.

Role of nucleoside transporters (NT) and adenosine receptors (AR) on sustained adenosine-induced EC apoptosis. Bovine PAEC were incubated for 24 h with V or 50 μM A plus 50 μM D in the absence or presence of NT inhibitors, dipyridamole (DPM; 10 μM) or nitrobenzylthioinosine (NBTI; 10 μM) (A–C), or of A2AR antagonist (DPMX, 10 μM), A2BR antagonist (MRS1754, 10 μM), A1R antagonist (DPCPX, 10 μM), or A3R antagonist (MRS1191, 10 μM) (D). Apoptosis was assessed by assaying cleaved, active form of caspase-3 (A), DAPI staining of apoptotic nuclei (B), and TUNEL staining (C). A and D: representative images from three independent experiments for each panel are shown. The immunoblots were stripped and reprobed for actin to control for protein loading. B and C: data are expressed as the ratio of apoptotic cells to the total counted cells × 100 (%). Values are means ± SE. About 600 cells in three high-power fields for each group in each independent experiment were analyzed by a blinded observer. n = 5. *P < 0.05 vs. V-treated EC. C: representative images for TUNEL staining are shown. Arrow indicates apoptotic, TUNEL-positive cells.
Sustained Ado exposure caused lung EC apoptosis via NT-facilitated intracellular uptake.
We have previously shown that sustained Ado exposure causes lung endothelial barrier dysfunction via NT-mediated intracellular Ado uptake (38). Similarly, inhibition of NTs with DPM completely prevented sustained Ado exposure-induced caspase-3 cleavage (Fig. 3A) and apoptosis (Fig. 3, B and C). A similar protective effect was seen when another NT inhibitor, NBTI, was used (Fig. 3A). However, antagonists for A1R (DPCPX), A2AR (DPMX), A2BR (MRS1754), and A3R (MRS1191) did not protect, but instead exacerbated, sustained Ado exposure-induced caspase-3 activation (Fig. 3D). These results indicate that sustained Ado exposure causes lung EC apoptosis via NT-mediated intracellular events, and that AR-mediated signaling may limit EC apoptosis.
Activation of p38 and JNK mediated sustained Ado exposure-induced EC apoptosis and mitochondrial defects.
Our laboratory has previously shown that sustained Ado exposure increased oxidative stress and activated the redox-sensitive proteins, p38 and JNK, via NT-mediated intracellular Ado uptake (38). To our surprise, neither antioxidant, NAC, nor NADPH oxidase inhibitor, apocynin, altered sustained Ado-induced caspase-3 cleavage (Fig. 4A). However, inhibition of p38 by SB203580 and inhibition of JNK by SP600125 blunted sustained Ado-induced caspase-3 cleavage (Fig. 4B) and apoptosis (Fig. 4C).
Fig. 4.

Effects of p38 and JNK on sustained adenosine exposure-induced EC apoptosis and mitochondrial (MT) defects. Bovine PAEC were treated with V or 50 μM A plus 50 μM D in the absence or presence of antioxidant, N-acetylcysteine (NAC; 12.5 mM), NADPH oxidase inhibitor, apocynin (Apo; 1 mM), p38 inhibitor, SB203580 (SB; 10 μM), JNK inhibitor, SP600125 (SP; 10 μM), or NT inhibitor DPM (10 μM) for 24 h. Apoptosis was assessed by procaspase-3 cleavage (A and B) and DAPI staining of apoptotic nuclei (C). D: MT morphology was assessed by fluorescence microscopy using MitoTracker Red CMXRos staining. E: MT membrane potential was also assessed by fluorescence microscopy of JC-1 staining. Representative images from three independent experiments for A, B, D, and E are shown. For A and B, the immunoblots were stripped and reprobed for actin to control for protein loading. For A, the immunoblot gels were rearranged by removing lanes that were not related to the point of this figure. C: *P < 0.05 vs. V-treated cells; ΦP < 0.05 vs. cells exposed to A+D. For E, the magnified images of the cells in the squares are shown.
Our laboratory has previously shown that activation of p38 and JNK by sustained Ado exposure occurs in mitochondria (38). Thus we hypothesized that sustained Ado exposure caused EC apoptosis via p38- and JNK-mediated mitochondrial defects. We first assessed mitochondria morphology and found that mitochondria in normal lung EC form an intricate filamentous network, as stained by MitoTracker Red CMXRos (Fig. 4D, V). Sustained Ado exposure disrupted the network, resulting in dysmorphic mitochondria (Fig. 4D, A+D). A similar structural mitochondrial defect has been linked to mitochondrial dysfunction (9). Interestingly, inhibition of p38 with SB203580 abolished sustained Ado-induced changes in mitochondrial morphology (Fig. 4D, SB+A+D). A similar protective effect was seen when JNK inhibitor, SP600125, was used (data not shown).
The loss of mitochondrial membrane potential is a hallmark for apoptosis. To further examine if sustained Ado exposure decreases mitochondrial membrane potential, we assessed the loss of mitochondrial membrane potential by JC-1 staining. In healthy cells, JC-1 exists as a monomer in the cytosol (green) and also accumulates as red aggregates in the mitochondria. In apoptotic cells, JC-1 exists in monomeric (green) form in cytosol, without red mitochondrial aggregates. Lung EC treated with vehicle for 48 h displayed numerous red aggregates (Fig. 4E, V), indicating intact mitochondrial membrane potential. These red aggregates were much less evident in EC treated with Ado plus DCF for 48 h (Fig. 4E, A+D), indicating loss of mitochondrial membrane potential. Sustained Ado-induced loss of mitochondrial membrane potential was abolished by NT inhibitor, DPM, JNK inhibitor, SP600125, and p38 inhibitor, SB203580 (Fig. 4E, DPM+A+D, SP+A+D, SB+A+D). These results suggest that sustained Ado exposure causes lung endothelial apoptosis and mitochondria defects via NT-mediated activation of p38 and JNK.
Sustained Ado exposure caused EC apoptosis via activation of mitochondria-mediated apoptotic pathway.
Our laboratory has previously shown that p38 and JNK were activated in mitochondria by sustained Ado exposure (38). Our present results demonstrate that inhibition of p38 and JNK blunted sustained Ado-induced EC apoptosis and mitochondria defects. Next, we assessed if sustained exposure to elevated Ado activates mitochondria-mediated apoptosis. We found that sustained exposure to elevated Ado for 24 and 48 h activated caspase-3 (Fig. 5A), and also activated caspase-9 (Fig. 5A), a marker for activation of mitochondria-mediated intrinsic pathway of apoptosis, but did not activate caspase-8 (Fig. 5A), a marker of extrinsic pathway of apoptosis. In addition, the levels of antiapoptotic protein, Bcl-2, was reduced, and the proapoptotic proteins, Bax and Bad, were elevated (Fig. 5B). The ratio of Bax to Bcl-2 was significantly increased (Fig. 5C). These results suggest that sustained Ado exposure causes lung EC apoptosis via alteration of Bcl-2 family proteins and subsequent mitochondria-mediated activation of caspase-9.
Fig. 5.

Effect of sustained adenosine exposure on MT-mediated apoptotic pathway. Bovine PAEC were treated with V or 50 μM A plus 50 μM D for 24 and 48 h. A: activation of caspase-9, caspase-8, and caspase-3 was assessed by immunoblot analysis of cleavage of procaspases. B: protein levels of Bcl-2, Bax, and Bad were also assessed. Individual immunoblots were stripped and reprobed for actin or GAPDH to control for protein loading. Representative images from three independent experiments for each panel are shown. C: densitometry was performed, and the ratio of Bax to Bcl-2 is presented. Values are means ± SE; n = 3. *P < 0.05 vs. V-treated cells for each time point.
Inhibition of xanthine oxidase had no effect on sustained Ado-induced caspase-3 activation.
Ado is metabolized to hypoxanthine by intracellular ADA, ultimately leading to generation of H2O2 if xanthine oxidase (XO) is activated. However, ADA inhibitor, DCF, has been shown to completely inhibit intracellular ADA activity and block conversion of exogenous [14C]Ado to hypoxanthine (30). Accordingly, we found that the XO inhibitor, allopurinol, did not alter sustained Ado-induced caspase-3 cleavage (Fig. 6). Thus our data confirm that intracellular ADA activity is inhibited by DCF in our model and that the XO-mediated pathway is unlikely to play a role in sustained Ado-induced endothelial apoptosis.
Fig. 6.

Effects of xanthine oxidase (XO) inhibitor on sustained adenosine exposure-induced EC apoptosis. Bovine PAEC were treated with V or 50 μM A plus 50 μM D in the absence or presence of allopurinol (AP; 100 μM) for 24 h, and apoptosis was assessed by procaspase-3 cleavage. Immunoblots were stripped and reprobed for actin to control for protein loading. Representative images from three independent experiments are shown.
DISCUSSION
Ado has been shown to be protective against apoptosis and tissue injury via activation of AR-mediated signaling (14, 18, 54, 55, 57). Our laboratory has previously shown that sustained Ado exposure causes pulmonary EC barrier dysfunction via NT-mediated intracellular Ado uptake (38). In this study, we further demonstrate that sustained Ado exposure causes pulmonary EC apoptosis via NT-dependent, p38- and JNK-mediated activation of mitochondrial apoptotic pathway. We also show that CS exposure increases lung tissue Ado levels in mice, an effect associated with lung EC apoptosis and early emphysema.
Lung endothelial apoptosis has been implicated in the pathogenesis of a variety of lung diseases, such as ALI, emphysema, and I/R lung injury (40). Plasma Ado is significantly elevated in patients with sepsis-induced ALI (43, 60) and tissue ischemia (59). Increased plasma Ado is associated with increased mortality in septic patients (43, 60). Ado levels are also increased in BAL fluid of human smokers (20). However, it is unknown whether lung tissue Ado levels are increased in smokers or patients with emphysema. ADA activity was decreased in plasma of rats exposed to CS for 4 wk (62) and in lungs of patients with chronic obstructive pulmonary disease (70), suggesting that lung tissue Ado levels may be elevated by CS exposure. In this study, we demonstrate that prolonged exposure (3 wk) to CS elevated lung tissue Ado levels in AKR mice, an effect associated with development of early emphysema and lung EC apoptosis. Extracellular ATP contributes to CS-induced lung inflammation and emphysema via purinergic receptors of inflammatory cells (16, 41, 47). ATP is a precursor of Ado. Whether elevated lung Ado plays a role in CS-induced lung endothelial apoptosis and emphysema remains to be determined. Nevertheless, our results demonstrate correlations between elevated lung tissue Ado and CS-induced emphysema and lung EC apoptosis. Studies from ADA-deficient mice have revealed that chronic elevation of Ado is associated with increased alveolar cell apoptosis and emphysema-like changes (8, 15, 69). Thus sustained elevation of lung Ado may contribute to CS-induced lung endothelial apoptosis and the development of emphysema.
Extracellular Ado can act via cell surface G protein-coupled AR and/or uptake into cells by NTs. Sustained Ado exposure in ADA-deficient mice enhanced alveolar cell apoptosis, an effect that was worsened in A2BR/ADA double-deficient mice (69), suggesting that prolonged Ado exposure-induced alveolar cell apoptosis is not due to activation of A2BR. Consistent with this in vivo effect, we show that inhibition of any one of the ARs exacerbated sustained Ado-induced apoptosis of cultured lung EC. These results suggest that AR-mediated signaling limits sustained Ado-induced EC apoptosis. Similar to other G-protein coupled receptors, prolonged exposure to Ado leads to desensitization and internalization of AR (24). Therefore, we speculate that sustained elevated Ado is taken up into cells by NTs, leading to enhancement of intracellular Ado levels that, in turn, triggers AR-independent effects. EC predominantly express equilibrative NT 1 (ENT1) and ENT2 with ENT1 expressed at twice the level of ENT2 (3). ENT1 has a 2.8-fold higher affinity for Ado than ENT2 (68). Concentrative NTs have very low affinity for Ado and limited expression in EC (35). We found that DPM and NBTI, highly specific inhibitors for ENT1 (∼1,000-fold less effective on ENT2) (5, 65), completely prevented sustained Ado exposure-induced EC apoptosis and loss of mitochondrial membrane potential, suggesting that ENT1 may play a major role in Ado transport into EC and may mediate sustained Ado-induced EC apoptosis. Specific molecular approaches are needed to confirm the role of ENT1 in future studies. Nevertheless, our results demonstrate that sustained Ado exposure causes lung EC apoptosis via NT-mediated intracellular uptake, rather than by AR-mediated signaling.
Our laboratory has previously demonstrated that elevated extracellular Ado causes lung endothelial apoptosis via increased ratio of intracellular S-adenosylhomocysteine (SAH) to S-adenosyl-l-methionine (SAM) due to inhibition of SAH hydrolase (56). Consistently, a recent study also showed that exogenous Ado causes endothelial apoptosis and oxidative stress via accumulation of intracellular SAH (58). The downstream signaling following accumulation of SAH mediating Ado-induced apoptosis remains unknown. SAM is synthesized exclusively in the cytosol (53) and then transported into mitochondria (2). Mitochondrial SAM is essential for maintenance of mitochondrial function due to its critical role as a precursor to glutathione. SAM has been shown to elevate glutathione levels in hepatocytes and prevent alcohol-induced mitochondrial dysfunction via attenuation of oxidative stress in rats (4, 12, 29). Our laboratory has shown that sustained exposure to elevated Ado increases reactive oxygen species (ROS) levels and activates p38 and JNK in lung EC mitochondria (38). Both p38 and JNK are redox-sensitive proteins activated upon oxidative stress (45). ROS-mediated p38 activation has been implicated in extracellular ATP-induced macrophage apoptosis (49) and H2O2-induced endothelial apoptosis (42). Activation of p38 has also been implicated in homocysteine-induced apoptosis of endothelial progenitor cells (6) and of cardiomyocytes (67). In this study, we show that inhibition of p38 and JNK prevents sustained Ado-induced mitochondrial defects and endothelial apoptosis. Our data suggest that sustained Ado exposure may increase intracellular SAH levels, thus reducing the availability of SAM to mitochondria, which leads to mitochondrial oxidative stress and subsequent activation of p38 and JNK in mitochondria, and ultimately resulting in endothelial apoptosis.
The antioxidant, NAC, and NADPH oxidase inhibitor, apocynin, were not able to rescue sustained Ado-induced apoptosis. We speculate that this may be due to the inability of these reagents to inhibit oxidative stress in mitochondria. We also show that the XO inhibitor, allopurinol, did not alter sustained Ado-induced caspase-3 cleavage, indicating that the XO-mediated pathway unlikely plays a role in sustained Ado-induced ROS generation and apoptosis.
Active p38 and JNK inhibit Bcl-2, thus causing apoptosis via direct phosphorylation of Bcl-2 (19, 23). Activation of p38 also mediates ischemia-induced apoptosis of isolated cardiomyocytes via increasing Bax mitochondrial translocation and accumulation (11). We show that sustained Ado exposure decreased Bcl-2, increased Bax and Bad, and significantly increased the ratio of Bax to Bcl-2. These effects were associated with activation of p38 and JNK, loss of mitochondrial membrane potential, caspase-9 activation, and apoptosis. Thus our data indicate that sustained exposure to Ado causes lung EC apoptosis via p38- and JNK-dependent activation of mitochondrial-mediated apoptotic pathway (Fig. 7).
Fig. 7.

Proposed model: Sustained exposure to elevated adenosine (Ado) causes endothelial apoptosis via NT-dependent activation of p38 and JNK and subsequent MT defects and ultimately activation of MT pathway of apoptosis. PM, plasma membrane. The solid lines indicate defined pathways, and the dashed lines indicate speculative pathways.
In summary, we show that CS exposure increased lung tissue Ado levels in mice, an effect associated with early emphysema and lung EC apoptosis. Using an in vitro model of sustained Ado exposure, we demonstrated that sustained Ado exposure caused lung EC apoptosis via NT-mediated intracellular Ado uptake, activation of p38 and JNK in mitochondria, subsequent mitochondria defects, and ultimately activation of the mitochondria-mediated apoptotic pathway. Our data reconcile the paradoxical effects of Ado on apoptosis in that prolonged exposure causes apoptosis via NT-mediated intracellular Ado uptake, whereas acute exposure is protective via activation of AR. Our data also suggest that sustained elevation of Ado in the lungs may contribute to CS-induced lung EC apoptosis and lung injury.
GRANTS
This study was supported with resources and the use of facilities at the Providence Veterans Affairs Medical Center and supported with Veterans Affairs BLR&D Merit Review (S. Rounds), National Heart, Lung, and Blood Institute Grant HL-64936 (S. Rounds), and American Thoracic Society/Pulmonary Hypertension Association research grant (Q. Lu).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Q.L. and S.R. conception and design of research; Q.L., P. Sakhatskyy, J.N., P. Shamirian, V.H., S.C., G.M.G., and M.P. performed experiments; Q.L., P. Sakhatskyy, J.N., P. Shamirian, V.H., and M.P. analyzed data; Q.L. and S.R. interpreted results of experiments; Q.L. prepared figures; Q.L. drafted manuscript; Q.L. and S.R. edited and revised manuscript; Q.L., P. Sakhatskyy, J.N., P. Shamirian, V.H., S.C., G.M.G., M.P., M.R.B., and S.R. approved final version of manuscript.
REFERENCES
- 1.Abadie Y, Bregeon F, Papazian L, Lange F, Chailley-Heu B, Thomas P, Duvaldestin P, Adnot S, Maitre B, Delclaux C. Decreased VEGF concentration in lung tissue and vascular injury during ARDS. Eur Respir J 25: 139–146, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Agrimi G, Di Noia MA, Marobbio CM, Fiermonte G, Lasorsa FM, Palmieri F. Identification of the human mitochondrial S-adenosylmethionine transporter: bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem J 379: 183–190, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Archer RG, Pitelka V, Hammond JR. Nucleoside transporter subtype expression and function in rat skeletal muscle microvascular endothelial cells. Br J Pharmacol 143: 202–214, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey SM, Robinson G, Pinner A, Chamlee L, Ulasova E, Pompilius M, Page GP, Chhieng D, Jhala N, Landar A, Kharbanda KK, Ballinger S, Darley-Usmar V. S-adenosylmethionine prevents chronic alcohol-induced mitochondrial dysfunction in the rat liver. Am J Physiol Gastrointest Liver Physiol 291: G857–G867, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Baldwin SA, Mackey JR, Cass CE, Young JD. Nucleoside transporters: molecular biology and implications for therapeutic development. Mol Med Today 5: 216–224, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Bao XM, Wu CF, Lu GP. Atorvastatin inhibits homocysteine-induced oxidative stress and apoptosis in endothelial progenitor cells involving Nox4 and p38MAPK. Atherosclerosis 210: 114–121, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Blackburn MR, Aldrich M, Volmer JB, Chen W, Zhong H, Kelly S, Hershfield MS, Datta SK, Kellems RE. The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J Biol Chem 275: 32114–32121, 2000 [DOI] [PubMed] [Google Scholar]
- 8.Blackburn MR, Volmer JB, Thrasher JL, Zhong H, Crosby JR, Lee JJ, Kellems RE. Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J Exp Med 192: 159–170, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Calfee CS, Matthay MA, Eisner MD, Benowitz N, Call M, Pittet JF, Cohen MJ. Active and passive cigarette smoking and acute lung injury following severe blunt trauma. Am J Respir Crit Care Med 183: 1660–1665, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capano M, Crompton M. Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J 395: 57–64, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cederbaum AI. Hepatoprotective effects of S-adenosyl-l-methionine against alcohol- and cytochrome P450 2E1-induced liver injury. World J Gastroenterol 16: 1366–1376, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chapman HA. A Fas pathway to pulmonary fibrosis. J Clin Invest 104: 1–2, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y. Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res 84: 1848–1855, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Chunn JL, Molina JG, Mi T, Xia Y, Kellems RE, Blackburn MR. Adenosine-dependent pulmonary fibrosis in adenosine deaminase-deficient mice. J Immunol 175: 1937–1946, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Cicko S, Lucattelli M, Muller T, Lommatzsch M, De Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R, Durk T, Zissel G, Boeynaems JM, Sorichter S, Ferrari D, Di Virgilio F, Virchow JC, Lungarella G, Idzko M. Purinergic receptor inhibition prevents the development of smoke-induced lung injury and emphysema. J Immunol 185: 688–697, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Crikis S, Lu B, Murray-Segal LM, Selan C, Robson SC, D'Apice AJ, Nandurkar HH, Cowan PJ, Dwyer KM. Transgenic overexpression of CD39 protects against renal ischemia-reperfusion and transplant vascular injury. Am J Transplant 10: 2586–2595, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Alimonte I, Ballerini P, Nargi E, Buccella S, Giuliani P, Di Iorio P, Caciagli F, Ciccarelli R. Staurosporine-induced apoptosis in astrocytes is prevented by A1 adenosine receptor activation. Neurosci Lett 418: 66–71, 2007 [DOI] [PubMed] [Google Scholar]
- 19.De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, Palamara AT, Russo T, Garaci E, Cozzolino F. Bcl-2 phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem 281: 21353–21361, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis 148: 91–97, 1993 [DOI] [PubMed] [Google Scholar]
- 21.Eckle T, Fullbier L, Wehrmann M, Khoury J, Mittelbronn M, Ibla J, Rosenberger P, Eltzschig HK. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol 178: 8127–8137, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Eltzschig HK, Faigle M, Knapp S, Karhausen J, Ibla J, Rosenberger P, Odegard KC, Laussen PC, Thompson LF, Colgan SP. Endothelial catabolism of extracellular adenosine during hypoxia: the role of surface adenosine deaminase and CD26. Blood 108: 1602–1610, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farley N, Pedraza-Alva G, Serrano-Gomez D, Nagaleekar V, Aronshtam A, Krahl T, Thornton T, Rincon M. p38 mitogen-activated protein kinase mediates the Fas-induced mitochondrial death pathway in CD8+ T cells. Mol Cell Biol 26: 2118–2129, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fredholm BB, APIJ , Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53: 527–552, 2001 [PMC free article] [PubMed] [Google Scholar]
- 25.Fujita M, Kuwano K, Kunitake R, Hagimoto N, Miyazaki H, Kaneko Y, Kawasaki M, Maeyama T, Hara N. Endothelial cell apoptosis in lipopolysaccharide-induced lung injury in mice. Int Arch Allergy Immunol 117: 202–208, 1998 [DOI] [PubMed] [Google Scholar]
- 26.Giordano RJ, Lahdenranta J, Zhen L, Chukwueke U, Petrache I, Langley RR, Fidler IJ, Pasqualini R, Tuder RM, Arap W. Targeted induction of lung endothelial cell apoptosis causes emphysema-like changes in the mouse. J Biol Chem 283: 29447–29460, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerassimov A, Hoshino Y, Takubo Y, Turcotte A, Yamamoto M, Ghezzo H, Triantafillopoulos A, Whittaker K, Hoidal JR, Cosio MG. The development of emphysema in cigarette smoke-exposed mice is strain dependent. Am J Respir Crit Care Med 170: 974–980, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Held HD, Uhlig S. Basal lung mechanics and airway and pulmonary vascular responsiveness in different inbred mouse strains. J Appl Physiol 88: 2192–2198, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Holguin F, Moss I, Brown LA, Guidot DM. Chronic ethanol ingestion impairs alveolar type II cell glutathione homeostasis and function and predisposes to endotoxin-mediated acute edematous lung injury in rats. J Clin Invest 101: 761–768, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holland MJ. Specificity of 2′-deoxycoformycin inhibition of adenosine metabolism in intact human skin fibroblasts. Res Commun Chem Pathol Pharmacol 51: 311–324, 1986 [PubMed] [Google Scholar]
- 31.Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 163: 737–744, 2001 [DOI] [PubMed] [Google Scholar]
- 32.Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106: 1311–1319, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawasaki M, Kuwano K, Hagimoto N, Matsuba T, Kunitake R, Tanaka T, Maeyama T, Hara N. Protection from lethal apoptosis in lipopolysaccharide-induced acute lung injury in mice by a caspase inhibitor. Am J Pathol 157: 597–603, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuwano K, Miyazaki H, Hagimoto N, Kawasaki M, Fujita M, Kunitake R, Kaneko Y, Hara N. The involvement of Fas-Fas ligand pathway in fibrosing lung diseases. Am J Respir Cell Mol Biol 20: 53–60, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Loffler M, Morote-Garcia JC, Eltzschig SA, Coe IR, Eltzschig HK. Physiological roles of vascular nucleoside transporters. Arterioscler Thromb Vasc Biol 27: 1004–1013, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Lu Q, Harrington EO, Hai CM, Newton J, Garber M, Hirase T, Rounds S. Isoprenylcysteine carboxyl methyltransferase modulates endothelial monolayer permeability: involvement of RhoA carboxyl methylation. Circ Res 94: 306–315, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Lu Q, Harrington EO, Newton J, Casserly B, Radin G, Warburton R, Zhou Y, Blackburn MR, Rounds S. Adenosine protected against pulmonary edema through transporter- and receptor A2-mediated endothelial barrier enhancement. Am J Physiol Lung Cell Mol Physiol 298: L755–L767, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Q, Newton J, Hsiao V, Shamirian P, Blackburn MR, Pedroza M. Sustained adenosine exposure causes lung endothelial barrier dysfunction via nucleoside transporter-mediated signaling. Am J Respir Cell Mol Biol 47: 604–613, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Q, Patel B, Harrington EO, Rounds S. Transforming growth factor-beta1 causes pulmonary microvascular endothelial cell apoptosis via ALK5. Am J Physiol Lung Cell Mol Physiol 296: L825–L838, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Q, Rounds S. Pulmonary endothelial cell death: implications for lung disease pathogenesis. In: The Pulmonary Endothelium: Function in Health and Disease, edited by Voelkel NF, Rounds S. Chichester, UK: Wiley, 2009, p. 243–260 [Google Scholar]
- 41.Lucattelli M, Cicko S, Muller T, Lommatzsch M, De Cunto G, Cardini S, Sundas W, Grimm M, Zeiser R, Durk T, Zissel G, Sorichter S, Ferrari D, Di Virgilio F, Virchow JC, Lungarella G, Idzko M. P2X7 receptor signaling in the pathogenesis of smoke-induced lung inflammation and emphysema. Am J Respir Cell Mol Biol 44: 423–429, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Machino T, Hashimoto S, Maruoka S, Gon Y, Hayashi S, Mizumura K, Nishitoh H, Ichijo H, Horie T. Apoptosis signal-regulating kinase 1-mediated signaling pathway regulates hydrogen peroxide-induced apoptosis in human pulmonary vascular endothelial cells. Crit Care Med 31: 2776–2781, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Martin C, Leone M, Viviand X, Ayem ML, Guieu R. High adenosine plasma concentration as a prognostic index for outcome in patients with septic shock. Crit Care Med 28: 3198–3202, 2000 [DOI] [PubMed] [Google Scholar]
- 44.Matot I, Weiniger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 adenosine receptors and mitogen-activated protein kinases in lung injury following in vivo reperfusion. Crit Care 10: R65, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta 1780: 1325–1336, 2008 [DOI] [PubMed] [Google Scholar]
- 46.Minelli A, Bellezza I, Tucci A, Rambotti MG, Conte C, Culig Z. Differential involvement of reactive oxygen species and nucleoside transporters in cytotoxicity induced by two adenosine analogues in human prostate cancer cells. Prostate 69: 538–547, 2009 [DOI] [PubMed] [Google Scholar]
- 47.Mortaz E, Braber S, Nazary M, Givi ME, Nijkamp FP, Folkerts G. ATP in the pathogenesis of lung emphysema. Eur J Pharmacol 619: 92–96, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Ng CS, Wan S, Yim AP. Pulmonary ischaemia-reperfusion injury: role of apoptosis. Eur Respir J 25: 356–363, 2005 [DOI] [PubMed] [Google Scholar]
- 49.Noguchi T, Ishii K, Fukutomi H, Naguro I, Matsuzawa A, Takeda K, Ichijo H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J Biol Chem 283: 7657–7665, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Petrache I, Natarajan V, Zhen L, Medler TR, Richter AT, Cho C, Hubbard WC, Berdyshev EV, Tuder RM. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med 11: 491–498, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quadri SM, Segall L, de Perrot M, Han B, Edwards V, Jones N, Waddell TK, Liu M, Keshavjee S. Caspase inhibition improves ischemia-reperfusion injury after lung transplantation. Am J Transplant 5: 292–299, 2005 [DOI] [PubMed] [Google Scholar]
- 52.Rangasamy T, Misra V, Zhen L, Tankersley CG, Tuder RM, Biswal S. Cigarette smoke-induced emphysema in A/J mice is associated with pulmonary oxidative stress, apoptosis of lung cells, and global alterations in gene expression. Am J Physiol Lung Cell Mol Physiol 296: L888–L900, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reytor E, Perez-Miguelsanz J, Alvarez L, Perez-Sala D, Pajares MA. Conformational signals in the C-terminal domain of methionine adenosyltransferase I/III determine its nucleocytoplasmic distribution. FASEB J 23: 3347–3360, 2009 [DOI] [PubMed] [Google Scholar]
- 54.Rivo J, Zeira E, Galun E, Einav S, Linden J, Matot I. Attenuation of reperfusion lung injury and apoptosis by A2A adenosine receptor activation is associated with modulation of Bcl-2 and Bax expression and activation of extracellular signal-regulated kinases. Shock 27: 266–273, 2007 [DOI] [PubMed] [Google Scholar]
- 55.Rivo J, Zeira E, Galun E, Matot I. Activation of A3 adenosine receptor provides lung protection against ischemia-reperfusion injury associated with reduction in apoptosis. Am J Transplant 4: 1941–1948, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Rounds S, Yee WL, Dawicki DD, Harrington E, Parks N, Cutaia MV. Mechanism of extracellular ATP- and adenosine-induced apoptosis of cultured pulmonary artery endothelial cells. Am J Physiol Lung Cell Mol Physiol 275: L379–L388, 1998 [DOI] [PubMed] [Google Scholar]
- 57.Simonis G, Wiedemann S, Joachim D, Weinbrenner C, Marquetant R, Strasser RH. Stimulation of adenosine A2b receptors blocks apoptosis in the noninfarcted myocardium even when administered after the onset of infarction. Mol Cell Biochem 328: 119–126, 2009 [DOI] [PubMed] [Google Scholar]
- 58.Sipkens JA, Hahn NE, Blom HJ, Lougheed SM, Stehouwer CD, Rauwerda JA, Krijnen PA, van Hinsbergh VW, Niessen HW. S-Adenosylhomocysteine induces apoptosis and phosphatidylserine exposure in endothelial cells independent of homocysteine. Atherosclerosis 221: 48–54, 2012 [DOI] [PubMed] [Google Scholar]
- 59.Sperlagh B, Doda M, Baranyi M, Hasko G. Ischemic-like condition releases norepinephrine and purines from different sources in superfused rat spleen strips. J Neuroimmunol 111: 45–54, 2000 [DOI] [PubMed] [Google Scholar]
- 60.Stringer KA, Serkova NJ, Karnovsky A, Guire K, Paine R, 3rd, Standiford TJ. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma (1)H-nuclear magnetic resonance quantitative metabolomics and computational analysis. Am J Physiol Lung Cell Mol Physiol 300: L4–L11, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang K, Rossiter HB, Wagner PD, Breen EC. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol 97: 1559–1566, 2004 [DOI] [PubMed] [Google Scholar]
- 62.Thome GR, Mazzanti CM, Ahmed M, Correa M, Spanevello RM, Maldonado PA, Luchese C, Cargnelutti D, Morsch VM, Duarte MM, Fiorenza AM, Nogueira CW, De Bona KS, Moretto MB, Da Luz SC, Mazzanti A, Schetinger MR. Activity of ectonucleotidases and adenosine deaminase in rats exposed to cigarette smoke. Inhal Toxicol 21: 906–912, 2009 [DOI] [PubMed] [Google Scholar]
- 63.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med 200: 1395–1405, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am J Physiol Lung Cell Mol Physiol 275: L1192–L1199, 1998 [DOI] [PubMed] [Google Scholar]
- 65.Visser F, Vickers MF, Ng AM, Baldwin SA, Young JD, Cass CE. Mutation of residue 33 of human equilibrative nucleoside transporters 1 and 2 alters sensitivity to inhibition of transport by dilazep and dipyridamole. J Biol Chem 277: 395–401, 2002 [DOI] [PubMed] [Google Scholar]
- 66.Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol 176: 4449–4458, 2006 [DOI] [PubMed] [Google Scholar]
- 67.Wang X, Cui L, Joseph J, Jiang B, Pimental D, Handy DE, Liao R, Loscalzo J. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J Mol Cell Cardiol 52: 753–760, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ward JL, Sherali A, Mo ZP, Tse CM. Kinetic and pharmacological properties of cloned human equilibrative nucleoside transporters, ENT1 and ENT2, stably expressed in nucleoside transporter-deficient PK15 cells. Ent2 exhibits a low affinity for guanosine and cytidine but a high affinity for inosine. J Biol Chem 275: 8375–8381, 2000 [DOI] [PubMed] [Google Scholar]
- 69.Zhou Y, Mohsenin A, Morschl E, Young HW, Molina JG, Ma W, Sun CX, Martinez-Valdez H, Blackburn MR. Enhanced airway inflammation and remodeling in adenosine deaminase-deficient mice lacking the A2B adenosine receptor. J Immunol 182: 8037–8046, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou Y, Murthy JN, Zeng D, Belardinelli L, Blackburn MR. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLos One 5: e9224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]