

Abstract
Obliterative bronchiolitis (OB), a fibrotic airway lesion, is the leading cause of death after lung transplantation. Type V collagen [col(V)] overexpression and IL-17-mediated anti-col(V) immunity are key contributors to OB pathogenesis. Here, we report a previously undefined role of IL-17 in inducing col(V) overexpression, leading to epithelial mesenchymal transition (EMT) and subsequent OB. We observed IL-17-mediated induction of col(V) α1 chains [α1 (V)] in normal airway epithelial cells in vitro and detected α1 (V)-specific antibodies in bronchoalveolar lavage fluid of lung transplant patients. Overexpression of IL-17 and col(V) was detected in OB lesions in patient lung biopsies and in a murine OB model. IL-17 is shown to induce EMT, TGF-β mRNA expression, and SMAD3 activation, whereas downregulating SMAD7 expression in vitro. Pharmacological inhibition of TGF-βRI tyrosine kinase, p38 MAPK, or focal adhesion kinase prevented col(V) overexpression and EMT. In murine orthotopic lung transplants, neutralizing IL-17 significantly decreased TGF-β mRNA and protein expression and prevented epithelial repair/OB. Our findings highlight a feed-forward loop between IL-17 and TGF-β, leading to induction of col(V) and associated epithelial repair, thus providing one possible link between autoimmunity and OB after lung transplantation.
Keywords: autoimmunity, p38 MAPK, focal adhesion kinase, small-airway epithelial cells, RLE-6TN, mouse transplant model, epithelial-mesenchymal transition
obliterative bronchiolitis (OB) is characterized by extensive peribronchiolar fibrosis with plugs of granulation tissues (fibroblasts and collagen) that occlude small airways. OB is the key reason that the 5-yr survival of lung transplant recipients is only 50%, the worst of all major solid organ transplants (42, 48).
Aberrant epithelial repair is a key event in the transplanted lung (1, 9) in which bronchioles lose resident epithelial cells and become occluded by granulation tissue. Abnormal epithelial repair eventually causes an epithelial-to-mesenchymal transition (EMT), a functional and phenotypic change of epithelial cells into spindle-shaped, migratory (43) and matrix-component-secreting mesenchymal cells (10, 41), and a process associated with lung fibrosis (15, 27). However, the direct connection between EMT and the in vivo phenomena of fibrosis and fibro-obliterative disease remains controversial.
We and others previously reported that OB is associated with dysregulation of two types of collagen: 1) marked increase in type V collagen [col(V)], a quantitatively minor lung collagen (8, 14, 40), and 2) a decrease in the major lung collagen type I [col(I)] (2, 53). We have shown that prospective monitoring of patients with human lung transplant revealed a critical role of col(V)-specific cellular immunity in OB pathogenesis (14, 40). Although overexpression of col(V), an otherwise quantitatively minor collagen, is involved in OB pathology, mechanisms leading to col(V) overexpression are unknown. Thus a mechanistic understanding of the triggers of col(V) overexpression could have clinical utility in earlier detection and treatment of fibrotic lung diseases, such as OB, and aid in averting one important complication of lung transplants.
The proinflammatory cytokine, IL-17A (referred to here as IL-17), has been implicated in the pathogenesis of multiple autoimmune diseases (3, 37, 38). We have reported that CD4+ T helper cells (TH-17 cells), which produce IL-17, play a central role in OB pathology (52) and that neutralizing IL-17 bioactivity prevents OB in a mouse orthotopic lung transplant model (19). However, in a clinical OB lesion or murine OB model, the mechanistic roles for IL-17 and TGF-β in the induction of col(V) overexpression and EMT have not been elucidated. Borthwick and colleagues (9–11) have described TGF-β-mediated EMT in epithelial cells from patients with lung transplants. TGF-β, a key initiator of EMT (31, 50), is a strong inducer of IL-17 (35), and IL-17, in turn, may be involved in lung fibrogenesis in TGF-β-dependent and independent pathways (36). Because IL-17 and col(V) overexpression are key contributors to OB pathogenesis, in this report on OB pathology, we have investigated the mechanistic role for IL-17 in col(V) overexpression and EMT.
MATERIALS AND METHODS
Cell culture.
Normal human small airway epithelial cells (SAEC; Clonetics, Cambrex Biosciences, Walkersville, MD) were grown in small airway basal media supplemented with growth factors (Clonetics). Immortalized normal rat lung alveolar type II epithelial cells (RLE-6TN; ATCC, Manassas, VA) were maintained in RPMI-1640 (Invitrogen, Carlsbad, CA) supplemented with 5% fetal calf serum, 100 U/ml penicillin/streptomycin and fungizone (Invitrogen). The above cells were seeded at 70% confluence and incubated in 5% CO2-95% air. Before stimulation, cells were growth arrested using 0.01% serum or 1:100 growth factors containing media for 16 h.
Antibodies and other reagents.
The antibodies used for immunoblotting or immunofluorescent labeling are as follows: α-SMA and vimentin (Dako, Carpenteria, CA), E-cadherin (E-CAD) (ECM Biosciences, Versailles, KY), zona occludens (ZO)-1 (Zymed, Carlsbad, CA), collagen type V (Lifespan Biosciences, Seattle, WA), S100A4 (Abcam, Cambridge, MA), GAPDH and phosphorylation-specific antibodies to Tyr-397-focal adhesion kinase (FAK) (Invitrogen), Thr-180/182-p38 MAPK, and Ser-423/425-SMAD3 (Cell Signaling, Boston, MA). Recombinant proteins used in this study were IL-17A, C, F (BD Biosciences, Franklin Lakes, NJ) and human platelet-derived TGF-β1 (Roche Diagnostics, Manneheim, Germany). All other reagents unless specified were from Sigma (St. Louis, MO). The soluble murine IL-17 receptor fusion protein was generously provided by Dr. Jay K. Kolls, Louisiana State University Health Sciences Center.
Immunofluorescence staining.
Cells were fixed in 4% formaldehyde for 15 min. Subsequent to permeabilization, cells were incubated with primary antibodies followed by rhodamine or FITC-labeled secondary antibodies (Jackson ImmunoResearch, West Grove, PA) for 1 h each. Nuclei were counterstained with DAPI. Cells were visualized by a Nikon i90 fluorescence microscope and images captured with NIS-Elements v2.0 (Nikon, Melville, NY).
Immunohistochemistry.
Sections obtained from paraffin-embedded, formalin-fixed lungs underwent antigen retrieval treatment, followed by peroxide and protein blocking (1× Power Block; Biogenex, San Ramon, CA). Sections were incubated with primary antibodies specific to IL-17A (1:200; R&D Systems, Minneapolis, MN), col(V) (Abcam; 1:150), S100A4 (Abcam; 1:150), mouse monoclonal TGF-β (1:150; R&D Systems) and then stained using a sensitive avidin-streptavidin-diaminobenzidine peroxidase kit (Biogenex) according to the manufacturer's instructions. The sections were counterstained with hematoxylin and mounted.
Western blotting of cell lysates and patient BAL antibodies.
Cell lysates were subjected to immunoblotting, as previously described (24). For conditioned media studies, conditioned media was normalized by loading equal protein concentrations (30 μg) for all samples under reducing conditions using lamelli buffer. For bronchoalveolar lavage (BAL) antibody studies, all subject consent protocols and procedures were approved by the Institutional Review Board, University of Wisconsin-Madison as described previously (14). Subject consent was obtained using IRB-approved, written, informed consent procedures at the University of Wisconsin Hospital and Clinics. IgG was affinity purified from the BAL fluid of patients and controls by passage over a protein G Sepharose column (Pharmacia, Piscataway, NJ). Recombinant col(V), treated or untreated with the proteinase bone morphogenetic protein 1 (BMP1), which processes col(V) to its mature matrix form, was prepared as previously described (23) and used as antigen (Fig. 2B) and size markers (Fig. 2A). Proteins were electrophoresed on 4–15% acrylamide gradient gels (Fig. 1B) or on 5% acrylamide gels with 3.5% stacking gels (Fig. 2B). Purified IgG was used to probe immunoblots for col(V), as reported (14).
Fig. 2.
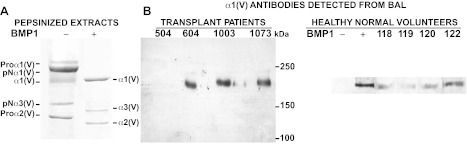
Lung transplant patients develop α1(V) antibodies. A: Coomassie blue-stained gel is shown of recombinant α1(V) α2(V) α3(V) procollagen heterotrimers cleaved (+) or not (-) with the protease bone morphogenetic protein 1 (BMP1) to produce the mature tissue form of each chain. B: immunoblots are shown of BMP1-cleaved α1(V) α2(V) α3(V) heterotrimers probed with purified antibodies from the bronchoalveolar lavage (BAL) fluid antibodies of 3 individual lung transplant patients. Patients are identified by sample numbers 504, 604, 1003, and 1073. Blots are aligned with the gel in B, right to show that only bands comigrating with α1(V) are recognized by patient antibodies. Normal controls are lavage fluid obtained from normal healthy nonsmoking volunteers. Note: Somewhat differential migration patterns of the col(V) chains in Fig. 1B and Fig. 2B are due to use of gradient (1B) or fixed concentration (2B) gels.
Fig. 1.
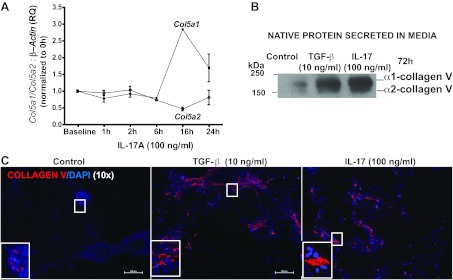
IL-17 induces the α1 chain of collagen type V [col(V)]. A: Col5a1 and Col5a2 expressions were detected by real-time PCR in RLE-6TN cells treated with IL-17 at the indicated time points. Error bars indicate means ± SE; n = 3. B: Western blotting of α1(V) and α2(V) chains of the native col(V) protein secreted into conditioned media obtained from C. C: using immunofluorescent labeling, col(V) was detected using rhodamine (red) at 72 h. Nuclei were counterstained with DAPI. Images were captured at ×10 magnification. RQ, relative quantitation.
Animal studies.
The Indiana University School of Medicine Animal Care and Use Committee (IACUC) approved the animal protocols used in this study. Mice were housed under specific pathogen-free conditions in plastic enclosed filter-top cages with hardwood shavings, five animals per cage. A 12-h:12-h light/dark cycle was maintained, and the mice had access to water and rodent laboratory chow ad libitum until they were euthanized. For the Lung transplant model, an orthotopic left lung minor-mismatch transplant model was utilized to induce OB as previously described (19). Left lung from 10-wk-old male C57BL/10 (donor) mice was transplanted onto weight-matched 10-wk-old male C57BL/6 (recipient) mice. On day 21 after lung transplants, mice were euthanized; lungs were harvested and processed for immunohistochemical staining or stored at −20°C until further analyzed.
Neutralization of IL-17A bioactivity.
Neutralization of circulating IL-17A and IL-17F was performed as previously described (19) using adenoviral vectors encoding the IL-17R:Fc fusion protein designated as Ad-IL-17R:Fc.
Real-time PCR.
Real-time PCR was performed on cDNA from cell lysates as described previously (19) using gene-specific primer pairs (Table 1). The semiquantitative real-time PCR data for each target gene was expressed as 2−ΔΔCT relative quantitation vs. endogenous β-actin, with error bars representing the SE for triplicate reactions.
Table 1.
Real-time PCR primers used in clinical lung tissues, murine OB model, and rat airway epithelial cells
| Species | Gene Name | Primer Sequence |
|---|---|---|
| Rat | TGF-β1 | Forward 5′-AAG AAG TCA CCC GCG TGC TA-3′ |
| Reverse 5′-TGT GTG ATG TCT TTG GTT TTG TCA-3′ | ||
| SMAD7 | Forward 5′-CCA ACT GCA GAC TGT CCA GA-3′ | |
| Reverse 5′-TTC TCC TCC CAG TAT GCC AC-3′ | ||
| Col5a1 | Forward 5′-GGA CTC GGC GGA ACA TT-3′ | |
| Reverse 5′-GGA GTT GAG GGA ACC AAA GAT-3′ | ||
| Col5a2 | Forward 5′-TGT GCG GGG AAG TGT AG-3′ | |
| Reverse 5′-CCA AGA GCA GCA GTA AGA T-3′ | ||
| β-Actin | Forward 5′-TGA CCC AGA TCA TGT TTG AGA CC-3′ | |
| Reverse 5′-GTG GTA CGA CCA GAG GCA TAC A-3′ | ||
| Mouse | IL-17A | Forward 5′-CTG TGT CTC TGA TGC TGT TG-3′ |
| Reverse 5′-ATG TGG TGG Tcc AGC TTT C-3′ | ||
| TGF-β1 | Forward 5′-TGA CGT CAC TGG AGT TGT ACG G-3′ | |
| Reverse 5′-GGT TCA TGT CAT GGA TGG TGC-3′ | ||
| β-Actin | Forward 5′-AGA GGG AAA TCG TGC CTC AC-3′ | |
| Reverse 5′-CAA TAG TGA TGA CCT GGC CGT-3′ | ||
| Human | IL-17A | Forward 5′-TCAACCCGATTGTCCACCAT-3′ |
| Reverse 5′-GAGTTTAGTCCGAAATGAGGCTG-3′ |
OB, obliterative bronchiolitis.
Wound-healing assay.
RLE-6TN cells were seeded in 24-well plates and cultured to 90% confluence. The cells were growth arrested for 16 h and then wounded by scratching with a pipette tip. RLE-6TNs were treated per described conditions for 72 h. Cells were dual labeled with fluorophores and imaged. The area of wound closure was measured using the NIH-Image J program.
Measurement of extracellular H2O2 release.
H2O2 release from cultured epithelial cells was assayed using a fluorometric method as previously described (24).
Statistical analyses.
Student's t-test and one-way ANOVA with Bonferroni post hoc test was performed using GraphPad Prism v3.0 for Windows, GraphPad Software (San Diego, CA) (24). Significance was defined as P < 0.05.
RESULTS
IL-17 mediates specific RNA and protein overexpression for the α1 chain of col(V).
We and others (8, 12–14) previously reported that autoimmune responses to col(V) are linked to the pathogenesis of lung fibrosis. We also have previously reported IL-17-dependent anti-col(V) cellular immune responses in patients with OB with lung transplants (as measured by the trans-vivo delayed-type hypersensitivity assay); we attributed this response to be possibly due to the overabundance of induced α1(V) chains noted in the OB lesions (14). Thus we sought to determine whether IL-17 might induce col(V) expression in airway epithelial cells. We observed robust, up to approximately threefold, upregulation of expression of the α1(V) chain gene Col5a1, with relatively minimal effects on expression of the α2(V) chain gene Col5a2, when RLE-6TN cells were treated with IL-17A (Fig. 1A). TGF-β is known to induce collagen synthesis and, as expected, stimulated col(V) expression in airway cells, as shown by Fig. 1B. Notably, IL-17 also induced col(V) expression, at levels comparable to those induced by TGF-β (Fig. 1B). Specifically, immunoblotting of the native col(V) chains secreted into conditioned media revealed strong induction of α1 (V) chains, with seemingly lesser induction of α2 (V) chains (Fig. 1B). Additionally, immunostaining of the cell layers from the same cultures revealed robust induction of col(V) expression in both TGF-β- and IL-17-treated cells (Fig. 1C). Collectively, the above data support the conclusion that IL-17 induces robust expression of the α1 chain of col(V) at both the mRNA level and at the level of production of the secreted protein.
Lung transplant recipients express antibodies specific to the α1(V) chain of col(V).
To characterize the nature of possible anti-col(V) humoral immune responses in lung transplantation, we next determined whether lung transplant recipients might also produce anti-col(V) antibodies and, if so, whether such antibodies would be specific to the α1(V) chain and/or to the other col(V) chains. Antibodies were isolated from BAL samples of patients of lung transplants. Patient samples shown (Fig. 2B) are representative specimens from the study population previously reported (14). As size markers and Western blot antigens, we employed recombinant heterotrimers containing α1(V), α2(V), and less abundant α3(V) col(V) chains, processed with the metalloproteinase BMP1 to produce the mature tissue forms of these chains (23) (Fig. 2, A and B). When we immunoprobed the BMP1-cleaved α1(V), α2(V), and α3(V) heterotrimers, we observed that purified antibodies from the BAL of three individual patients of lung transplant detected α1(V) but not α2(V) or α3(V) chains (Fig. 2B). Of the 15 patients analyzed, 10 of them (∼67%) had strong antibody reactivity to α1(V) detected by Western blot analysis in at least one posttransplant BAL sample (data not shown). In contrast, we detected only very low levels of antibodies to α1(V) in BAL samples from normal healthy volunteers (Fig. 2B). Taken together, these findings indicate that 1) IL-17 induces col(V) in airway epithelial cells and 2) patients of lung transplant have a heightened local humoral antibody response specific to the α1 chain of col(V) compared with normal healthy control BAL donors.
Col(V) is highly expressed in clinical OB lesions and in a murine OB model after lung transplant.
Prior reports demonstrated that induction of tolerance to col(V) protects against OB mediated by col(V)-specific reactive Th17 cells, but the col(V) distribution in the clinical OB lesions has not been illustrated (13, 53). Therefore, immunohistochemistry was utilized to determine the localization of col(V) in clinical OB. We observed strong signals of col(V) deposition in regions in and around the fibrotic foci in lungs of patients with OB (Fig. 3A). In contrast, and consistent with prior studies, col(V) was only faintly detectable in normal tissues (Fig. 3A). The demographics of the OB patients are shown in Table 2. In the orthotopic murine OB model, significant fibrosis was observed at day 21 as shown by trichrome staining (Fig. 3B). The transplant lung had significant expression of col(V) in the fibrotic lesions of OB and around the airway epithelium (Fig. 3B); in contrast, the native lung had a thin band of col(V) confined to the subepithelial airway connective tissue (Fig. 3B). These data illustrate overexpression of col(V) in the OB lesions of clinical biopsies and our murine OB model.
Fig. 3.
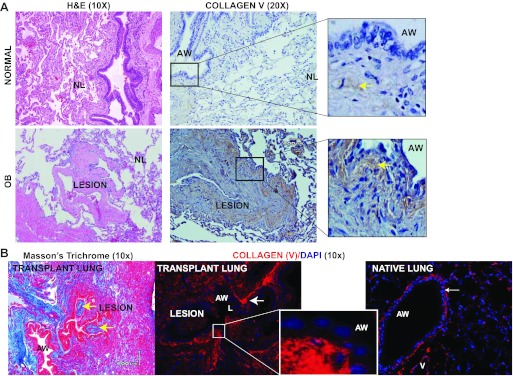
Col(V) is expressed in clinical obliterative bronchiolitis (OB) lesions and in a murine OB model of orthotopic lung transplant. A: representative immunohistochemical staining showing expression of col(V) in 1 of 3 patients with OB, and in normal lungs. Hematoxylin and eosin staining reveals the respective lung architectures. Inset in OB: dense collagen V deposition in the fibrotic plugs. Inset in normal lung: epithelial and subepithelial tissues with weak staining for col(V). B: Trichrome staining of representative lungs reveals fibrotic lesions (yellow arrows) in the lumen of the airway at day 21. Immunostaining of col(V) expression in transplant and native lung. Inset: profuse col(V) (rhodamine-red) accumulation in the fibrotic lesions of OB. Nuclei were counterstained using DAPI. NL, normal lung architecture; AW, airway.
Table 2.
Demographics of patients with OB
| Sample# | Age | Sex | Days after transplant |
|---|---|---|---|
| 1 | 63 | M | 4,354 |
| 2 | 61 | F | 326 |
| 3 | 64 | F | 2,145 |
IL-17 induces EMT in normal airway epithelium.
Among the six members of the IL-17 family, isoforms A, C, and F have been implicated in fibrogenesis (19, 28, 51). To determine whether IL-17 might induce EMT and, if so, identify the specific isoform involved, normal primary human SAEC, similar to those involved in OB, were cultured in the presence or absence of IL-17A, C, or F, followed by an assessment of E-CAD expression. Cells cultured in the presence of TGF-β served as a positive control. As expected, TGF-β downregulated E-CAD (Fig. 4, A and B). Notably, IL-17A was found to significantly downregulate E-CAD expression in a dose-dependent manner on the sixth day of culture (with only modest decreases observed for IL-17C or IL-17F) (Fig. 4, A and B). To confirm the temporal pattern of IL-17-mediated EMT, we determined E-CAD and α-SMA expression on days 2, 3, 4, 5, and 6. E-CAD expression began to decrease by day 4, whereas we observed induction of α-SMA expression by day 2 (Fig. 4C). α-SMA expression is known to be expressed constitutively at very low levels in epithelial cells (22). S100A4, also known as fibroblast-specific protein-1 (11 kDa), belongs to the calmodulin-S100-troponin C superfamily of binding proteins that has been widely reported to identify fibroblasts or cells with a fibroblast phenotype (33, 45). To determine whether IL-17 would induce similar changes in other lung-derived epithelial cells, immortalized normal rat alveolar type II cells (RLE-6TN) were cultured in the presence of IL-17. IL-17 induced increase in S100A4 expression in RLE-6TNs within 24 h (Fig. 4E). IL-17 induced dose-dependent downregulation of E-CAD and upregulation of α-SMA by 72 h in these same cells (Fig. 4D). Together, these results demonstrate that IL-17-mediated EMT may be a conserved response in rat and human lung epithelial cells.
Fig. 4.
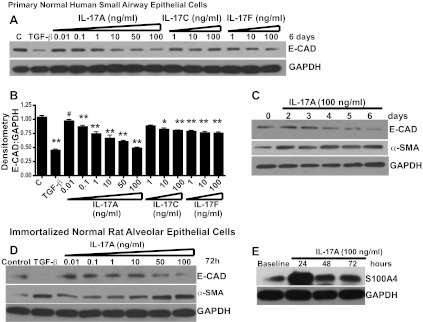
IL-17A induces epithelial mesenchymal transition (EMT) in airway epithelial cells. A: E-cadherin (E-CAD) protein expression as determined by Western blotting. B: densitometry normalized to GAPDH in a dose-response treatment of normal human small airway epithelial cells (SAECs) with IL-17A, IL-17C, and IL-17F; values represent means ± SE; compared with control; #P < 0.05; *P < 0.01; **P < 0.001; n = 3. C: Western blotting analysis of E-CAD and α-smooth muscle actin (α-SMA) protein expression temporally regulated by IL-17A (100 ng/ml) in normal human SAECs. D: IL-17 induced differential expression of E-CAD, α-SMA. E: S100A4 in rat lung epithelial cells as detected by protein expressions in RLE-6TN.
IL-17-mediated EMT is observed in OB lesions after human lung transplant and in lungs from a murine OB model after lung transplant.
Although reports from other investigators have demonstrated EMT in epithelial cells isolated from airway brushings in patients after lung transplant (9, 10), EMT has not been demonstrated in OB lesions. Downregulation of the epithelial marker, ZO-1, and upregulation of α-SMA in epithelial cells are some of the typical phenotypic characteristics of EMT. We observed robust expression of α-SMA, with diminished expression of ZO-1, in the same lesions (Fig. 5A). To further explore the process of IL-17-mediated EMT in vivo, immunohistochemistry was utilized to detect S100A4 expression in the fibro-proliferative lesions. Notably, S100A4-positive epithelial cells were detected in OB lesions (Fig. 5B), in contrast to native lungs, which had very low or no S100A4 expression. Dual labeling of lung tissue sections with antibodies to α-SMA and ZO-1 revealed an occasional colocalization of these markers in airway epithelial cells associated with OB lesions (Fig. 5C). In contrast, the native lung had normal lung architecture that presented with a relatively thin band of connective tissue that stained positive for α-SMA detected below the airway epithelium and no detectable α-SMA and ZO-1 colocalization in the same cell.
Fig. 5.

IL-17A induces EMT in airway epithelial cells located in clinical OB lesions and in a murine model of orthotopic lung transplant. A: OB lesions were immunostained for epithelial marker, zona occludens [ZO-1-rhodamine (red)] and mesenchymal marker, α-SMA-FITC (green). Inset: colocalization of the proteins (yellow arrow) in the fibrotic plugs. Nuclei were counterstained with DAPI. Representative lesions are presented subsequent to examining lung biopsies from 3 different patients. B: immunostaining of S100A4 expression in transplant and native lung. Yellow arrows indicate S100A4 detection in the lesions, and red arrows indicate S100A4 expression in the airway epithelium around the fibrotic plugs (inset). C: colocalization of ZO-1 and α-SMA using fluorescent labeling in the transplant and native lung. Inset: colocalization of EMT markers (yellow arrow). Nuclei were counterstained using DAPI. L, lumen.
IL-17 induces TGF-β and associated signaling pathways.
Among its many roles, TGF-β contributes to immunity and modulates inflammation, and the data, above, demonstrated that IL-17 mediated EMT is comparable to TGF-β. Thus it is possible that both IL-17 and TGF-β contribute to EMT induction and col(V) upregulation. To investigate possible interactions between IL-17 and TGF-β in EMT induction, we next examined the possibility of effects of IL-17 on TGF-β expression. We observed approximately fivefold upregulation of TGF-β transcripts at 6-h incubation of RLE-6TN cells in the presence of IL-17 (Fig. 6A), with simultaneous significant downregulation of transcript levels of SMAD7, an inhibitor of TGF-β signaling. TGF-β signals via two heterodimeric transmembrane receptors, the type II and type I receptors. Activation of the type I receptor tyrosine kinase (ALK5, activin receptor-like kinase-5) in turn activates SMAD2/3, receptor-regulated SMADs of the TGF-β signaling pathway. Thus it was of interest that we observed that even low doses of IL-17 (1 or 10 ng/ml) caused SMAD3 activation (Fig. 6, B and C).
Fig. 6.
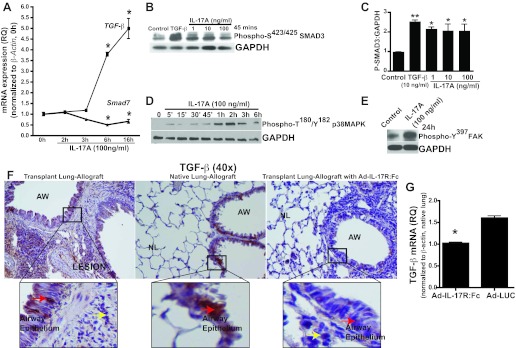
Neutralization of IL-17 bioactivity suppresses IL-17A-induced TGF-β in murine OB model of orthotopic lung transplant. A: qPCR analyses of TGF-β and SMAD7 mRNA expression in response to IL-17 in RLE-6TN cells. Values represent means ± SE (n = 3; *P < 0.001 compared with baseline). B: Western blotting analysis of activated SMAD3 protein (P-SMAD3) expression in epithelial cells treated with several doses of IL-17A. C: densitometry of P-SMAD3 normalized to GAPDH. Values represent means ± SE (n = 3; **P < 0.01; *P < 0.05, compared with control; 1-way ANOVA; post hoc: Dunnett's test). D: temporal upregulation of activated p38 MAPK. E: focal adhesion kinase (FAK) expression with IL-17A treatment as determined by Western blotting in RLE-6TNs at indicated time points. F: immunostaining of TGF-β expression in the epithelial cells of allograft transplant and native lungs of mice 21 days after orthotopic lung transplant. Lower TGF-β expression in transplant lungs of mice pretreated with soluble murine IL-17 receptor fusion protein (Ad:IL-17R:Fc) 72 h before transplantation. G: mRNA expression of TGF-β in whole lung homogenates was determined by real-time PCR. Values represent means ± SE and normalized to β-actin and right lung (native lung); Ad:IL-17R:Fc group: n = 4 and Ad:LUC: n = 3. (*P <0.0001 compared with control).
We next examined the roles of protein kinases reported to be associated with TGF-β-mediated EMT. Therapeutically altering TGF-β activity via specific kinase (p38 MAPK, FAK) inhibitors to ameliorate EMT and fibrotic lung disease (7, 16, 17, 47) is a topic of intense research and multiple clinical trials. p38 MAPK is required for TGF-β-driven EMT (5, 7, 47), whereas IL-17-mediated p38 MAPK activation has been reported in human bronchial epithelial cells (30), and FAK has been reported in TGF-β-mediated EMT (16, 17). We found IL-17 to mediate robust early phosphorylation of p38 MAPK at Thr180/Tyr182, which peaked at 2 h in RLE-6TN cells (Fig. 6D), whereas FAK phosphorylation at Tyr197 occurred at 24 h (Fig. 6E).
We next hypothesized that systemic blockade of IL-17 will block downstream signaling events leading to EMT in lung transplant-associated OB. As previously reported (19), overexpression of murine soluble IL-17 receptor fusion protein (Ad-IL17R:Fc) 72 h before orthotopic lung transplantation protects mice from developing OB. Thus we hypothesized that systemic blockade of IL-17 might block downstream signaling events leading to EMT in this system. To determine whether expression of IL-17 and TGF-β are linked in vivo, we next investigated the expression levels of TGF-β in response to the neutralization of circulating IL-17, in the mouse model of orthotopic lung transplantation-associated OB. Immunohistochemical staining showed that overexpression of soluble murine IL-17 receptor fusion protein (Ad-IL17R:Fc) 72 h before orthotopic lung transplantation resulted in decreased TGF-β expression levels in airway epithelial cells of the transplanted lung, compared with high TGF-β expression in both transplant and native lungs of the allograft controls (Fig. 6F). Quantitative real-time PCR analyses of TGF-β expression in lung homogenates (Fig. 6G) revealed significant downregulation in mice overexpressing Ad-IL17R:Fc, compared with higher levels in mice expressing the firefly luciferase gene (Ad-LUC, control). These data collectively suggest that IL-17 induces TGF-β expression in epithelial cells both in vitro and in vivo and that neutralization of IL-17 bioactivity in the murine OB model suppresses TGF-β expression. Collectively, these observations suggest that IL-17-mediated EMT may precede fibrosis in the murine orthotopic lung transplant model of OB.
IL-17-mediated EMT, associated mesenchymal functions, and col(V) expression are blocked by p38 MAPK/FAK signaling pathways.
To determine the upstream signaling involved in IL-17-mediated EMT, we employed specific pharmacological inhibitors against ALK5, p38 MAPK, and FAK/Src. Interestingly, pharmacological inhibitor of ALK5 blocked both IL-17 and TGF-β-mediated induction of α-SMA (Fig. 7A), suggesting that, similar to TGF-β signaling, IL-17 either signals through ALK5 or induces TGF-β, which, in turn, signals via ALK5. Interestingly, combined treatment with IL-17 and TGF-β appeared to have a synergistic effect on EMT, as there was a slight increase in the induction in α-SMA expression, compared with levels achieved using either factor alone (Fig. 7A). Treatment with inhibitors against p38 MAPK (SB202190- 5 μM) and FAK/Src (PP2, 5 μM) resulted in dose-dependent suppression of IL-17-mediated EMT, as reflected in levels of the mesenchymal marker, α-SMA (Fig. 7B).
Fig. 7.
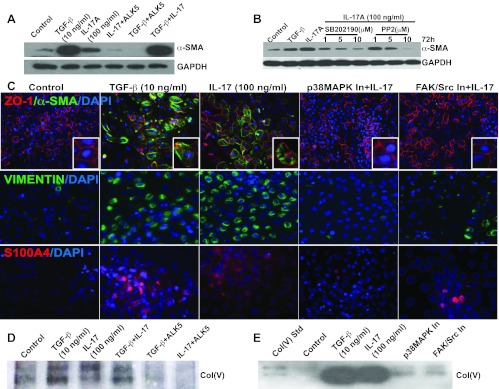
Blockade of IL-17-mediated TGF-β associated pathways prevents EMT and col(V) expression in vitro. A: Western blotting analysis of α-SMA protein expression in RLE-6TN cells pretreated with pharmacological inhibitor against ALK5 In (SB431542; 2.5 μM; ALK5 In) for 1 h and then treated with IL-17 (100 ng/ml) or TGF-β (10 ng/ml) or both IL-17 and TGF-β for 72 h. B: α-SMA protein expression was determined by Western blotting in RLE-6TNs pretreated with pharmacological inhibitors against p38 MAPK (SB202190; 5 μM) and FAK (PP2; 5 μM) at the indicated doses for 1 h and then stimulated with IL-17A (100 ng/ml) for 72 h. TGF-β was used as the positive control. C: RLE-6TNs were treated as described in B and dual-labeled for E-CAD (red) and α-SMA (green) at 72 h. Using immunofluorescent labeling, Vimentin (green; at 48 h) and S100A4 (red; at 24 h) were detected. Nuclei were counterstained with DAPI. Images were captured at ×20 magnification. To further investigate col(V)-related signaling, RLE-6TNs were treated as described in A (Fig. 7D) and B (Fig. 7E). Conditioned media were normalized for equal protein concentration by loading 30 μg of the proteins for all samples and then immunoprobed for col(V).
Immunofluorescence images of untreated cells showed intact epithelial membrane labeled with ZO-1 with a cobblestone appearance and lack of expression of α-SMA (Fig. 7C). Treatment with IL-17 or TGF-β for a period of 72 h caused the disruption or near loss of ZO-1, with concomitant organized appearance of stress fibers, detected by staining for α-SMA. Treatment with inhibitors specific to p38 MAPK and FAK/Src significantly suppressed EMT-related changes. Similar effects were observed with the mesenchymal marker, vimentin, detected at 48 h (Fig. 7C) and the early EMT marker, S100A4, detected at 24 h (Fig. 7C).
We also investigated the possibility of synergistic effects due to the combined treatment of IL-17 and TGF-β on col(V) expression. Although the induction of col(V) expression due to either IL-17 or TGF-β was significant and comparable, we observed only a modest increase in col(V) induction upon combined treatment with both IL-17 and TGF-β. Interestingly, blockade with 2 μM of pharmacological inhibitor specific to ALK5 (a TGF-β-type I receptor tyrosine kinase) resulted in suppression of col(V) expression induced by IL-17. This effect was comparable to blockade of TGF-β-mediated col(V) induction (Fig. 7D). It was also of interest that immunoblotting of conditioned media from cells treated with inhibitors in the presence of IL-17 revealed that pretreatment with pharmacological inhibitors specific to p38 MAPK and FAK/Src block IL-17-induced upregulation of col(V) levels (Fig. 7E). These data collectively suggest that IL-17 signaling may be via TGF-β/ALK5 and support a role for IL-17-mediated p38 MAPK and FAK activation involved in col(V) overexpression and EMT.
Mesenchymal cells contribute to cardinal functions such as migration/tissue remodeling (44) and peroxide production (24). We also performed the “scratch” or “wound closure” assay, a commonly used mesenchymal function indicator. A uniform cobblestone-like monolayer of epithelial cells was “wounded” using a pipette tip (Fig. 8A). The cells were rinsed with PBS and then treated with TGF-β or IL-17. The cells acquired a spindle-shaped, motile phenotype, characteristic of mesenchymal cells, and expressed α-SMA. The capacity of these epithelial cells to migrate and express α-SMA was blocked using pharmacological inhibitors for p38 MAPK, FAK, and ALK5. Assessment of the percent area of wound closure (Fig. 8B) confirmed that treatment with IL-17 or TGF-β resulted in almost complete wound closure. Pretreatment with pharmacological inhibitors against p38 MAPK, FAK, and ALK5 abrogated the capacity of the epithelial cells to acquire a spindle-shaped, motile mesenchymal phenotype and cause wound closure. TGF-β-induced EMT is associated with production of H2O2 in the newly formed mesenchymal cells (24). Because our data showed that IL-17-mediated EMT is dependent on TGF-β-associated signaling, we next determined whether IL-17 would induce H2O2 production comparable to that induced by TGF-β. IL-17 was found to induce H2O2 comparable to TGF-β, and this process was dependent on p38 MAPK and FAK activation (Fig. 8C). Collectively, these data suggest that IL-17-mediated EMT and associated mesenchymal functions are dependent on TGF-β-associated pathways, including p38 MAPK and FAK activation.
Fig. 8.
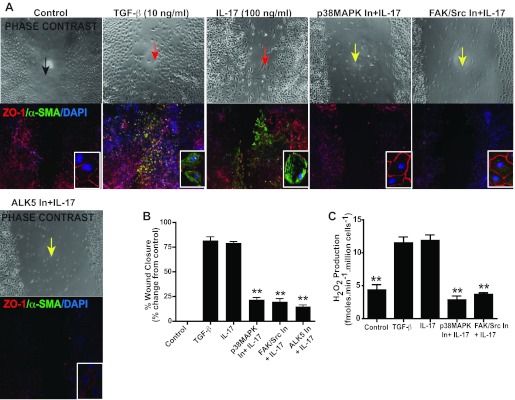
IL-17-mediated EMT is dependent on p38 MAPK and FAK. RLE-6TNs were pretreated with pharmacological inhibitors against p38 MAPK (SB202190; 5 μM) and FAK (PP2; 5 μM) and ALK5 (SB431542; 2.5 μM; ALK5 In) for 1 h and then stimulated with IL-17A (100 ng/ml) for 72 h. TGF-β was used as the positive control. A: “scratch” or wound closure assay: RLE-6TNs were treated as described in C and then given a baseline scratch. At 72 h, cells were formalin fixed, imaged, and immunostained by fluorescent labeling for E-CAD and α-SMA expression. Nuclei were counterstained with DAPI. Images were captured at ×10 magnification (inset: images show representative cells within the wound area). B: surface area of wound closure was measured using NIH ImageJ. Values represent means ± SE, n = 3 per group. (**P <0.001 compared with TGF-β and IL-17). C: extracellular H2O2 release from RLE-6TNs. Cells were treated as described in A for 5 days. Values represent means ± SE, n = 3 per group. *P <0.001 compared with control.
IL-17 is expressed in clinical OB lesions and in murine OB model after lung transplant.
We finally sought to confirm whether IL-17 is expressed in proximity to OB lesions derived from clinical lung transplant specimens. Notably, IL-17 was detected adjacent to OB lesions as shown in Fig. 9A (bottom), whereas IL-17 was not detected in normal lung tissues (Fig. 9A, top). We further confirmed significantly higher levels of IL-17A mRNA in OB lung biopsies from transplant patients, compared with normal lung tissues (Fig. 9B). To determine whether our observations in human lung tissues would be consistent with results in our orthotopic murine allograft model, we analyzed protein and mRNA expression of IL-17A in normal and OB tissue from this model. We detected IL-17 expression patterns in transplanted lung by immunostaining, and these were comparable to the pattern in human OB lung (Fig. 9C). We did not detect significant IL-17A expression in the right, normal lung. We then quantitatively analyzed the mRNA expression of IL-17A over a period of 7, 14, and 21 days in the transplanted murine lung (Fig. 9D). The above data collectively suggest that IL-17-expressing cells are present proximal to human OB lesions, and this observation is comparable to the murine orthotopic lung allograft model.
Fig. 9.
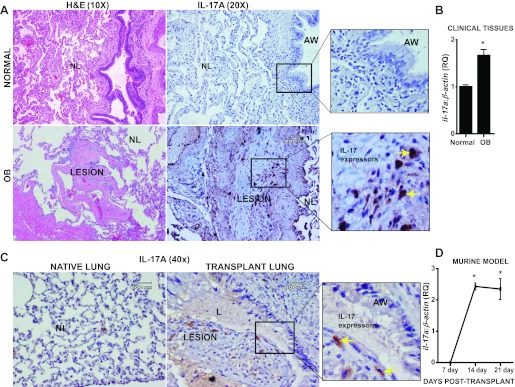
IL-17A is expressed in clinical OB lesions after lung transplant. A: immunohistochemical staining showing expression of IL-17A in a representative human subject with OB and normal lungs. Hematoxylin and eosin staining reveals the respective lung architecture. Inset in OB: IL-17 expressing cells in the fibrotic plugs. Inset in normal lung: epithelial and subepithelial tissues but no staining for IL-17. Representative lesions are presented subsequent to examining lung biopsies from 3 different patients. B: mRNA expression of IL-17A normalized to β-actin in human lung tissues. Values represent mean ± SE; *P < 0.05; normal = 4, OB = 3. C: immunohistochemical staining showing expression of IL-17A in a representative mouse orthotopic allograft model in both native (right) and allograft (left) lungs. Transplant lung shows IL-17 expressing cells in the fibrotic plugs (yellow arrows). Native lung shows epithelial and subepithelial tissues but no staining for IL-17. Representative lesions are presented subsequent to examining lung biopsies from 3 different mice. D: mRNA expression of IL-17A normalized to β-actin in mouse orthotopic lung allograft model at days 7, 14, and 21 days after lung transplant. Values represent means ± SE; *P < 0.05; n = 4.
DISCUSSION
This study shows that IL-17A, a proinflammatory cytokine (but not IL-17C or IL-17F), is a mediator of the novel biomarker col(V), a minor collagen that can be found in the apical region of epithelial cells under normal conditions. Epithelial cells deposit large amounts of col(V) into the extracellular space in response to IL-17 induction. We report evidence of IL-17-expressing cells and col(V) in lungs of patients with OB. Specifically, IL-17 appears to preferentially induce the α1 chain of col(V). We describe humoral immune response against this specific chain of col(V) and on the mechanism underlying IL-17-mediated col(V) overexpression.
IL-17 mediates TGF-β expression, the effects of which are seemingly complemented by downregulation of the inhibitory protein SMAD7 and activation of the receptor-regulated SMAD3. IL-17-mediated EMT is dependent on the TGF-β signaling pathway and is demonstrated here to also be influenced by p38 MAPK and FAK. Systemic neutralization of IL-17 in our OB mouse model resulted in decreased TGF-β expression and protection against EMT.
Col(V), which can be considered a cryptic antigen, is normally intercalated within fibrils of col(I) (34). As such, col(V) is expressed at low levels in perivascular interstitial spaces and on the apical, but not basolateral, surfaces of airway epithelial cells (26). We have previously reported de novo generation of antibodies to col(V) in patients with lung transplants with OB (49) and that this was IL-17 dependent (14). Notably, we also observed humoral immune responses, as we detected significant expression of antibodies against α1(V) chain in patients with lung transplants. Interestingly, we observed positive reactivity of α1(V) to be higher in patients with lung transplant compared with normal healthy volunteers. Tiriveedhi and colleagues (46) have shown that circulating antibodies to α1(V) developed in BOS+ patients with lung transplant and that this fraction may be the immunodominant portion in the pathogenesis of BOS (46). We have reported α1(V) antibodies detected locally through lavage fluid. Although col(V) is constitutively found on the apical surface of airway epithelium, it is not secreted by such cells into the extracellular milieu (26). We observed robust expression of α1(V) in response to IL-17 and TGF-β treatment, which is the same α-chain recognized as the antigen. Employing the hydroxy proline assay, we observed significantly higher total collagen synthesis upon IL-17 and TGF-β treatment in primary rat lung epithelial cells (L2), and this effect was blocked by inhibitors specific to p38 MAPK and FAK (data not shown). The deposition of substation col(V) into extracellular matrix by the epithelium, in response to IL-17A or TGF-β, suggests col(V), seemingly as a potential novel biomarker in the pathogenesis of OB after lung transplantation.
In clinical transplant allografts, Lama and colleagues (32) have demonstrated evidence of donor-derived lung-resident mesenchymal stem cells and that these cells establish interactions with both airway and alveolar epithelium and secrete growth factors necessary for proliferation and differentiation (4). IL-17A has recently been highlighted as a profibrotic cytokine in murine models of idiopathic pulmonary fibrosis (51) and has been reported to have profibrotic effects in renal epithelial cells (18) and human mesenchymal stem cells (25). Among the six isoforms of the IL-17 family, IL-17A and IL-17F are produced by T helper-17 lymphocytes (TH-17 cells), which have been implicated in autoimmunity (6, 8, 29) and fibrogenesis (21, 51). Our present studies indicate that IL-17A is a potent inducer of EMT; in contrast, IL-17C and IL-17F fail to induce EMT in these cells. Here we observed that IL-17-mediated EMT is comparable in both normal primary human small airway epithelial and, using a well-documented EMT cell line, RLE-6TNs (20, 39, 50, 54). These data provide more evidence demonstrating IL-17-mediated EMT similar to its effect in tumor-derived epithelial cells (36). Because IL-17 induces the production of IL-6, we also investigated whether IL-6 would mediate EMT similar to IL-17 and TGF-β. However, IL-6 did not have any effect on alveolar type II epithelial cells (data not shown).
Whereas TGF-β is necessary for IL-17 synthesis in TH-17 cells (6), the present study provides evidence for the role for TGF-β and associated pathways in the murine model of OB. In a prior report (36), in silica-induced inflammation and pulmonary fibrosis, IL-17 induces TGF-β expression in airway epithelial cells. Thus there appears to be mutually reinforcing cross-talk between two potent proinflammatory cytokines with key roles in fueling the processes of EMT and lung fibrosis. We present observations supporting the conclusion that IL-17-mediated col(V) expression and EMT are dependent on TGF-β and that this may be an autocrine effect. We also present data indicating that combined treatment with TGF-β and IL-17 causes only a minor synergistic effect and does not significantly augment levels of EMT and col(V) expression achieved by either factor alone. The exact mechanism by which IL-17 causes induction of TGF-β is not yet elucidated. However, it is interesting to speculate that activation of SMAD, p38 MAPK, or FAK might be directly in response to IL-17 via its receptor. Therefore, developing pharmacological targets to block IL-17 and the pathways responsible for TGF-β induction may have clinical utility. Our data support the hypothesis that IL-17-mediated col(V) expression and EMT may occur via the induction of TGF-β and associated signaling pathways, such as p38 MAPK and FAK, consistent with overlaps in effects on signaling pathways by the two cytokines.
Our studies provide evidence of IL-17 expressing cells in human OB tissues. This was also quantitatively confirmed using mRNA analyses. Notably, results in our orthotopic murine allograft model were consistent with results from human OB lesions. However, we were unable to characterize the cell types involved. Our data do not conclusively suggest that the cellular source of IL-17 in these lesions are not CD3- or CD4-positive T lymphocytes (data not shown). There is a possibility that these cells may also be monocytes or macrophages. Further systematic studies are required to characterize the specific cells or cell types that synthesize and secrete IL-17.
Limitations of our study include the lack of reliably specific antibodies against the individual chains of col(V). Additionally, use of transgenic mice with epithelium-specific reporter expression and conditional overexpression of col(V) would aid in demonstrating the effects of overexpression of col(V) and EMT in our murine OB model. Our future experiments will need to be focused in these directions and should further unravel the mechanisms behind col(V) dysregulation and downstream signaling, including establishing the role of col(V) and the α1(V) chain as novel biomarkers after lung transplant.
In summary, the present data support our hypothesis that IL-17 influences adaptive and innate immunity that promotes fibrosis. Our observations suggest that TGF-β and IL-17 are involved in a feed-forward loop that drives the pathogenesis of fibrotic lung diseases by linking autoimmune responses and EMT with fibrosis (Fig. 10). In this loop, col(V) acts as an autoantigen and seemingly as a potential novel biomarker. IL-17 likely drives EMT, leading to fibrotic pathogenesis by pathways listed as follows: 1) eliciting anti-col(V) immunity, 2) upregulating col(V) expression, 3) inducing EMT, 4) inducing TGF-β expression, and 5) signaling downstream activation of p38 MAPK and FAK in the epithelial cells. TGF-β, in turn, exacerbates the condition by possibly creating an environment that supports TH-17 development. How Th17 cells are released from the normal constraints of Treg cells that normally limit tissue-specific autoimmunity in vivo is still an open question. Targeting protein kinases such as p38 MAPK and FAK seems to represent an important strategy for emerging classes of therapeutic drugs for controlling inflammatory responses and blunting profibrotic signals. In addition, it is interesting to speculate that the mechanisms described here, in relation to fibrotic lung disease, may be extrapolated to other IL-17-dependent autoimmune diseases and cancer.
Fig. 10.

Schematic representation of events leading to IL-17-mediated EMT after lung transplantation. In a post-lung-transplant environment, IL-17 is produced leading to improper resolution of inflammation and tissue damage. A normal airway epithelium has apical-basal polarity with expressions of ZO-1 and E-CAD at the adherent junctions, coupled with presence of col(V) bound to the apical surface of the membrane. The effects of IL-17 on the epithelium result in 1) induction of EMT by the loss of epithelial proteins, ZO-1, and E-CAD at the junctions, coupled with acquisition of early EMT marker, S100A4, and organization of stress fibers at the cytoskeleton (α-SMA), to form a spindle-shaped mesenchymal cell; 2) secretion of col(V) fragments into the matrix, thus juxtaposing col(V) as a seemingly potential novel EMT marker; 3) induction of TGF-β, a known inducer of IL-17 and EMT. The effects of TGF-β are as follows: 1) key player in the maturation of TH-17 cells to generate more IL-17; 2) induction of EMT; and 3) upregulates col(V) secretion. Col(V), a potential novel EMT marker and a key contributor of OB, is upregulated by both IL-17 and TGF-β. The secreted fragments in the extracellular milieu causes autoimmune responses resulting in more IL-17 synthesis likely via col(V)-specific TH-17 effector cells. In a transplanted lung, IL-17 induces EMT via TGF-β, with col(V) contributing toward this feed-forward loop, resulting in progression of obliterative bronchiolitis.
GRANTS
This study was supported by the Biomedical Research Grant and KL2 RR025760-04, Indiana University School of Medicine, the National Institute of Health-NHLBI HL109288 (R. Vittal) and by the National Institutes of Health-NHLBI HL067177 (D. Wilkes); NIH-NIAMS AR047746 (D. Greenspan), NIH-NIAID 1P01AI084853 (D. Wilkes, W. Burlingham, and D. Greenspan).
DISCLOSURES
D. Wilkes is a cofounder of ImmuneWorks, a biotechnology company involved in developing therapeutics for various forms of lung diseases, and D. Greenspan is a scientific advisor.
AUTHOR CONTRIBUTIONS
Author contributions: R.V. and D.S.W. conception and design of research; R.V., L.F., E.A.M., B.G., H.G., H.L.B., and C.Z. performed experiments; R.V. analyzed data; R.V., D.S.G., O.W.C., and D.S.W. interpreted results of experiments; R.V. prepared figures; R.V. drafted manuscript; R.V., D.S.G., W.B., and D.S.W. edited and revised manuscript; R.V. and D.S.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thanks Lana Christian of Createwrite for writing assistance.
REFERENCES
- 1. Adams BF, Brazelton T, Berry GJ, Morris RE. The role of respiratory epithelium in a rat model of obliterative airway disease. Transplantation 69: 661–664, 2000 [DOI] [PubMed] [Google Scholar]
- 2. Alho HS, Inkinen KA, Salminen US, Maasilta PK, Taskinen EI, Glumoff V, Vuorio EI, Ikonen TS, Harjula AL. Collagens I and III in a porcine bronchial model of obliterative bronchiolitis. Am J Respir Crit Care Med 164: 1519–1525, 2001 [DOI] [PubMed] [Google Scholar]
- 3. Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P, Naves R, Han M, Zhong F, Castellanos JG, Mair R, Christakos A, Kolkowitz I, Katz L, Killestein J, Polman CH, de Waal Malefyt R, Steinman L, Raman C. T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med 16: 406–412, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Badri L, Walker NM, Ohtsuka T, Wang Z, Delmar M, Flint A, Peters-Golden M, Toews GB, Pinsky DJ, Krebsbach PH, Lama VN. Epithelial interactions and local engraftment of lung-resident mesenchymal stem cells. Am J Respir Cell Mol Biol 45: 809–816, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 115: 3193–3206, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol 8: 345–350, 2007 [DOI] [PubMed] [Google Scholar]
- 7. Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL. Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38 MAPK and epithelial plasticity. J Biol Chem 276: 46707–46713, 2001 [DOI] [PubMed] [Google Scholar]
- 8. Bobadilla JL, Love RB, Jankowska-Gan E, Xu Q, Haynes LD, Braun RK, Hayney MS, Munoz del Rio A, Meyer K, Greenspan DS, Torrealba J, Heidler KM, Cummings OW, Iwata T, Brand D, Presson R, Burlingham WJ, Wilkes DS. Th-17, monokines, collagen type V, and primary graft dysfunction in lung transplantation. Am J Respir Crit Care Med 177: 660–668, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borthwick LA, McIlroy EI, Gorowiec MR, Brodlie M, Johnson GE, Ward C, Lordan JL, Corris PA, Kirby JA, Fisher AJ. Inflammation and epithelial to mesenchymal transition in lung transplant recipients: role in dysregulated epithelial wound repair. Am J Transplant 10: 498–509, 2010 [DOI] [PubMed] [Google Scholar]
- 10. Borthwick LA, Parker SM, Brougham KA, Johnson GE, Gorowiec MR, Ward C, Lordan J, Corris P, Kirby JA, Fisher AJ. Epithelial to mesenchymal transition (EMT) and airway remodelling after human lung transplantation. Thorax 64: 770–777, 2009 [DOI] [PubMed] [Google Scholar]
- 11. Borthwick LA, Sunny SS, Oliphant V, Perry J, Brodlie M, Johnson GE, Ward C, Gould K, Corris PA, De Soyza A, Fisher AJ. Pseudomonas aeruginosa accentuates epithelial to mesenchymal transition in the airway. Eur Respir J 37: 1237–1247, 2010 [DOI] [PubMed] [Google Scholar]
- 12. Braun RK, Martin A, Shah S, Iwashima M, Medina M, Byrne K, Sethupathi P, Wigfield CH, Brand DD, Love RB. Inhibition of bleomycin-induced pulmonary fibrosis through pre-treatment with collagen type V. J Heart Lung Transplant 29: 873–879, 2010 [DOI] [PubMed] [Google Scholar]
- 13. Braun RK, Molitor-Dart M, Wigfield C, Xiang Z, Fain SB, Jankowska-Gan E, Seroogy CM, Burlingham WJ, Wilkes DS, Brand DD, Torrealba J, Love RB. Transfer of tolerance to collagen type V suppresses T-helper-cell-17 lymphocyte-mediated acute lung transplant rejection. Transplantation 88: 1341–1348, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burlingham WJ, Love RB, Jankowska-Gan E, Haynes LD, Xu Q, Bobadilla JL, Meyer KC, Hayney MS, Braun RK, Greenspan DS, Gopalakrishnan B, Cai J, Brand DD, Yoshida S, Cummings OW, Wilkes DS. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest 117: 3498–3506, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH. Integrin alpha6beta4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest 121: 2855–2862, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cicchini C, Laudadio I, Citarella F, Corazzari M, Steindler C, Conigliaro A, Fantoni A, Amicone L, Tripodi M. TGFbeta-induced EMT requires focal adhesion kinase (FAK) signaling. Exp Cell Res 314: 143–152, 2008 [DOI] [PubMed] [Google Scholar]
- 17. Deng B, Yang X, Liu J, He F, Zhu Z, Zhang C. Focal adhesion kinase mediates TGF-beta1-induced renal tubular epithelial-to-mesenchymal transition in vitro. Mol Cell Biochem 340: 21–29, 2010 [DOI] [PubMed] [Google Scholar]
- 18. Dudas PL, Sague SL, Elloso MM, Farrell FX. Proinflammatory/Profibrotic Effects of Interleukin-17A on Human Proximal Tubule Epithelium. Nephron Exp Nephrol 117: e114–e123, 2010 [DOI] [PubMed] [Google Scholar]
- 19. Fan L, Benson HL, Vittal R, Mickler EA, Presson R, Fisher AJ, Cummings OW, Heidler KM, Keller MR, Burlingham WJ, Wilkes DS. Neutralizing IL-17 prevents obliterative bronchiolitis in murine orthotopic lung transplantation. Am J Transplant 11: 911–922, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Felton VM, Borok Z, Willis BC. N-acetylcysteine inhibits alveolar epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 297: L805–L812, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feng W, Li W, Liu W, Wang F, Li Y, Yan W. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp Mol Pathol 87: 212–218, 2009 [DOI] [PubMed] [Google Scholar]
- 22. Garcia CM, Kwon GP, Beebe DC. alpha-Smooth muscle actin is constitutively expressed in the lens epithelial cells of several species. Exp Eye Res 83: 999–1001, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gopalakrishnan B, Wang WM, Greenspan DS. Biosynthetic processing of the Pro-alpha1(V)Pro-alpha2(V)Pro-alpha3(V) procollagen heterotrimer. J Biol Chem 279: 30904–30912, 2004 [DOI] [PubMed] [Google Scholar]
- 24. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, Pennathur S, Martinez FJ, Thannickal VJ. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med 15: 1077–1081, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, Lee Y, Kim HH. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ 16: 1332–1343, 2009 [DOI] [PubMed] [Google Scholar]
- 26. Iwata T, Philipovskiy A, Fisher AJ, Presson RG, Jr, Chiyo M, Lee J, Mickler E, Smith GN, Petrache I, Brand DB, Burlingham WJ, Gopalakrishnan B, Greenspan DS, Christie JD, Wilkes DS. Anti-type V collagen humoral immunity in lung transplant primary graft dysfunction. J Immunol 181: 5738–5747, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420–1428, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kawaguchi M, Fujita J, Kokubu F, Huang SK, Homma T, Matsukura S, Adachi M, Hizawa N. IL-17F-induced IL-11 release in bronchial epithelial cells via MSK1-CREB pathway. Am J Physiol Lung Cell Mol Physiol 296: L804–L810, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177: 566–573, 2006 [DOI] [PubMed] [Google Scholar]
- 30. Laan M, Lotvall J, Chung KF, Linden A. IL-17-induced cytokine release in human bronchial epithelial cells in vitro: role of mitogen-activated protein (MAP) kinases. Br J Pharmacol 133: 200–206, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LaGamba D, Nawshad A, Hay ED. Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn 234: 132–142, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Lama VN, Smith L, Badri L, Flint A, Andrei AC, Murray S, Wang Z, Liao H, Toews GB, Krebsbach PH, Peters-Golden M, Pinsky DJ, Martinez FJ, Thannickal VJ. Evidence for tissue-resident mesenchymal stem cells in human adult lung from studies of transplanted allografts. J Clin Invest 117: 989–996, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lawson WE, Polosukhin VV, Zoia O, Stathopoulos GT, Han W, Plieth D, Loyd JE, Neilson EG, Blackwell TS. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am J Crit Care Med 171: 899–907, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Linsenmayer TF, Gibney E, Igoe F, Gordon MK, Fitch JM, Fessler LI, Birk DE. Type V collagen: molecular structure and fibrillar organization of the chicken alpha 1(V) NH2-terminal domain, a putative regulator of corneal fibrillogenesis. J Cell Biol 121: 1181–1189, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441: 231–234, 2006 [DOI] [PubMed] [Google Scholar]
- 36. Mi S, Li Z, Yang HZ, Liu H, Wang JP, Ma YG, Wang XX, Liu HZ, Sun W, Hu ZW. Blocking IL-17A promotes the resolution of pulmonary inflammation and fibrosis via TGF-beta1-dependent and -independent mechanisms. J Immunol 187: 3003–3014, 2011 [DOI] [PubMed] [Google Scholar]
- 37. Muramatsu R, Kubo T, Mori M, Nakamura Y, Fujita Y, Akutsu T, Okuno T, Taniguchi J, Kumanogoh A, Yoshida M, Mochizuki H, Kuwabara S, Yamashita T. RGMa modulates T cell responses and is involved in autoimmune encephalomyelitis. Nat Med 17: 488–494, 2011 [DOI] [PubMed] [Google Scholar]
- 38. Nakano K, Yamaoka K, Hanami K, Saito K, Sasaguri Y, Yanagihara N, Tanaka S, Katsuki I, Matsushita S, Tanaka Y. Dopamine induces IL-6-dependent IL-17 production via D1-like receptor on CD4 naive T cells and D1-like receptor antagonist SCH-23390 inhibits cartilage destruction in a human rheumatoid arthritis/SCID mouse chimera model. J Immunol 186: 3745–3752, 2011 [DOI] [PubMed] [Google Scholar]
- 39. Ramos C, Becerril C, Montano M, Garcia-De-Alba C, Ramirez R, Checa M, Pardo A, Selman M. FGF-1 reverts epithelial-mesenchymal transition induced by TGF-β1 through MAPK/ERK kinase pathway. Am J Physiol Lung Cell Mol Physiol 299: L222–L231, 2010 [DOI] [PubMed] [Google Scholar]
- 40. Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, Steward N, Aloush A, Hachem R, Trulock E, Meyers B, Patterson GA, Mohanakumar T. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant 30: 624–631, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sidhu SS, Yuan S, Innes AL, Kerr S, Woodruff PG, Hou L, Muller SJ, Fahy JV. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc Natl Acad Sci USA 107: 14170–14175, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Studer SM, Levy RD, McNeil K, Orens JB. Lung transplant outcomes: a review of survival, graft function, physiology, health-related quality of life and cost-effectiveness. Eur Respir J 24: 674–685, 2004 [DOI] [PubMed] [Google Scholar]
- 43. Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, Fazli L, Wada R, Huang J, Vessella RL, An J, Horvath S, Gleave M, Rettig MB, Wainberg ZA, Reiter RE. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med 16: 1414–1420, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M, Cao X. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15: 757–765, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, Sherrill TP, Plieth D, Neilson EG, Blackwell TS, Lawson WE. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med 180: 657–665, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tiriveedhi V, Angaswamy N, Brand D, Weber J, Gelman AG, Hachem R, Trulock EP, Meyers B, Patterson G, Mohanakumar T. A shift in the collagen V antigenic epitope leads to T helper phenotype switch and immune response to self-antigen leading to chronic lung allograft rejection. Clin Exp Immunol 167: 158–168, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vidyasagar A, Reese S, Acun Z, Hullett D, Djamali A. HSP27 is involved in the pathogenesis of kidney tubulointerstitial fibrosis. Am J Physiol Renal Physiol 295: F707–F716, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilkes DS. Chronic lung allograft rejection and airway microvasculature: Is HIF-1 the missing link? J Clin Invest 121: 2155–2157, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilkes DS, Heidler KM, Yasufuku K, Devito-Haynes L, Jankowska-Gan E, Meyer KC, Love RB, Burlingham WJ. Cell-mediated immunity to collagen V in lung transplant recipients: correlation with collagen V release into BAL fluid. J Heart Lung Transplant 20: 167, 2001 [DOI] [PubMed] [Google Scholar]
- 50. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. Am J Pathol 166: 1321–1332, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, Wynn TA. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J Exp Med 207: 535–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoshida S, Haque A, Mizobuchi T, Iwata T, Chiyo M, Webb TJ, Baldridge LA, Heidler KM, Cummings OW, Fujisawa T, Blum JS, Brand DD, Wilkes DS. Anti-type V collagen lymphocytes that express IL-17 and IL-23 induce rejection pathology in fresh and well-healed lung transplants. Am J Transplant 6: 724–735, 2006 [DOI] [PubMed] [Google Scholar]
- 53. Zheng L, Ward C, Snell GI, Orsida BE, Li X, Wilson JW, Williams TJ, Walters EH. Scar collagen deposition in the airways of allografts of lung transplant recipients. Am J Respir Crit Care Med 155: 2072–2077, 1997 [DOI] [PubMed] [Google Scholar]
- 54. Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol 297: L1120–L1130, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]