

Abstract
Repair of the lung epithelium after injury is integral to the pathogenesis and outcomes of diverse inflammatory lung diseases. We previously reported that β-catenin signaling promotes epithelial repair after inflammatory injury, but the β-catenin target genes that mediate this effect are unknown. Herein, we examined which β-catenin transcriptional coactivators and target genes promote epithelial repair after inflammatory injury. Transmigration of human neutrophils across cultured monolayers of human lung epithelial cells resulted in a fall in transepithelial resistance and the formation of discrete areas of epithelial denudation (“microinjury”), which repaired via cell spreading by 96 h. In mice treated with intratracheal (i.t.) LPS or keratinocyte chemokine, neutrophil emigration was associated with increased permeability of the lung epithelium, as determined by increased bronchoalveolar lavage (BAL) fluid albumin concentration, which decreased over 3–6 days. Activation of β-catenin/p300-dependent gene expression using the compound ICG-001 accelerated epithelial repair in vitro and in murine models. Neutrophil transmigration induced epithelial expression of the β-catenin/p300 target genes Wnt-induced secreted protein (WISP) 1 and cysteine-rich (Cyr) 61, as determined by real-time PCR (qPCR) and immunostaining. Purified neutrophil elastase induced WISP1 upregulation in lung epithelial cells, as determined by qPCR. WISP1 expression increased in murine lungs after i.t. LPS, as determined by ELISA of the BAL fluid and qPCR of whole lung extracts. Finally, recombinant WISP1 and Cyr61 accelerated repair, and Cyr61-neutralizing antibodies delayed repair of the injured epithelium in vitro. We conclude that β-catenin/p300-dependent expression of WISP1 and Cyr61 is critical for epithelial repair and represents a potential therapeutic target to promote epithelial repair after inflammatory injury.
Keywords: neutrophils, lung injury, lung epithelium, transmigration, CCN matricellular proteins, β-catenin
in many inflammatory lung diseases, neutrophils emigrate from the pulmonary microvasculature and across the epithelium into the airspaces (67). This acute inflammatory response is associated with injury to the lung epithelium characterized by increased permeability (21, 22). The initial period of injury is followed by a repair phase in which an anatomically and functionally intact epithelium is restored through a complex process involving spreading, proliferation, and differentiation of epithelial cells (re-epithelialization) (13). Epithelial repair is an important determinant of the pathogenesis and resolution of diverse inflammatory lung diseases, including acute lung injury (ALI), chronic obstructive pulmonary disease (COPD), and asthma (13, 68).
We have previously reported that β-catenin signaling is activated in the lung epithelium in response to neutrophil transmigration and plays a critical role in epithelial repair after inflammatory injury (72), and other reports corroborate this finding (17, 64, 75). In canonical Wnt/β-catenin signaling, β-catenin translocates to the nucleus and binds to the T cell factor (TCF)/lymphoid enhancer factor (LEF) family of transcription factors, enhancing expression of specific target genes, many of which are involved in cell proliferation; hence, β-catenin signaling has been implicated in development, tumorigenesis, and tissue repair (12, 52). Proteinase-mediated cleavage of E-cadherin may result in release of β-catenin from the adherens junctions, leading to an increase in nuclear β-catenin and transcription of β-catenin-dependent genes (28, 47, 48), and the activation of β-catenin signaling in response to neutrophil migration may depend on neutrophil elastase (72). The particular pattern of gene expression induced by β-catenin depends on the recruitment of specific transcriptional coactivators, including p300 or cAMP response element-binding protein (CBP), to the β-catenin/TCF complex (49, 61). Importantly, inhibition of the β-catenin/CBP interaction promotes the binding of β-catenin to its alternate coactivator, p300, resulting in expression of β-catenin/p300-dependent target genes (49). Our previous studies suggested that inhibition of β-catenin/p300 signaling delayed epithelial repair after inflammatory injury (72).
Wnt-induced secreted protein (WISP) 1 (CCN4) and cysteine-rich 61 (Cyr61) (CCN1) are matricellular proteins of the CCN family that regulate survival, proliferation, and migration of diverse cell types and thus play a role in wound repair, angiogenesis, and tumorigenesis (6, 33, 41). The CCN proteins have been implicated in the pathogenesis of diverse diseases involving inflammation and tissue repair, including atherosclerosis, arthritis, fibrosis, and diabetic nephropathy and retinopathy (reviewed in Ref. 33). In the lung, expression of WISP1 is increased in alveolar type (AT) II cells and induces ATII cell proliferation and epithelial-mesenchymal transition (EMT) in murine models of pulmonary fibrosis (26, 38). Additionally, Cyr61 is upregulated and promotes cell survival and proliferation of the lung epithelium after hyperoxic injury (30, 31). Expression of both WISP1 and Cyr61 is regulated by β-catenin signaling (65).
To identify specific target genes that mediate β-catenin-signaling in epithelial repair after inflammatory injury, we employed ICG-001, a compound that promotes β-catenin/p300-dependent transcriptional events by enhancing the β-catenin/p300 interaction at the expense of the β-catenin/CBP interaction (49). We observed that β-catenin/p300 signaling accelerates repair of the lung epithelium after inflammatory injury. Moreover, we demonstrated that WISP1 and Cyr61 are β-catenin/p300 dependent, are upregulated in response to neutrophil transmigration or neutrophil elastase, and play a role in epithelial repair. β-catenin/p300 signaling and the CCN proteins WISP1 and Cyr61 represent potential therapeutic targets to promote epithelial repair after inflammatory injury.
MATERIALS AND METHODS
Cell culture.
Calu-3 human lung epithelial cells (American Type Culture Collection) or Calu-3 cells transduced with lentiviral vectors expressing green fluorescent protein (GFP) (Calu-3-GFP), shRNA to β-catenin, or a nonsilencing (control) shRNA, generated as previously described (72), were grown in DMEM (Invitrogen) containing 44 mM NaHCO3, 1 mM sodium pyruvate, 4 mM L-alanyl-glutamine, 90 μg/ml streptomycin, 40 μg/ml penicillin, and 10% FBS (Hyclone). Human small airway epithelial cells (SAEC; Lonza) were grown in small airway basal medium supplemented with small airway growth medium SingleQuots (Lonza). For transmigration experiments, 0.75 × 106 Calu-3 cells (passages 9–25) or 0.5 × 106 SAEC cells (passage 3) were seeded on the undersurface of 1.12 cm2 polycarbonate membranes (3-μm pore size) in Transwell inserts (Corning), coated with bovine collagen (Advanced BioMatrix) for SAEC, as previously described (55). Cells were grown to confluence (6–8 days). In selected experiments, 0.25 × 106 Calu-3 cells were seeded into 24-well plates, treated with 1 μg/ml mitomycin for 3 days, trypsinized, and counted.
Neutrophil transmigration.
Human neutrophils were isolated from healthy donors as previously described (25) and in accordance with an approved Institutional Review Board protocol at National Jewish Health. Neutrophils were induced to migrate across the epithelial monolayer as previously described (72). Briefly, neutrophils were suspended (60–120 × 106/ml) in modified Hanks' balanced salt solution (HBSS) without calcium or magnesium (HBSS−−). Epithelial monolayers were washed twice with HBSS with calcium and magnesium (HBSS++), and 300 μl of HBSS++ was added to the upper chamber (corresponding to the basolateral surface of epithelial cells) followed by addition of 1 ml of 1 μM N-Formyl-Met-Leu-Phe (fMLP; Sigma) to the bottom (apical) chamber. Control cells were treated with 1 μM fMLP in the bottom chamber. Baseline transepithelial resistance (TER) was measured with an Evometer (World Precision Instruments). Subsequently, 6–12 × 106 neutrophils or buffer were added to the upper chamber, and transmigration was allowed to proceed in the physiological basolateral-to-apical direction for 90–120 min at 37°C. The monolayers were washed three times in HBSS++ and cultured in media at 37°C, 5% CO2. In selected experiments, 1–10 μM ICG-001 (15) or 0.01–0.001% DMSO, 1 μg/ml recombinant human (rh)WISP (R&D Systems), 3 μg/ml rhCyr61 (produced as described in Refs. 36, 71) or 3 μg/ml BSA, 20 μg/ml Cyr61 or control IgG antibody, or 1 μg/ml cytochalasin or 1 μg/ml mitomycin (as previously described in Ref. 60) was added to the cells immediately after or 24 h after migration. At the indicated time points, TER was measured, and fluorescent images of live cell cultures were obtained by stratified random sampling using a Zeiss Axiovert 200 microscope at ×2.5 magnification. With the use of NIH ImageJ software, the total cross-sectional area of the defects was measured. Experiments in which baseline TER was <1,000 Ω-cm2 or wounds repaired >85% by 48 h were excluded from analysis. At the indicated time points, total RNA was extracted from epithelial cells by TRIzol (Invitrogen) using the QIAcube (QIAGEN).
Preparation of epithelial cell supernatants.
Following transmigration, supernatants on the apical surface of the epithelial monolayer (bottom chamber) from five wells were pooled. Protease inhibitors (1 mM PMSF, 0.5 mM benzamidine, 10 μg/ml aprotinin, and 10 μg/ml leupeptin) were added, and samples were centrifuged at 300 g for 5 min to remove cell debris. Cell-free supernatants were then concentrated using Amicon Ultra-4 centrifugal filters with a 10-kDa size exclusion (Millipore) by centrifugation at 7,197 g for 20 min at 4°C. Concentrated samples were boiled in Laemmli buffer.
Elastase stimulation.
SAEC were grown to 80% confluence and treated with 1 mM EDTA at 37°C for 3 min followed by 0.1 U/ml human leukocyte elastase (Elastin Products) diluted in HBSS++ at 37°C for 1 h. Monolayers were washed free of elastase and incubated in media for 2 h. Total RNA was extracted from epithelial cells by TRIzol (Invitrogen) using the QIAcube (QIAGEN).
Expression microarray analysis.
At 4 h after transmigration, epithelial cells were separated from neutrophils using magnetic-activated cell sorting with EPCAM MicroBeads (Miltenyi), as previously described (72). RNA from four separate experiments was pooled, reverse transcribed into cDNA, labeled, and hybridized on Agilent 44k human whole genome arrays (G4112F) as previously described (GEO GSE31697) (72).
Scratch wound.
Calu-3-GFP cells were grown to confluence, and wounds were created by scraping a cross in the monolayer with a pipette tip. ICG-001 1 μM or 0.001% DMSO was added to the culture media immediately after scratch wounding. At the indicated time points, fluorescent images of the wound were obtained using a Zeiss Axiovert 200 microscope at ×2.5 magnification. With the use of NIH ImageJ software, the area of the wound within a 5-mm2 area centered on the cross was measured.
Immunoblotting.
Calu-3 cells (0.5 × 106) were seeded in 24-well tissue culture plates and treated with 10 μM ICG-001. Twenty-four hours later, the cells were lysed in buffer (25 mM Tris, 10% glycerol, 1% IGEPAL, pH 7.5) supplemented with Complete Mini Protease Inhibitor Cocktail (Roche). Epithelial cell lysates were analyzed by SDS-PAGE and immunoblotting for WISP1 (Abcam 10737), GAPDH (Abcam 8245), or E-cadherin (Chemicon, DECMA-1). Densitometry was performed using NIH ImageJ software.
Immunofluorescence.
At the indicated time points after transmigration, epithelial cell monolayers were fixed in 4% paraformaldehyde in PBS (20 min, 24°C), permeabilized in 0.2% Triton X-100 in PBS (2 min, 24°C) and blocked with 5% goat serum in PBS (30 min, 24°C). Monolayers were incubated in primary antibody to active β-catenin (Millipore; 8E7), WISP1 (Abcam 10737), Cyr61 (Abcam 24448), or p300 (Abcam 54894 or 59240) at a 1:50–1:100 dilution in PBS with 5% goat serum and 0.2% Triton X-100 overnight at 4°C or Alexa 594-phalloidin (Invitrogen) at a 1:40 dilution in PBS with 1% BSA (25 min, 24°C), and then incubated with an Alexa 488- or 555- or 594-conjugated secondary antibody (Invitrogen) at 1:200 for 1 h at 24°C. In the indicated experiments, epithelial monolayers were treated with 10 μM 5-bromo-2-deoxyuridine (BrdU) (Sigma) and fixed in 4% paraformaldehyde with 1.5% sucrose in PBS 2 h later. Monolayers were incubated in 2 M HCl (1 h, 37°C) followed by 0.1 M sodium tetraborate, pH 8.5 (12 m, 24°C). Cells were permeabilized and blocked with 0.5% Triton and 5% donkey serum in PBS (1 h, 24°C), incubated in anti-BrdU antibody (AbD Serotec; BU1/75) at a 1:200 dilution (1 h, room temperature), and then incubated with a Cy3-conjugated anti-rat secondary antibody (Jackson ImmunoResearch Laboratories 712-166-150) at a 1:100 dilution (1 h, room temperature). Filters were mounted on slides using Vecta Shield mounting solution containing DAPI (Vector Laboratories). Images were photographed using a Zeiss Axiovert 200 or Confocal LSM 700 microscope at ×20 magnification. Fluorescence intensity was analyzed by random sampling of cells adjacent to and away from the wounds using NIH ImageJ software. Internuclear distances between adjacent cells at and away from the wound edge were calculated from DAPI images as previously described (5, 18).
Murine models of ALI.
All animal protocols were approved by the Animal Care and Use Committee at National Jewish Health. Mice were maintained in a pathogen-free environment on a 12-h:12-h light/dark cycle with full access to food and water. Female C57BL/6 mice, age 8–12 wk, were treated with 20 μg LPS (Escherichia coli 0111:B4; List Biological Laboratories) in 50 μl saline or 50 μl saline intratracheally (i.t.) and euthanized 2–8 days later. In separate experiments, C57BL/6 mice were treated with 1 μg recombinant murine keratinocyte chemokine (KC) (R&D Systems) in 0.1% human serum albumin (HSA) in 50 μl saline or 0.1% HSA in 50 μl saline by i.t. instillation and euthanized 12–96 h later. In selected experiments, mice were treated with 1.25 mg ICG-001 (manufactured as previously described in Ref. 15) in 28 μl DMSO or 28 μl DMSO subcutaneously 2 h after KC treatment. Bronchoalveolar lavage (BAL) cell counts were performed as previously described (53). Albumin (Bethyl Laboratories) and WISP1 (R&D) ELISAs were performed on BAL fluid. Lungs were frozen in liquid nitrogen, and RNA was isolated using the mirVana miRNA Isolation Kit (Invitrogen).
Real-time PCR.
RNA was reverse transcribed into cDNA using the Quantitect Kit (Qiagen) according to the manufacturer's instructions. cDNA was analyzed by qPCR using primers for hCyr61: 5′-CCC GTT TTG GTA GAT TCT GG-3′ and 5′-GCT GGA ATG CAA CTT CGG-3′; hHHPRT: 5′-TGC TCG AGA TGT GAT GAA GGA G-3′ and 5′-TGA TGT AAT CCA GCA GGT CAG C-3′; and hWISP1: 5′-GTA TGT GAG GAC GAC GCC AAG-3′ and 5′-GGC TAT GCA GTT CCT GTG CC-3′. TaqMan probes (mWISP1: Mm00457574_m1; mCyr61: Mm00487498_m1; m18S: Mm03928990; mGUSB: Mm00446956_m1) were purchased from Assays-on-Demand (Applied Biosystems). qPCR was performed for 40 cycles on the CFX96 (Bio-Rad) using iQ SYBR Green Supermix (Bio-Rad) or Amplitaq Gold (Applied Biosystems). Relative mRNA expression levels were calculated using the 2−ΔΔCt method (43).
Statistical analysis.
Data are expressed as means ± SE. Data were analyzed from n ≥ 4 independent experiments; in vitro experiments were performed in duplicate or triplicate. Statistical analysis was performed by Student's paired or unpaired t-test or Wilcoxon signed-rank test where indicated. Multiple comparisons were performed by one- or two-way ANOVA with Tukey or Bonferroni (post hoc) test for determination of differences between groups. For the neutrophil transmigration wound repair assays, statistical analysis was performed on the area of denudation at 48 h after migration, expressed as a percentage of the area of initial injury at 24 h, with the treatment group expressed as a percentage of the control group for each experiment. P < 0.05 was considered significant. GraphPad PRISM software was used for all statistical calculations.
RESULTS
Inflammatory injury followed by repair of the lung epithelium in vitro and in murine models.
To model the events occurring during an acute inflammatory response in the lung, human neutrophils were induced to transmigrate across monolayers of human lung epithelial (Calu-3) cells in the physiological basolateral-to-apical direction by a gradient of the chemoattractant fMLP. Neutrophil transmigration induced a decrease in TER (Fig. 1A) and the formation of discrete circular areas of epithelial denudation (microinjury) in the epithelial monolayer (Fig. 1, B and C). Over the ensuing 96 h, TER returned to baseline (Fig. 1A), and the defects in the monolayer re-epithelialized (Fig. 1, B and C), signifying repair of the injured epithelium. To determine the contribution of cell spreading and proliferation to re-epithelialization, neutrophils were induced to transmigrate across epithelial monolayers as described above. After 30 h, the epithelial cells were labeled with BrdU for 2 h, fixed, and stained with anti-BrdU antibodies or phalloidin. Epithelial cells at the wound edge did not exhibit evidence of increased proliferation (Fig. 1D) but did demonstrate enhanced F-actin staining in the leading edge of cells migrating into the denuded area (Fig. 1E). During the period of wound repair, internuclear distances between cells contiguous to the wound were greater than between cells distant from the wound (Fig. 1F), indicative of cell spreading. Inhibition of cytoskeletal assembly with cytochalasin D (60) delayed wound closure (Fig. 1G). By contrast, there was no effect on re-epithelialization of the monolayers in response to treatment with mitomycin (Fig. 1H), which effectively inhibited cell proliferation (Fig. 1I). Taken together, these observations suggest that, in this model system, closure of the epithelial wounds occurs primarily by cell spreading.
Fig. 1.
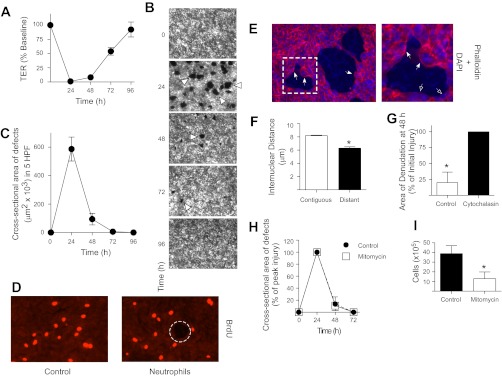
Inflammatory injury followed by repair of the lung epithelium in vitro. A–H: neutrophils were induced to migrate across Calu-3 or Calu-3-green fluorescent protein (GFP) cells for 120 min by a gradient of N-Formyl-Met-Leu-Phe (fMLP) (1 μM). A: transepithelial resistance (TER) was measured. B and C: fluorescent images (×2.5) were acquired at the specified time points, and the total cross-sectional area of epithelial defects in 5 randomly selected high-powered fields (HPF) was measured. Arrowheads indicate epithelial wounds. D: at 30 h after migration, epithelial monolayers were cultured with 10 μM 5-bromo-2-deoxyuridine (BrdU) for 2 h, fixed, and stained with anti-BrdU antibodies. The dashed circle indicates an epithelial wound. E: at 30 h after migration, epithelial monolayers were fixed and stained with phalloidin and DAPI. Closed arrows indicate enhanced F-actin staining in the leading edge of cells migrating into the denuded area; open arrows indicate baseline F-actin staining in cells that do not appear to be migrating into the denuded area. The boxed region indicates the area magnified in the inset (right). F: at 30 h after migration, epithelial monolayers were fixed and stained with DAPI. Fixed monolayers were analyzed for internuclear distances. At 24 h after migration, epithelial monolayers were cultured with 1 μg/ml cytochalasin (G) or 1 μg/ml mitomycin (H). In G, area of denudation at 48 h is expressed as a percentage of the area of initial injury at 24 h, with the control group expressed as a percentage of the treatment group. Fluorescent images (×2.5) were acquired at the specified time points, and the total cross-sectional area of epithelial defects in 5 randomly selected HPF was measured. I: Calu-3 cells were seeded into 24-well plates, treated with 1 μg/ml mitomycin for 3 days, and trypsinized and counted. n ≥ 4. *P ≤ 0.05. Error bars represent SE.
To assess the effects of neutrophil transmigration into the lung on epithelial permeability in intact animals, two murine models of neutrophilic inflammation associated with acute lung injury were used (45). Intratracheal administration of LPS induced robust neutrophil influx into the airspaces. This was associated with increased lung permeability, as measured by increased albumin levels in BAL fluid, which decreased over 6–8 days but did not return to baseline (Fig. 2A). In the second model, i.t. administration of the CXC chemokine KC induced a rapid and vigorous neutrophil influx and increased epithelial permeability as assessed by increased albumin levels in BAL fluid (Fig. 2B). This was followed by a progressive decrease in BAL albumin levels over time to baseline (Fig. 2B), signifying epithelial repair.
Fig. 2.

Inflammatory injury followed by repair of the lung epithelium in vivo. C57BL/6 mice were treated with LPS (A) 20 μg in 50 μl saline or 50 μl saline or keratinocyte chemokine (KC) (B) 1 μg in 50 μl saline + 0.1% human serum albumin (HSA) or 50 μl 0.1% HSA i.t. and euthanized at the indicated time points. Bronchoalveolar lavage (BAL) cell count and differential were performed, and albumin concentrations were measured by ELISA. Error bars represent SE.
β-Catenin/p300 signaling accelerates repair of the lung epithelium in vitro.
Neutrophil transmigration across monolayers of cultured lung epithelial cells resulted in translocation of epithelial β-catenin from the intercellular junctions to the nucleus, particularly at the sites at which neutrophils penetrated the monolayer, leading to epithelial denudation (Fig. 3A). In this system, we have previously shown that activation of β-catenin signaling plays an important role in epithelial repair (72). Because the particular subset of β-catenin target genes expressed in a given context depends on the specific coactivator recruited to the β-catenin/TCF complex, we examined whether β-catenin/p300- or β-catenin/CBP-dependent gene transcription mediates epithelial repair in this model. To assess this, we examined p300 expression and found that it was enhanced in the epithelial cells adjacent to the wounds (Fig. 3B) in the same cells that demonstrate increased nuclear β-catenin (Fig. 3C). We next utilized ICG-001, a small molecule that enhances the β-catenin/p300 interaction at the expense of the β-catenin/CBP interaction and therefore selectively induces transcription of a subset of β-catenin target genes associated with β-catenin/p300 signaling (49). Twenty-four hours after neutrophil transmigration, at the peak of injury (Fig. 1, A–C), ICG-001 was added to the epithelial monolayer. We observed that treatment with ICG-001, presumably via activation of β-catenin/p300 signaling, accelerated epithelial repair, as measured by return of TER to basal levels and re-epithelialization of the denuded monolayer (Fig. 3, D and E). To determine whether activation of the β-catenin/p300 pathway would also accelerate repair of the lung epithelium after mechanical injury, we used the scratch wound model. Immediately after the scratch wound was made, the monolayer was treated with ICG-001. As illustrated in Fig. 3F, treatment with ICG-001 also significantly accelerated wound healing after this mechanical injury.
Fig. 3.

β-Catenin/p300 signaling accelerates repair of the lung epithelium after inflammatory injury and mechanical wounding in vitro. Neutrophils were induced to migrate across Calu-3 (A–C) or Calu-3-GFP (D and E) cells for 90–120 min. At 24 h after migration, the epithelial monolayer was fixed and stained for active β-catenin (A) or p300 (B) or active β-catenin and p300 (C). Arrows indicate nuclear β-catenin and/or p300. Boxed region indicates the area magnified in the inset (right). D and E: ICG-001 was added to the culture media at 24 h after migration, as described in materials and methods. At the indicated time points, TER (D) was measured, and fluorescent images (×2.5) (E) were acquired. The total cross-sectional area of epithelial defects in 5 randomly selected HPF was measured. Arrowheads indicate epithelial wounds. Area is normalized to control in each experiment. In these experiments, the media were changed at 24 h after migration to remove secreted growth factors, which likely prevented the TER from returning to baseline in the control samples. F: scratch wounds were made on confluent monolayers of Calu-3-GFP cells, followed immediately by the addition of ICG-001 to the culture media. The area of defect within a 5-mm2 region centered on the intersection of the scratches was measured. *P < 0.05; n ≥ 4. Error bars represent SE.
β-catenin/p300 signaling accelerates repair of the lung epithelium in murine models of ALI.
Given that β-catenin/p300 signaling accelerates repair of injured lung epithelium in vitro, and because we have previously shown that inhibition of β-catenin/p300 signaling delayed repair of the alveolar epithelium in animal models (72), we investigated whether activation of β-catenin/p300 signaling with ICG-001 would accelerate repair after inflammatory injury in vivo. Because i.t. KC induces transient epithelial injury followed by repair, as demonstrated by complete resolution of permeability (Fig. 2B), this model is ideal for evaluating factors that may modulate epithelial repair. In mice treated with i.t. KC, activation of β-catenin/p300 signaling via administration of ICG-001 accelerated epithelial repair, as assessed by the resolution of lung permeability (Fig. 4). Notably, ICG-001 did not affect the magnitude of initial lung injury, as measured by initial permeability (data not shown).
Fig. 4.

β-catenin/p300 signaling accelerates repair of the lung epithelium after inflammatory injury in murine models. C57BL/6 mice were treated with KC 1 μg followed by 1.25 mg ICG-001 in 28 μl DMSO or 28 μl DMSO s.c. 2 h later and euthanized at the indicated time points. Albumin concentrations in the BAL fluid were measured by ELISA. *P < 0.05. Error bars represent SE.
WISP1 is a β-catenin/p300 target gene.
Having established that β-catenin/p300 signaling accelerates repair of the lung epithelium after inflammatory injury in vitro and in vivo, we sought to determine which target genes mediate this effect. Based on the results of a recent genome-wide microarray, which revealed that neutrophil transmigration results in a 39.5-fold increase in epithelial expression of WISP1 (Fig. 5A) (72), a β-catenin target gene (65) known to promote cell survival (63), migration (29), and proliferation (38), we hypothesized that WISP1 mediates the reparative effects of β-catenin/p300 signaling. Accordingly, we examined whether activation of β-catenin/p300 resulted in increased WISP1 expression. Calu-3 cells were treated with ICG-001, and whole cell lysates were immunoblotted for WISP1. This analysis revealed significant upregulation of WISP1 protein levels (Fig. 5B) in response to β-catenin/p300 activation.
Fig. 5.

Neutrophil transmigration or β-catenin/p300 signaling induces Wnt-induced secreted protein (WISP1) expression in lung epithelial cells. A: neutrophils were induced to migrate across Calu-3 cells for 90 min followed by incubation in media for 4 h. Epithelial cell cDNA from 4 separate experiments was pooled and analyzed using a whole genome expression microarray (Agilent). The 10 most highly upregulated genes are shown. B: Calu-3 cells were treated with ICG-001 10 μM for 24 h and lysed. Cell lysates were analyzed by SDS-PAGE and immunoblotting for WISP1 and GAPDH. WISP1 protein levels were calculated relative to GAPDH levels by densitometry.
Expression of WISP1 and Cyr61 is increased in the lung epithelium in response to neutrophil transmigration in vitro and is further increased by β-catenin/p300 activation.
To determine the contribution of β-catenin/p300-regulated WISP1 or Cyr61 to epithelial repair after inflammatory injury, we induced neutrophils to migrate across lung epithelial cells cultured on Transwell inserts and analyzed subsequent responses in the epithelial cells in the presence or absence of ICG-001. Neutrophil transmigration resulted in an increase in WISP1 and Cyr61 mRNA and protein expression in Calu-3 cells (Fig. 6, A and C) (72) and primary cultures of human small airway epithelial cells (SAEC) (Fig. 6, B and D). Enhancement of β-catenin/p300 signaling with ICG-001 resulted in a further increase in expression of WISP1 and Cyr61 in response to neutrophil transmigration (Fig. 6E). Conversely, shRNA knockdown of β-catenin (72) attenuated WISP1 upregulation in response to neutrophil transmigration (Fig. 6F), demonstrating that WISP1 expression under these conditions was β-catenin dependent. Notably, although activation of β-catenin/p300 signaling or knockdown of β-catenin affected WISP1/Cyr61 expression throughout the monolayer, the effect was especially pronounced at the sites of neutrophil penetration and injury (Fig. 6, C, E, and F).
Fig. 6.

Neutrophil transmigration results in upregulation of WISP1 and cysteine-rich 61 (Cyr61) in lung epithelial cells. Neutrophils (polymorphonuclear leukocytes, PMN) were induced by a gradient of fMLP (1 μM) to migrate for 90 min across Calu-3 cells (A, C, and E) or primary human small airway epithelial cells (SAEC) (B and D), or Calu-3 cells (F) transfected with a nonsilencing shRNA or shRNA to β-catenin. E: ICG-001 (10 μM) was added to the monolayer after migration. A and B: real-time quantitative PCR analysis of WISP1 or Cyr61 expression was performed on epithelial cDNA isolated 2 h after the end of migration. C–F: epithelial monolayers were fixed 28 h after migration and immunostained for WISP1 or Cyr61. E: fluorescence intensity was analyzed in the epithelial cells contiguous (C) to the denuded area and distant (D) from the sites of injury using ImageJ software. Arrows indicated enhanced epithelial WISP1 or Cyr61 expression at the sites of injury. *P ≤ 0.05, n ≥ 4. Error bars represent SE.
Neutrophil elastase increases WISP1 expression in the lung epithelium.
To determine whether neutrophil elastase mediates the upregulation of WISP1 in response to neutrophil transmigration, we treated primary human SAEC with purified human neutrophil elastase. Elastase resulted in increased expression of WISP1 (Fig. 7A). Because β-catenin-dependent transcriptional events have previously been shown to result from protease-mediated cleavage of E-cadherin from the apical junctional complexes (28, 47, 48), we explored whether cleavage of junctional E-cadherin by elastase might contribute to enhanced WISP1 expression. After neutrophil transmigration across SAEC, apical supernatants were concentrated and analyzed by immunoblotting using an antibody that recognizes the extracellular domain of E-cadherin. As demonstrated in Fig. 7B, neutrophil transmigration resulted in cleavage of an ∼50-kDa extracellular fragment of E-cadherin.
Fig. 7.

Neutrophil elastase triggers WISP1 upregulation and neutrophil transmigration results in cleavage of E-cadherin from the epithelial cell surface. A: primary human SAEC were treated with 0.1 U/ml purified elastase for 1 h and then incubated in media for 2 h. Epithelial cDNA was analyzed by qPCR for WISP1. B: neutrophils were induced to migrate across SAEC for 90 min. Epithelial cell supernatants were harvested from the apical surface, concentrated by ultracentrifugation through a 10-kDa MW filter, separated by SDS/PAGE, and immunoblotted with an antibody that recognizes the extracellular domain of E-cadherin (DECMA-1). *P ≤ 0.05, n ≥ 4.
WISP1 is upregulated in the lung epithelium in response to neutrophil transmigration in animal models.
Having determined that WISP1 is upregulated in the lung epithelium in response to neutrophil migration in vitro, we sought to determine whether expression of WISP1 was increased in animal models of lung injury. Because i.t. LPS results in a prolonged period of inflammation and injury and therefore would be likely to be accompanied by a measurable increase in modulators of repair, we assessed whether treatment with i.t. LPS resulted in increased expression of the CCN proteins. We observed that neutrophil transmigration in response to i.t. LPS induced upregulation of WISP1 mRNA levels in the lung, as measured by qPCR of whole lung extracts (Fig. 8A) and WISP1 protein levels in the BAL, as measured by ELISA (Fig. 8B). There was a trend toward upregulation of Cyr61 mRNA in response to neutrophil transmigration that did not achieve statistical significance (Fig. 8A).
Fig. 8.

Neutrophilic inflammation is associated with upregulation of WISP1 in animal models. C57BL/6 mice were treated with LPS 20 μg in 50 μl saline or 50 μl of saline and euthanized at 4–5 days. A: lungs were snap frozen, and RNA was extracted, reverse transcribed to cDNA, and subjected to qPCR using primers for WISP1 and Cyr61. B: WISP1 ELISA was performed on BAL fluid. *P ≤ 0.05. Error bars represent SE. Difference in Cyr61 mRNA expression was not significant between control and LPS-treated lungs.
WISP1 and Cyr61 play a role in repair of the lung epithelium after inflammatory injury in vitro.
Because β-catenin/p300 signaling regulates repair of the lung epithelium after neutrophil-mediated injury, and neutrophil transmigration resulted in the upregulation of WISP1 and Cyr61, likely via β-catenin/p300 signaling, we sought to determine whether the role of β-catenin/p300 signaling in repair of the lung epithelium was mediated by the CCN proteins. Epithelial monolayers were treated with rhWISP1, rhCyr61, or a neutralizing antibody to Cyr61 at 24 h after migration. We observed that rhWISP1 (Fig. 9A) and rhCyr61 (Fig. 9B) accelerated repair of the lung epithelium, as measured by re-epithelialization of the denuded monolayer or return of TER to baseline, respectively. Conversely, inhibition of Cyr61 with a neutralizing antibody prevented epithelial repair as assessed by recovery of TER (Fig. 9C).
Fig. 9.

WISP1 and Cyr61 accelerate repair of the lung epithelium after inflammatory injury. Neutrophils were induced to migrate across monolayers of Calu-3-GFP cells. At 24 h after migration, epithelial monolayers were treated with rhWISP1 (A), rhCyr61 (B), or Cyr61-neutralizing antibody (C), as described in materials and methods. A: fluorescent images (×2.5) were acquired at the specified time points and the total cross-sectional area of epithelial defects in 5 HPF was measured. Area is normalized to control in each experiment. Arrowheads indicate epithelial wounds. B and C: TER was measured. In these experiments, the media were changed at 24 h after migration to remove secreted growth factors, which likely prevented the TER from returning to baseline in the control samples. *P < 0.05; n ≥ 4. Error bars represent SE.
DISCUSSION
Epithelial injury is central to the pathogenesis of diverse inflammatory lung diseases and may be in part attributable to neutrophil-mediated cytotoxicity during transmigration across the epithelium (73). Importantly, timely repair of the injured epithelium is critical to the clinical outcomes in these diseases (13, 68). We previously reported that β-catenin signaling is important for repair of the lung epithelium after inflammatory injury (72), but the β-catenin target genes that mediate this effect were not identified. In the present study, we examined whether enhancing β-catenin/p300 signaling can accelerate epithelial repair and whether this effect is mediated by the CCN proteins WISP1 and Cyr61. Herein, we demonstrate that activation of β-catenin/p300 signaling, using the small molecule compound ICG-001, accelerates repair of the lung epithelium after neutrophil transmigration both in vitro and in animal models. Furthermore, we provide supporting evidence that WISP1 and Cyr61 expression are β-catenin/p300 dependent, that neutrophil transmigration or purified neutrophil elastase upregulates WISP1 and Cyr61 expression, and that WISP1 and Cyr61 accelerate repair of the lung epithelium and the resolution of permeability. In sum, our data suggest that during neutrophil transmigration, elastase-mediated cleavage of E-cadherin is associated with release of junctional β-catenin and increased nuclear β-catenin that binds to the transcriptional coactivator p300 and induces expression of WISP1 and Cyr61, which play a role in epithelial repair. These observations have important clinical implications for inflammatory lung diseases, such as ALI and COPD (13, 68).
The data presented here, together with our previous work, support the novel concept that neutrophils, although capable of inducing epithelial injury under certain circumstances, paradoxically can also activate reparative pathways. This phenomenon has been described in the lung and other organs in an emerging literature (1, 9). Our work suggests that neutrophil transmigration activates β-catenin signaling via elastase-mediated cleavage of E-cadherin, with consequent destabilization of the cadherin-catenin complex, leading to an increase in nuclear β-catenin (72) and upregulation of the CCN proteins. During transepithelial neutrophil migration, the membranes of migrating neutrophils and adjacent epithelial cells form a protected space in which neutrophil proteinases can attain high concentrations (54), suggesting the possibility that proteinase-dependent signaling pathways might activate reparative responses in adjacent epithelial cells. In this context, it is notable that the activation of β-catenin and the upregulation of p300 and the CCN proteins are localized effects, restricted to the epithelial cells in closest proximity to the migrating neutrophils (Figs. 3, A and B, and 6C), which are presumably the cells responsible for repair. Similarly, β-catenin/p300 signaling induces CCN protein expression throughout the monolayer, but particularly at the sites of neutrophil transmigration (Figs. 5B, 6E).
Our findings are consistent with the established role of β-catenin signaling and the CCN matricellular proteins in epithelial repair processes (30, 31, 33, 38). Indeed, β-catenin and the CCN proteins are known to be involved in tissue repair after injury in various organs, including the lung, where they have been implicated in limiting injury and/or promoting repair in acute lung injury (2, 7, 14, 17, 30, 31, 64) and COPD (37, 75). However, the effects of CCN proteins may be context dependent because WISP1 may enhance ventilator-induced lung injury via augmentation of macrophage proinflammatory responses (42). Moreover, β-catenin (11, 35, 50, 74) and the CCN proteins (26, 38, 46) have also been implicated in EMT and fibrosis in the lung and other organs (62) although Cyr61 can also limit the fibrotic response by inducing cellular senescence (32). As EMT may be defined more accurately as changes in morphology and motility rather than bona fide conversion of epithelial cells into fibroblasts (57), our data demonstrating the role of β-catenin and the CCN proteins in cell spreading and migration are consistent with this literature. We speculate that acute neutrophilic inflammation triggers physiological cell spreading and migration of epithelial cells via β-catenin signaling involving CCN growth factors, whereas, in the setting of repetitive cycles of injury and repair, these same pathways become deregulated, resulting in an unchecked profibrotic response.
The CCN matricellular proteins exert their effects on cells via binding to integrins and heparan sulfate proteoglycans, thereby triggering intracellular signaling cascades that ultimately regulate cell survival, proliferation, migration, and spreading. As matricellular proteins, CCN proteins can also modulate the expression, activity, and bioavailability of other growth factors (10, 32). Thus our data showing that WISP1 and Cyr61 are involved in epithelial repair are consistent with established properties of these proteins. The precise mechanisms by which WISP1 and Cyr61 promote epithelial repair, whether via cell spreading, proliferation, or reassembly of tight junctions (3, 13, 16), and the identity of the specific integrins and intracellular signaling pathways through which these CCN proteins act is unknown and merits further investigation. However, inasmuch as the wounds in our in vitro model close by cell spreading rather than proliferation (Fig. 1), which is consistent with other in vitro models of epithelial wound repair (5, 18, 19), we hypothesize that WISP1 promotes re-epithelialization primarily via cell spreading and migration (Fig. 9A), processes known to be regulated by CCN proteins (24, 29). Cyr61 may be involved in reformation of the intercellular junctions after injury on the basis of the effects on recovery of TER but not re-epithelialization observed after treatment with rhCyr61- or Cyr61-neutralizing antibodies (Fig. 9, B and C). The failure of Cyr61 to accelerate re-epithelialization of the denuded monolayer may reflect the complex biology of this protein, which has been shown to induce either survival and proliferation or apoptosis and senescence depending on the context, cell type, and identity of bound integrins (41). Finally, the CCN proteins also modulate inflammation (39), including immune cell migration (59), so complex bidirectional interactions likely exist between the epithelial cells, which express CCN growth factors in response to inflammatory injury, and the infiltrating inflammatory cells.
In hematopoietic and embryonic stems cells, the β-catenin/p300-dependent gene expression profile initiates cell differentiation, whereas a switch to β-catenin/CBP-mediated gene expression leads to self-renewal and the maintenance of pluripotency (49, 56). In the lung epithelium, previous studies have suggested that β-catenin/p300 signaling may be responsible for the proper termination of cell proliferation with accelerated differentiation, whereas aberrant CBP coactivator usage results in improper repair and the development of pulmonary fibrosis (27). The results herein, showing that β-catenin/p300 signaling enhances cell spreading, support this paradigm, as cell differentiation includes the spreading of a cuboidal ATII cell into a flattened ATI cell during repair. If pathways activated by acute neutrophilic inflammation enhance physiological epithelial repair (via p300) and avert pathological responses (via CBP), neutrophil-dependent responses could be exploited pharmacologically to treat, not only acute inflammatory diseases, but also chronic fibrotic diseases of the airways and parenchyma that are characterized by dysfunctional epithelial repair.
WISP1 is known to be regulated by β-catenin binding to either TCF or CREB sites on the WISP1 promoter (66, 70), and our data suggest that WISP1 expression is regulated by the β-catenin/p300 transcriptional complex. We speculate that, in response to neutrophil transmigration or treatment with ICG-001, which liberates transcriptionally active β-catenin from CBP, β-catenin complexes with p300 and TCF and/or CREB, and this complex binds the WISP1 promoter and induces WISP1 expression.
Epithelial denudation due to neutrophil transmigration in our model is likely attributable in part to apoptosis of epithelial cells (22), consistent with observations in other models of inflammatory lung injury (51). It is possible that injured or even apoptotic cells at the site of neutrophil transmigration may signal to more distant surviving cells via calcium waves or paracrine mediators, as has been previously demonstrated (4, 58), responses that could effectuate repair. As the CCN proteins are known to regulate apoptosis (41, 63), an additional mechanism by which they might regulate wound repair is via decreased apoptosis of epithelial cells at the wound edge, an established mechanism for differential wound repair (20). Although neutrophil transmigration induces epithelial cell death and denudation of the epithelium in our models, the stimuli used (1 μM fMLP in vitro or 1 μg KC in vivo) are relatively modest, and the injury induced is relatively mild and repairs rapidly. The role of β-catenin and the CCN proteins in repair of the epithelium after more severe injury in response to other pathological stimuli such as live bacteria or acid aspiration is unknown and will be the subject of future studies in our laboratory. We anticipate that, during a more prolonged repair phase in response to more severe injury, epithelial cell proliferation and differentiation might be important mechanisms for re-epithelialization and that the CCN proteins might play an important role in these processes.
With regard to repair of the alveolar epithelium in ALI, the classic paradigm has been that, after death of susceptible ATI cells, surviving ATII cells spread into denuded areas, proliferate, and eventually differentiate into ATI cells to restore a normal alveolar epithelium (3, 16, 57). It is likely that progenitor cells also give rise to new ATII cells after injury (8, 34, 40). Recent in vitro studies reveal that ATI cells may be capable of spreading, migration, and proliferation (23), suggesting that they may also play an active role in repair. If ATI cells do play a role in alveolar repair, studies should be undertaken to determine whether β-catenin/CCN signaling is involved.
One limitation of the present work is the use of Calu-3 cells, which are a neoplastic airway cell line. However, these cells are able to form robust tight junctions and generate TER and are thus useful in the study of the mechanisms of epithelial injury and repair. Importantly, we have confirmed key findings in primary human SAEC, and our previous work suggests that the biological behavior of the Calu-3 cells in our model is similar to that of primary alveolar epithelial cells both in vitro and in animal models (72).
In conclusion, we have provided strong evidence that neutrophil transmigration across the lung epithelium induces β-catenin/p300-mediated expression of the CCN growth factors WISP1 and Cyr61, perhaps via elastase-mediated cleavage of E-cadherin, leading to an increase in nuclear β-catenin, and that this pathway is critical to repair of the injured epithelium. On the basis of our work, we propose that neutrophils, while inducing epithelial injury, simultaneously promote reparative pathways. Our data are consistent with the established role of β-catenin and the CCN growth factors in cell survival, spreading, and proliferation. Importantly, epithelial repair after injury is critical to clinical outcomes in many lung diseases (13, 67, 69), and clinical trials of agents targeting β-catenin/p300 signaling (ClinicalTrials.gov NCT01302405) and CCN proteins (44) have demonstrated safety and plausibility. If pathways activated by acute neutrophilic inflammation enhance physiological repair and impede pathological responses of the epithelium, these pathways are promising therapeutic targets not only to accelerate the resolution of lung permeability in acute inflammatory diseases such as ALI but also to mitigate dysfunctional epithelial repair and progressive fibrosis in diseases such as COPD and pulmonary fibrosis.
GRANTS
This work was supported by NIH HL103772 (R. Zemans), HL090669 (G. Downey), a Young Clinical Scientist Award from the Flight Attendant Medical Research Institute (R. Zemans), a Parker B. Francis Fellowship (R. Zemans), and funds from National Jewish Health.
DISCLOSURES
M. Kahn has a financial interest in Prism Pharmaceuticals of greater than 5%.
AUTHOR CONTRIBUTIONS
Author contributions: R.L.Z., N.B., L.F.L., M.K., and G.P.D. conception and design of research; R.L.Z., J.M., Y.A., N.B., and S.K.Y. performed experiments; R.L.Z., J.M., Y.A., and S.K.Y. analyzed data; R.L.Z., M.K., and G.P.D. interpreted results of experiments; R.L.Z. and J.M. prepared figures; R.L.Z. drafted manuscript; R.L.Z., L.F.L., M.K., and G.P.D. edited and revised manuscript; R.L.Z., J.M., N.B., S.K.Y., L.F.L., M.K., and G.P.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge Kenneth Malcolm for technical assistance and Peter M. Henson for thoughtful discussions.
REFERENCES
- 1. Aarbiou J, Ertmann M, van Wetering S, van Noort P, Rook D, Rabe KF, Litvinov SV, van Krieken JH, de Boer WI, Hiemstra PS. Human neutrophil defensins induce lung epithelial cell proliferation in vitro. J Leukoc Biol 72: 167–174, 2002 [PubMed] [Google Scholar]
- 2. Adamson A, Perkins S, Brambilla E, Tripp S, Holden J, Travis W, Guinee D., Jr Proliferation, C-myc, and cyclin D1 expression in diffuse alveolar damage: potential roles in pathogenesis and implications for prognosis. Hum Pathol 30: 1050–1057, 1999 [DOI] [PubMed] [Google Scholar]
- 3. Adamson IY, Bowden DH. The type 2 cell as progenitor of alveolar epithelial regeneration. A cytodynamic study in mice after exposure to oxygen. Lab Invest 30: 35–42, 1974 [PubMed] [Google Scholar]
- 4. Ashino Y, Ying X, Dobbs LG, Bhattacharya J. [Ca(2+)](i) oscillations regulate type II cell exocytosis in the pulmonary alveolus. Am J Physiol Lung Cell Mol Physiol 279: L5–L13, 2000 [DOI] [PubMed] [Google Scholar]
- 5. Atabai K, Ishigaki M, Geiser T, Ueki I, Matthay MA, Ware LB. Keratinocyte growth factor can enhance alveolar epithelial repair by nonmitogenic mechanisms. Am J Physiol Lung Cell Mol Physiol 283: L163–L169, 2002 [DOI] [PubMed] [Google Scholar]
- 6. Babic AM, Kireeva ML, Kolesnikova TV, Lau LF. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc Natl Acad Sci USA 95: 6355–6360, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bui KC, Buckley S, Wu F, Uhal B, Joshi I, Liu J, Hussain M, Makhoul I, Warburton D. Induction of A- and D-type cyclins and cdc2 kinase activity during recovery from short-term hyperoxic lung injury. Am J Physiol Lung Cell Mol Physiol 268: L625–L635, 1995 [DOI] [PubMed] [Google Scholar]
- 8. Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH. Integrin alpha6beta4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest 121: 2855–2862, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheek JM, McDonald RJ, Rapalyea L, Tarkington BK, Hyde DM. Neutrophils enhance removal of ozone-injured alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol 269: L527–L535, 1995 [DOI] [PubMed] [Google Scholar]
- 10. Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol 41: 771–783, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chilosi M, Poletti V, Zamo A, Lestani M, Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, Cancellieri A, Maestro R, Semenzato G, Doglioni C. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol 162: 1495–1502, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell 127: 469–480, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 298: L715–L731, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Douglas IS, Diaz del Valle F, Winn RA, Voelkel NF. Beta-catenin in the fibroproliferative response to acute lung injury. Am J Respir Cell Mol Biol 34: 274–285, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eguchi M, Nguyen C, Lee SC, Kahn M. ICG-001, a novel small molecule regulator of TCF/beta-catenin transcription. Med Chem 1: 467–472, 2005 [DOI] [PubMed] [Google Scholar]
- 16. Evans MJ, Dekker NP, Cabral-Anderson LJ, Freeman G. Quantitation of damage to the alveolar epithelium by means of type 2 cell proliferation. Am Rev Respir Dis 118: 787–790, 1978 [DOI] [PubMed] [Google Scholar]
- 17. Flozak AS, Lam AP, Russell S, Jain M, Peled ON, Sheppard KA, Beri R, Mutlu GM, Budinger GR, Gottardi CJ. Beta-catenin/T-cell factor signaling is activated during lung injury and promotes the survival and migration of alveolar epithelial cells. J Biol Chem 285: 3157–3167, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garat C, Kheradmand F, Albertine KH, Folkesson HG, Matthay MA. Soluble and insoluble fibronectin increases alveolar epithelial wound healing in vitro. Am J Physiol Lung Cell Mol Physiol 271: L844–L853, 1996 [DOI] [PubMed] [Google Scholar]
- 19. Geiser T, Atabai K, Jarreau PH, Ware LB, Pugin J, Matthay MA. Pulmonary edema fluid from patients with acute lung injury augments in vitro alveolar epithelial repair by an IL-1beta-dependent mechanism. Am J Respir Crit Care Med 163: 1384–1388, 2001 [DOI] [PubMed] [Google Scholar]
- 20. Geiser T, Ishigaki M, van Leer C, Matthay MA, Broaddus VC. H(2)O(2) inhibits alveolar epithelial wound repair in vitro by induction of apoptosis. Am J Physiol Lung Cell Mol Physiol 287: L448–L453, 2004 [DOI] [PubMed] [Google Scholar]
- 21. Ginzberg HH, Cherapanov V, Dong Q, Cantin A, McCulloch CA, Shannon PT, Downey GP. Neutrophil-mediated epithelial injury during transmigration: role of elastase. Am J Physiol Gastrointest Liver Physiol 281: G705–G717, 2001 [DOI] [PubMed] [Google Scholar]
- 22. Ginzberg HH, Shannon PT, Suzuki T, Hong O, Vachon E, Moraes T, Abreu MT, Cherepanov V, Wang X, Chow CW, Downey GP. Leukocyte elastase induces epithelial apoptosis: role of mitochondial permeability changes and Akt. Am J Physiol Gastrointest Liver Physiol 287: G286–G298, 2004 [DOI] [PubMed] [Google Scholar]
- 23. Gonzalez RF, Allen L, Dobbs LG. Rat alveolar type I cells proliferate, express OCT-4, and exhibit phenotypic plasticity in vitro. Am J Physiol Lung Cell Mol Physiol 297: L1045–L1055, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grzeszkiewicz TM, Lindner V, Chen N, Lam SC, Lau LF. The angiogenic factor cysteine-rich 61 (CYR61, CCN1) supports vascular smooth muscle cell adhesion and stimulates chemotaxis through integrin alpha(6)beta(1) and cell surface heparan sulfate proteoglycans. Endocrinology 143: 1441–1450, 2002 [DOI] [PubMed] [Google Scholar]
- 25. Haslett C, Guthrie LA, Kopaniak MM, Johnston RB, Jr, Henson PM. Modulation of multiple neutrophil functions by preparative methods or trace concentrations of bacterial lipopolysaccharide. Am J Pathol 119: 101–110, 1985 [PMC free article] [PubMed] [Google Scholar]
- 26. Heise RL, Stober V, Cheluvaraju C, Hollingsworth JW, Garantziotis S. Mechanical stretch induces epithelial-mesenchymal transition in alveolar epithelia via hyaluronan activation of innate immunity. J Biol Chem 286: 17435–17444, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Henderson WR, Jr, Chi EY, Ye X, Nguyen C, Tien YT, Zhou B, Borok Z, Knight DA, Kahn M. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci USA 107: 14309–14314, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, Gulino-Debrac D. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem 278: 14002–14012, 2003 [DOI] [PubMed] [Google Scholar]
- 29. Hou CH, Chiang YC, Fong YC, Tang CH. WISP-1 increases MMP-2 expression and cell motility in human chondrosarcoma cells. Biochem Pharmacol 81: 1286–1295, 2011 [DOI] [PubMed] [Google Scholar]
- 30. Jin Y, Kim HP, Cao J, Zhang M, Ifedigbo E, Choi AM. Caveolin-1 regulates the secretion and cytoprotection of Cyr61 in hyperoxic cell death. FASEB J 23: 341–350, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin Y, Kim HP, Ifedigbo E, Lau LF, Choi AM. Cyr61 protects against hyperoxia-induced cell death via Akt pathway in pulmonary epithelial cells. Am J Respir Cell Mol Biol 33: 297–302, 2005 [DOI] [PubMed] [Google Scholar]
- 32. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12: 676–685, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov 10: 945–963, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, Crowley D, Bronson RT, Jacks T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121: 823–835, 2005 [DOI] [PubMed] [Google Scholar]
- 35. Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, Frank JA, Brumwell AN, Wheeler SE, Kreidberg JA, Chapman HA. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest 119: 213–224, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kireeva ML, Mo FE, Yang GP, Lau LF. Cyr61, a product of a growth factor-inducible immediate-early gene, promotes cell proliferation, migration, and adhesion. Mol Cell Biol 16: 1326–1334, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kneidinger N, Yildirim AO, Callegari J, Takenaka S, Stein MM, Dumitrascu R, Bohla A, Bracke KR, Morty RE, Brusselle GG, Schermuly RT, Eickelberg O, Konigshoff M. Activation of the WNT/beta-catenin pathway attenuates experimental emphysema. Am J Respir Crit Care Med 183: 723–733, 2011 [DOI] [PubMed] [Google Scholar]
- 38. Konigshoff M, Kramer M, Balsara N, Wilhelm J, Amarie OV, Jahn A, Rose F, Fink L, Seeger W, Schaefer L, Gunther A, Eickelberg O. WNT1-inducible signaling protein-1 mediates pulmonary fibrosis in mice and is upregulated in humans with idiopathic pulmonary fibrosis. J Clin Invest 119: 772–787, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kular L, Pakradouni J, Kitabgi P, Laurent M, Martinerie C. The CCN family: a new class of inflammation modulators? Biochimie 93: 377–388, 2011 [DOI] [PubMed] [Google Scholar]
- 40. Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, Wang de Y, Lim B, Chow VT, Crum CP, Xian W, McKeon F. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147: 525–538, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lau LF. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci 68: 3149–3163, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li HH, Li Q, Liu P, Liu Y, Li J, Wasserloos K, Chao W, You M, Oury TD, Chhinder S, Hackam DJ, Billiar TR, Leikauf GD, Pitt BR, Zhang LM. WNT1-Inducible Signaling Pathway Protein 1 Contributes to Ventilator-Induced Lung Injury. Am J Respir Cell Mol Biol 47: 528–535, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408, 2001 [DOI] [PubMed] [Google Scholar]
- 44. Mageto YFK, Brown K, Fong A, Raghu G. Safety and tolerability of human monoclonal antibody FG-3019, anti-connective tissue growth factor, in patients with idiopathic pulmonary fibrosis (Abstract). Chest 126: 7735-a, 2004 [Google Scholar]
- 45. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matute-Bello G, Wurfel MM, Lee JS, Park DR, Frevert CW, Madtes DK, Shapiro SD, Martin TR. Essential role of MMP-12 in Fas-induced lung fibrosis. Am J Respir Cell Mol Biol 37: 210–221, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mayerle J, Schnekenburger J, Kruger B, Kellermann J, Ruthenburger M, Weiss FU, Nalli A, Domschke W, Lerch MM. Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology 129: 1251–1267, 2005 [DOI] [PubMed] [Google Scholar]
- 48. McGuire JK, Li Q, Parks WC. Matrilysin (matrix metalloproteinase-7) mediates E-cadherin ectodomain shedding in injured lung epithelium. Am J Pathol 162: 1831–1843, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miyabayashi T, Teo JL, Yamamoto M, McMillan M, Nguyen C, Kahn M. Wnt/beta-catenin/CBP signaling maintains long-term murine embryonic stem cell pluripotency. Proc Natl Acad Sci USA 104: 5668–5673, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Morrisey EE. Wnt signaling and pulmonary fibrosis. Am J Pathol 162: 1393–1397, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakamura M, Matute-Bello G, Liles WC, Hayashi S, Kajikawa O, Lin SM, Frevert CW, Martin TR. Differential response of human lung epithelial cells to fas-induced apoptosis. Am J Pathol 164: 1949–1958, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nick JA, Young SK, Brown KK, Avdi NJ, Arndt PG, Suratt BT, Janes MS, Henson PM, Worthen GS. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J Immunol 164: 2151–2159, 2000 [DOI] [PubMed] [Google Scholar]
- 54. Owen CA, Campbell MA, Sannes PL, Boukedes SS, Campbell EJ. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel, non-oxidative mechanism by which neutrophils focus and preserve catalytic activity of serine proteinases. J Cell Biol 131: 775–789, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Parkos CA, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured intestinal epithelium. Dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. J Clin Invest 88: 1605–1612, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rebel VI, Kung AL, Tanner EA, Yang H, Bronson RT, Livingston DM. Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci USA 99: 14789–14794, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA 108: E1475–E1483, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rosenblatt J, Raff MC, Cramer LP. An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol 11: 1847–1857, 2001 [DOI] [PubMed] [Google Scholar]
- 59. Rother M, Krohn S, Kania G, Vanhoutte D, Eisenreich A, Wang X, Westermann D, Savvatis K, Dannemann N, Skurk C, Hilfiker-Kleiner D, Cathomen T, Fechner H, Rauch U, Schultheiss HP, Heymans S, Eriksson U, Scheibenbogen C, Poller W. Matricellular signaling molecule CCN1 attenuates experimental autoimmune myocarditis by acting as a novel immune cell migration modulator. Circulation 122: 2688–2698, 2010 [DOI] [PubMed] [Google Scholar]
- 60. Savla U, Olson LE, Waters CM. Mathematical modeling of airway epithelial wound closure during cyclic mechanical strain. J Appl Physiol 96: 566–574, 2004 [DOI] [PubMed] [Google Scholar]
- 61. Teo JL, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Adv Drug Deliv Rev 62: 1149–1155, 2010 [DOI] [PubMed] [Google Scholar]
- 62. Venkatachalam K, Venkatesan B, Valente AJ, Melby PC, Nandish S, Reusch JE, Clark RA, Chandrasekar B. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-alpha (TNF-alpha)-stimulated cardiac fibroblast proliferation but inhibits TNF-alpha-induced cardiomyocyte death. J Biol Chem 284: 14414–14427, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Venkatesan B, Prabhu SD, Venkatachalam K, Mummidi S, Valente AJ, Clark RA, Delafontaine P, Chandrasekar B. WNT1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell Signal 22: 809–820, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Villar J, Cabrera NE, Casula M, Valladares F, Flores C, Lopez-Aguilar J, Blanch L, Zhang H, Kacmarek RM, Slutsky AS. WNT/beta-catenin signaling is modulated by mechanical ventilation in an experimental model of acute lung injury. Intensive Care Med 37: 1201–1209, 2011 [DOI] [PubMed] [Google Scholar]
- 65. Vlad A, Rohrs S, Klein-Hitpass L, Muller O. The first five years of the Wnt targetome. Cell Signal 20: 795–802, 2008 [DOI] [PubMed] [Google Scholar]
- 66. Wang H, Zhang R, Wen S, McCafferty DM, Beck PL, MacNaughton WK. Nitric oxide increases Wnt-induced secreted protein-1 (WISP-1/CCN4) expression and function in colitis. J Mol Med 87: 435–445, 2009 [DOI] [PubMed] [Google Scholar]
- 67. Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 342: 1334–1349, 2000 [DOI] [PubMed] [Google Scholar]
- 68. Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 1376–1383, 2001 [DOI] [PubMed] [Google Scholar]
- 69. Ware LB, Matthay MA. Maximal alveolar epithelial fliud clearance in clinical acute lung injury: an excellent predictor of survival and the duration of mechanical ventilation (Abstract). Am J Respir Crit Care Med 159: A694, 1999 [Google Scholar]
- 70. Xu L, Corcoran RB, Welsh JW, Pennica D, Levine AJ. WISP-1 is a Wnt-1- and beta-catenin-responsive oncogene. Genes Dev 14: 585–595, 2000 [PMC free article] [PubMed] [Google Scholar]
- 71. Yang GP, Lau LF. Cyr61, product of a growth factor-inducible immediate early gene, is associated with the extracellular matrix and the cell surface. Cell Growth Differ 2: 351–357, 1991 [PubMed] [Google Scholar]
- 72. Zemans RL, Briones N, Campbell M, McClendon J, Young SK, Suzuki T, Yang IV, De Langhe S, Reynolds SD, Mason RJ, Kahn M, Henson PM, Colgan SP, Downey GP. Neutrophil transmigration triggers repair of the lung epithelium via beta-catenin signaling. Proc Natl Acad Sci USA 108: 15990–15995, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zemans RL, Colgan SP, Downey GP. Transepithelial migration of neutrophils: mechanisms and implications for acute lung injury. Am J Respir Cell Mol Biol 40: 519–535, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhou B, Liu Y, Kahn M, Ann DK, Han A, Wang H, Nguyen C, Flodby P, Zhong Q, Krishnaveni MS, Liebler JM, Minoo P, Crandall ED, Borok Z. Interactions between beta-catenin and transforming growth factor-beta signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J Biol Chem 287: 7026–7038, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhu M, Tian D, Li J, Ma Y, Wang Y, Wu R. Glycogen synthase kinase 3beta and beta-catenin are involved in the injury and repair of bronchial epithelial cells induced by scratching. Exp Mol Pathol 83: 30–38, 2007 [DOI] [PubMed] [Google Scholar]