

Abstract
Cigarette smoking attenuates acetylcholine (ACh)-induced cutaneous vasodilation in humans, but the underlying mechanisms are unknown. We tested the hypothesis that smokers have impaired nitric oxide (NO)- and cyclooxygenase (COX)-dependent cutaneous vasodilation to ACh infusion. Twelve young smokers, who have smoked more than 5.2 ± 0.7 yr with an average daily consumption of 11.4 ± 1.2 cigarettes, and 12 nonsmokers were tested. Age, body mass index, and resting mean arterial pressure were similar between the groups. Cutaneous vascular conductance (CVC) was evaluated as laser-Doppler flux divided by mean arterial pressure, normalized to maximal CVC (local heating to 43.0°C plus sodium nitroprusside administration). We evaluated the increase in CVC from baseline to peak (CVCΔpeak) and area under the curve of CVC (CVCAUC) during a bolus infusion (1 min) of 137.5 μM ACh at four intradermal microdialysis sites: 1) Ringer (control), 2) 10 mM NG-nitro-l-arginine methyl ester (l-NAME; NO synthase inhibitor), 3) 10 mM ketorolac (COX inhibitor), and 4) combination of l-NAME + ketorolac. CVCΔpeak and CVCAUC at the Ringer site in nonsmokers were greater than in smokers (CVCΔpeak, 42.9 ± 5.1 vs. 22.3 ± 3.5%max, P < 0.05; and CVCAUC, 8,085 ± 1,055 vs. 3,145 ± 539%max·s, P < 0.05). In nonsmokers, CVCΔpeak and CVCAUC at the l-NAME site were lower than the Ringer site (CVCΔpeak, 29.5 ± 6.2%max, P < 0.05; and CVCAUC, 5,377 ± 1,109%max·s, P < 0.05), but in smokers, there were no differences between the Ringer and l-NAME sites (CVCΔpeak, 16.8 ± 4.3%max, P = 0.11; and CVCAUC, 2,679 ± 785%max·s, P = 0.30). CVCΔpeak and CVCAUC were reduced with ketorolac in nonsmokers (CVCΔpeak, 13.3 ± 3.6%max, P < 0.05; and CVCAUC, 1,967 ± 527%max·s, P < 0.05) and smokers (CVCΔpeak, 7.8 ± 1.8%max, P < 0.05; and CVCAUC, 1,246 ± 305%max·s, P < 0.05) and at the combination site in nonsmokers (CVCΔpeak, 15.9 ± 3.1%max, P < 0.05; and CVCAUC, 2,660 ± 512%max·s, P < 0.05) and smokers (CVCΔpeak, 11.5 ± 2.6%max, P < 0.05; and CVCAUC, 1,693 ± 409%max·s, P < 0.05), but the magnitudes were greater in nonsmokers (P < 0.05). These results suggest that impaired ACh-induced skin vasodilation in young smokers is related to diminished NO- and COX-dependent vasodilation.
Keywords: cyclooxygenase, acetylcholine, cigarette, skin, microvascular
almost 6 million people die from tobacco use and exposure each year (50). Given that smoking is a major independent risk factor for hypertension, myocardial infarction, and atherosclerosis (2, 48) and that chronic exposure to cigarette smoking changes the structure and function of conduit arteries (39), it is not surprising that the majority of tobacco-related deaths are due to cardiovascular disease (8). In addition, chronic smoking has been reported to attenuate the function of the microcirculation, as acetylcholine (ACh)-induced vasodilation, which is an index of endothelial function and has been used in clinical studies (47), is impaired in human skin in smokers compared with controls when ACh is administered using the iontophoresis method (6, 7, 18, 35). Given that microvascular dysfunction is a crucial step in the complications that lead to cardiovascular disease (27), advancing our understanding of the mechanisms behind how smoking affects skin microvascular function would provide strategies to ameliorate dysfunction of the microcirculation and thus tobacco-related deaths. Currently, the mechanisms behind the impaired ACh-induced skin vasodilation in smokers are unknown.
ACh stimulates the endothelium, producing several substances that can induce a direct vasodilation of the smooth muscle: nitric oxide (NO), formed via NO synthase (NOS); prostacyclin (PGI2), produced via the cyclooxygenase (COX) pathway; and endothelial-dependent hyperpolarizing factors (EDHFs) that stimulate calcium-activated potassium channels and thus hyperpolarize vascular smooth muscle. Importantly, the substance(s) responsible for vasodilation to ACh varies across vascular beds (41). In healthy human skin, ACh-induced cutaneous vasodilation is attenuated by administration of NOS and/or COX inhibitors (17, 21, 26), suggesting that both NO and COX pathways contribute to ACh-induced cutaneous vasodilation. As such, the impaired ACh-induced cutaneous vasodilation in smokers may be due to decreased contribution(s) of NO and/or the COX pathway to vasodilation. Supporting this notion, previous studies in vivo have shown that acute or chronic exposure to cigarette smoke extract or compounds found in cigarette smoke, including nicotine, carbon monoxide, and free radicals, lowers the bioavailability of NO (14, 36) and PGI2 (1, 5, 19, 32, 37).
Using the above information as background, we hypothesized that impaired ACh-mediated cutaneous vasodilation in smokers is due to diminished NO- and COX-dependent cutaneous vasodilation.
MATERIALS AND METHODS
Subjects.
Twelve nonsmokers and twelve smokers participated in this study, which was approved by the Institutional Review Board at The University of Oregon and conformed to the guidelines set forth by the Declaration of Helsinki. The characteristics of the subjects are shown in Table 1. Verbal and written informed consent was obtained from all subjects before their participation in the study. Smokers were defined as having smoked for at least 1 yr with an average daily cigarette consumption of > 6. Subjects were excluded if they had a history of hypertension, heart disease, diabetes, or autonomic disorders. All subjects were not currently taking prescription medications with the exception of oral contraceptives. All subjects abstained from the use of all medications, including nonsteroidal anti-inflammatory agents, and alcohol, caffeine, and exercise for at least 24 h before the study. The smokers abstained from smoking for at least 12 h before the study to avoid any acute effects of smoking on ACh-mediated skin vasodilation (6). Female subjects were only studied during the early follicular phase (within 6 days of the start of menstruation) or placebo phase if taking contraceptives, minimizing the effects of female sex hormones.
Table 1.
Characteristics of subjects
| Nonsmokers | Smokers | P Value | |
|---|---|---|---|
| Number of subjects (male/female) | 12 (8/4) | 12 (9/3) | |
| Age, yr | 22.3 ± 0.8 | 22.1 ± 0.9 | 0.84 |
| Height, m | 1.76 ± 0.03 | 1.72 ± 0.04 | 0.37 |
| Body weight, kg | 77.9 ± 4.3 | 73.9 ± 4.3 | 0.52 |
| Body mass index | 24.9 ± 0.8 | 24.8 ± 0.8 | 0.96 |
| Average years smoked | — | 5.2 ± 0.7 | |
| Average cigarettes/day | — | 11.4 ± 1.2 | |
| Arterial blood pressure, mmHg | |||
| Systolic | 123.0 ± 2.6 | 125.0 ± 3.3 | 0.63 |
| Diastolic | 69.6 ± 2.1 | 71.0 ± 2.4 | 0.67 |
| Mean | 87.4 ± 1.9 | 89.0 ± 2.3 | 0.61 |
Values are means ± SE.
Instrumentation.
Upon arrival at the laboratory, subjects were placed in a semi-supine position and instrumented with four intradermal microdialysis fibers (10 mm, 20 kDa cutoff membrane MD2000, Bioanalytical Systems) on the ventral side of the forearm in the dermal layer of the skin as follows. A 25-gauge needle was first inserted into the unanesthetized skin using aseptic technique with at least 4.0 cm between each site. The entry and exit points were about ∼2.5 cm apart. The microdialysis fiber was then threaded through the lumen of the needle, after which the needle was withdrawn keeping the fiber in place. Microdialysis fibers were secured with tape. Each fiber was attached to the outlet port of four liquid switch stopcocks (CMA Microdialysis, Solma, Sweden). Lactated Ringer solution was perfused through each microdialysis fiber at a rate of 2.0 μl/min (CMA 1025 microdialysis pump, CMA Microdialysis) until the start of drug infusions.
Measurements.
Arterial blood pressure was measured via automated brachial oscillation (CardioCap 5, Datex-Ohemda, Tewksbury, MA) during the protocol. Mean arterial blood pressure was calculated as diastolic arterial blood pressure plus one-third pulse pressure. To obtain an index of skin blood flow, cutaneous red blood cell flux was measured with a single-point laser-Doppler flowmeter probe placed in a local heater (Temperature Monitor SHO2, Moor Instruments, Axminster, UK) over each microdialysis fiber. Cutaneous vascular conductance (CVC) was calculated as cutaneous red blood cell flux divided by mean arterial pressure.
Experimental protocol.
A time of 60–90 min was required to recover from the trauma response caused by the microdialysis fiber placement. After the recovery, the experimental protocol started. Microdialysis fibers were randomly assigned to receive 10 mM NG-nitro-l-arginine methyl ester (l-NAME; Tocris, Ellisville, MO) to inhibit NO production by NOS or 10 mM ketorolac (Keto; Sigma-Aldrich, St. Louis, MO) to nonspecifically inhibit COX, thereby inhibiting all prostanoid and thromboxane products through COX. Both NOS and COX were inhibited in a third microdialysis site with 10 mM l-NAME + 10 mM Keto. l-NAME (10 mM) maximally inhibits NO production as previously shown (20, 28, 29). Keto (10 mM) was sufficient to inhibit production of vasoactive substances from COX as previous works have demonstrated (17, 21, 25). All pharmacological agents were dissolved in lactated Ringer solution. A continuous infusion of Ringer solution was perfused through the fourth microdialysis fiber to act as a control site. All infusions remained constant throughout the protocol with the exception of the 1-min ACh infusion (see below for more detail), which was accomplished by switching the stopcock. To ensure adequate NOS and COX inhibition, all pharmacological agents were perfused at a rate of 2.0 μl/min for at least 75 min before baseline recordings.
Baseline was recorded for at least 10 min, followed by ACh infusion procedures. The dose of 137.5 μM ACh (Sigma-Aldrich) was first administered at a rate of 2.0 μl/min on one of four skin sites for 1 min. We previously confirmed that this dose of ACh induces similar cutaneous vasodilation as observed during whole body heating at rest (17). Others have demonstrated that a similar dose of ACh (100 μM) induced a similar amount of cutaneous vasodilation (26). At least 2 min after the first ACh infusion, the same ACh concentration was applied to another of the four skin sites, and this procedure continued until all four sites received ACh. Thereafter, this four-site ACh infusion was duplicated as a second trial. The first and second ACh infusions within the same skin sites were separated by at least 20 min. Liquid switch stopcocks were used to obtain precise volumes of infusions. Arterial blood pressure measurements were taken at baseline, peak, and return to baseline during each ACh infusion. After completion of both ACh infusions, infusion of 56 mM sodium nitroprusside (Nitropress, Ciba Pharmaceuticals, East Hanover, NJ) at a rate of 2.0 μl/min and local heating of the skin to 43.0°C were applied to all skin sites to induce maximal cutaneous vasodilation (CVCmax). This normalization is necessary to minimize the effect of site-to-site heterogeneity in the level of skin blood flow (27).
Data acquisition and analyses.
Data were recorded and stored on a computer using Windaq data acquisition software (Dataq Instruments, Akron, OH). All CVC data were expressed as percentages of maximal CVC (%CVCmax). Initial baseline CVC (CVCbaseline) at each site was obtained during the 10-min baseline measurement. The increase in CVC during ACh infusion from the baseline to 30-s averaged peak value (CVCΔpeak) was evaluated. We also evaluated the area under the curve of the ACh response (CVCAUC; expressed as %CVCmax·s). The beginning and ending points for the CVCAUC analysis are the time the infusion began and the 10th min of infusion. Five minutes of baseline was taken before the start of the infusion, which was used to calculate CVCAUC and CVCΔpeak, thus excluding the influence of differing baselines of CVC across skin sites or groups. There was approximately a 4-min delay from the start of infusion to the initiation of vasodilation. Since CVC values were stable from preinfusion to the initiation of vasodilation, we did not consider the delay for the calculation of CVCAUC and CVCΔpeak. Because CVC responses to ACh infusion were highly reproducible between the first and second trials for CVCΔpeak (r = 0.97) and CVCAUC (r = 0.92), averaged values were used for data analyses.
Statistical analyses.
A two-way repeated-measures analysis of variance was conducted in each group with factors of drug: Ringer, l-NAME, Keto, and combination (Combo) and time (Fig. 1). A two-way, mixed-model, repeated-measures analysis of variance was conducted with factors of smoking habit (nonsmoker and smoker) and drug (Ringer, l-NAME, Keto, and Combo) (Fig. 2, and Table 2). When a significant main effect was detected, significant differences of paired variables between groups or drug sites were determined by t-test with correction of Holm method so that α-level was kept at 0.05. The Holm statistical model, which is a modified Bonferroni method, has been developed to increase statistical power since the original Bonferroni method is too conservative. The optimality of the Holm method for these analyses has been previously validated (12). t-tests were also used to determine significant differences in other comparisons including physical characteristics (Table 1) and whether CVCΔpeak or CVCAUC is different from baseline values before ACh infusion. If CVCAUC is not significantly different from baseline, it suggests no vasodilation to ACh administration, whereas if CVCAUC value is positive and significantly different from baseline, it is suggested that there is ACh-induced vasodilation. Pearson's product moment correlation coefficients were used to relate CVCAUC or CVCΔpeak with smoking years, daily numbers of cigarettes consumed, age, body weight, body mass index (BMI), and arterial blood pressures. The level of significance was set at α = 0.05. Values are presented as mean ± standard error (SE).
Fig. 1.
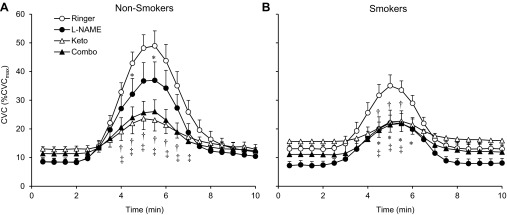
Averaged tracings for individual drug treatment sites to infusion of 137.5 μM acetylcholine in nonsmokers (A) and smokers (B). CVC, cutaneous vascular conductance; CVCmax, maximal CVC; l-NAME, nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester; Keto, nonspecifically cyclooxygenase inhibitor ketorolac; Combo, combination of l-NAME and ketorolac. *P < 0.05, Ringer vs. l-NAME within group; †P < 0.05, Ringer vs. Keto within group; ‡P < 0.05, Ringer vs. Combo within group.
Fig. 2.
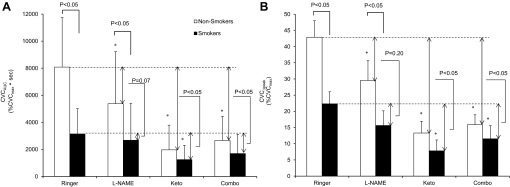
Cutaneous vasodilation responses to 1-min 137.5 μM acetylcholine administration in all drug treatment sites evaluated as area under the curve (CVCAUC; A) and baseline to peak value (CVCΔpeak; B). Values are means ± SE. *P < 0.05, vs. Ringer site within group. Comparisons are made between smokers and nonsmokers, between the CVC for each site, and between the differences in CVC from the Ringer site in the corresponding group.
Table 2.
CVCbaseline and CVCmax
| CVCbaseline, mV/MAP·100 |
CVCbaseline, %maximal |
CVCmax, mV/MAP·100 |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Nonsmokers | Smokers | P value | Nonsmokers | Smokers | P value | Nonsmokers | Smokers | P value | |
| Ringer | 23 ± 4 | 25 ± 4 | 0.41 | 8.8 ± 1.4 | 14.2 ± 2.2 | 0.05 | 272 ± 30 | 200 ± 28 | 0.09 |
| l-NAME | 22 ± 4 | 17 ± 3 | 0.16 | 10.2 ± 2.4 | 8.1 ± 1.8 | 0.49 | 257 ± 43 | 217 ± 17 | 0.40 |
| Keto | 28 ± 6 | 28 ± 6 | 0.48 | 12.8 ± 2.6 | 14.2 ± 2.6 | 0.71 | 242 ± 35 | 201 ± 22 | 0.33 |
| Combo | 31 ± 5 | 27 ± 5 | 0.27 | 12.0 ± 2.0 | 13.1 ± 1.9 | 0.67 | 280 ± 33 | 201 ± 26 | 0.07 |
| Pooled across the 4 sites | 26 ± 7 | 24 ± 7 | 0.56 | 11.0 ± 3.2 | 12.4 ± 3.6 | 0.34 | 263 ± 76 | 205 ± 59 | 0.01 |
Values are means ± SE; CVCbaseline, baseline cutaneous vascular conductance; CVCmax, maximal cutaneous vascular conductance; l-NAME, nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester; Keto, nonspecific cyclooxygenase inhibitor ketorolac; Combo, combination of l-NAME and ketorolac.
RESULTS
Physical characteristics.
The physical characteristics of the subjects are presented in Table 1. No significant differences were observed between the groups for age, height, body weight, BMI, and blood pressure variables.
ACh-induced cutaneous vasodilation.
Figure 1 shows the ACh-induced changes in CVC in nonsmokers and smokers. After administration of ACh, CVC increased, after which it went back to baseline values in all sites. CVC at the Ringer site during minutes 4–7 was significantly higher compared with the other three sites, both in nonsmokers and smokers. Figure 2A indicates averaged data for CVCAUC in both groups. As predicted, CVCAUC at the Ringer site was significantly attenuated in smokers compared with nonsmokers. l-NAME administration significantly reduced CVCAUC relative to that at the Ringer site in nonsmokers. By contrast, l-NAME administration did not affect CVCAUC compared with the Ringer site in smokers. Administrations of Keto and/or combination of l-NAME and Keto significantly lowered CVCAUC compared with the Ringer site in both groups; however, these reductions were significantly less in the smokers. Irrespective of skin sites or groups, CVCAUC was always significantly higher than baseline, suggesting that ACh-induced vasodilation was observed in all cases and that the double blockade did not abolish the response to ACh. Results of CVCΔpeak were similar to those of CVCAUC (Fig. 2B).
Baseline and maximal cutaneous vascular tone.
Table 2 displays results of CVCbaseline and CVCmax. No differences in CVCbaseline were observed between the groups at all sites, as well as between the groups for the pooled data across the four sites, though we observed a near significant difference in CVCbaseline between the groups at the Ringer site when expressed as %maximal between the groups (P = 0.05). Although a trend was observed in each site, CVCmax in smokers was not significantly different from that in nonsmokers in any individual site due to the high degree of variability. However, when the data were pooled across all four sites, pooled CVCmax in smokers was significantly lower than that in nonsmokers.
Correlation analyses.
Although CVCAUC at the l-NAME, Keto, and Combo sites displayed large individual variations irrespective of group (nonsmoker or smoker), they were not significantly correlated with age, body weight, BMI, or arterial blood pressures across subjects (n = 24). Thus the observed individual variation cannot be attributed to any single factor. In smokers, CVCAUC at the l-NAME, Keto, and Combo sites were not significantly correlated with years of smoking or daily number of cigarettes consumed. Similar correlation results were also observed for CVCΔpeak.
DISCUSSION
We are the first to investigate the mechanisms by which chronic cigarette smoking attenuates ACh-induced cutaneous vasodilation in humans using the microdialysis technique. The main findings of the present study are that 1) ACh-induced cutaneous vasodilation in smokers is attenuated compared with nonsmokers, 2) inhibition of NOS attenuates the ACh-induced cutaneous vasodilation in nonsmokers but not in smokers, and 3) inhibition of COX reduces the ACh-induced cutaneous vasodilation in both nonsmokers and smokers with a smaller reduction being observed in smokers.
The mechanisms of ACh-mediated cutaneous vasodilation.
The present study showed that inhibiting NO production significantly lowered CVCAUC and CVCΔpeak in nonsmokers (Fig. 2, A and B). Additionally, CVCAUC and CVCΔpeak were attenuated by administrating Keto in nonsmokers (Fig. 2, A and B). These results suggest that both NO and COX pathways contribute to ACh-mediated cutaneous vasodilation. This observation is consistent with some (21, 26), but not all (17), previous studies. The reason for the discrepancy is currently unclear, but large individual variation in the response when NO production is blocked with l-NAME, as observed in the present study, might be the reason. On the other hand, even when both NO and COX pathways were blocked, a significant ACh-induced cutaneous vasodilation remained in the present study in both groups (Fig. 2, A and B), similar to previous studies (17, 21–23, 26). The involvement of EDHFs in ACh-induced vasodilation is suggested in rat hepatic (3) and retinal (30) arteries. Also, the involvement of EDHFs in the cutaneous vasculature in humans is suggested for the hyperemic response to external pressure (9), cutaneous reactive hyperemia following arterial occlusion (24), and thermal hyperemia induced by local skin heating (4). Therefore, we speculate that EDHFs may be also involved in ACh-induced cutaneous vasodilation in humans. However, given that ACh binds to muscarinic receptors in the smooth muscle cells causing vasoconstriction, the residual vasodilation with l-NAME and Keto is not necessarily attributable to EDHFs. Further studies specifically investigating calcium-activated potassium channels (EDHFs activate the channels and cause hyperpolarization and vasodilation) are required to determine a role for EDHFs in ACh-induced cutaneous vasodilation.
The response to combined l-NAME and Keto infusions on CVCΔpeak and CVCAUC (Fig. 2, A and B) accounts for a large portion of the vasodilation to ACh at the Ringer site; however, the summed effects of l-NAME and Keto were greater than that of the Combo site. This finding is consistent with the concept of cross talk between the NO, COX, and EDHF pathways. In support of this suggestion, the contribution of EDHFs to ACh-induced vasodilation is increased when both the NO and COX pathways are inhibited (43). Since this EDHF-induced vasodilation is blocked by sulfaphenazole, an inhibitor of cytochrome P-450 (43), it is speculated that blocking the COX pathway leaves more available arachadonic acid to be metabolized by P-450, causing EDHF-dependent vasodilation. Also, our results may potentially be due to the dependence of the COX system on the NO system as NO activates COX enzymes (42).
Chronic smoking-induced attenuation of ACh induced cutaneous vasodilation.
CVCAUC and CVCΔpeak values at the Ringer site were significantly lower in smokers than in nonsmokers (Fig. 2, A and B), suggesting that chronic smoking attenuates ACh-induced cutaneous vasodilation, which is consistent with the results from previous studies using the iontophoresis method (6, 7, 18, 35). More importantly, the present study is the first to explore the underlying mechanisms of the impaired cutaneous vasodilation response to ACh in smokers.
l-NAME reduced CVCAUC and CVCΔpeak in nonsmokers relative to the Ringer site, whereas no effect was observed in smokers (Fig. 2, A and B), suggesting that chronic smoking impairs NO-mediated vasodilation, contributing to the impairment of ACh-induced vasodilation in the cutaneous vessels. Cigarette smoke is a rich source of reactive oxygen species (ROS), such as superoxide (38). Superoxide significantly lowers NO availability in forearm circulation (14). Also, a linkage between ROS and the NO pathway is suggested in human skin since ascorbate (an antioxidant) augments cutaneous active vasodilation during whole body heating in hypertensive humans, and this effect can be decreased by l-NAME (16). Therefore, a chronic increase in ROS by cigarette smoking seems to be a major factor for the impaired NO-mediated cutaneous vasodilation in smokers. Another possibility is that chronic smoking induces an increase in plasma endothelin-1 (34), which may have attenuated the NO-dependent vasodilation in the skin vessels through direct interaction between cyclic guanosine monophosphate and the intracellular pathway activated by endothelin receptor type A, as suggested in rabbit cerebral arteries (11).
Keto administration lowered CVCAUC and CVCΔpeak in both groups, but smaller reductions were observed in smokers (Fig. 2, A and B). It is therefore suggested that smokers have impaired COX-dependent cutaneous vasodilation, contributing to the attenuated ACh-induced cutaneous vasodilation. Consistent with this notion, acute or chronic exposure to cigarette smoke extract or nicotine reduced the formation of PGI2 in animal arteries (1, 19, 37), humans umbilical vessels (5, 19), and human urine (32). On the other hand, superoxide production from chronic smoking might increase COX-dependent vasoconstricting prostanoids (e.g., thromboxane A2), as demonstrated in rat aortic rings (40), counteracting the effect of PGI2-induced vasodilation and contributing to the impaired COX-mediated cutaneous vasodilation.
When we blocked both NO and COX, there were no significant differences in CVCAUC and CVCΔpeak between the groups (Fig. 2, A and B). With the assumption that the remaining vasodilation is mediated by EDHFs, our results may indicate that EDHF-dependent cutaneous vasodilation is not impaired in smokers. Inconsistent with this notion, however, a recent cross-sectional study by Theken et al. (49) showed that compared with nonsmokers, smokers have lower plasma level of epoxyeicosatrienoic acids, which is a type of EDHF that has been shown to be involved in human skin in thermal hyperemia induced by local skin heating (4). Direct further studies are clearly needed to elucidate the effects of chronic smoking on EDHF-mediated cutaneous vasodilation in humans.
Since aging (15, 17, 29), hypertension (44), and disease status (45, 46) influence skin endothelial function, we recruited healthy young smokers and matched controls to avoid these confounding factors. Also, earlier studies have investigated the acute (10, 35, 36) and chronic (6, 7, 18, 35) influences of cigarette smoking and the compounds found in cigarette smoke on skin vessel function, but importantly, the effect of smoking is different depending on whether one smokes cigarettes acutely or chronically (6). To exclude the acute effect, we had the subjects not smoke for more than 12 h before the study. Therefore, the observed attenuation of ACh-induced cutaneous vasodilation in smokers of the present study is not likely caused by the above factors.
Chronic cigarette smoking and premature skin aging.
Aging causes skin microvascular dysfunction as NO-mediated cutaneous vasodilation during local heating (29), mild whole body heating (15), and COX-induced cutaneous vasodilation to ACh administration (17) were attenuated in aged humans compared with young ones. Similarly, the present study showed that NO- and COX-dependent cutaneous vasodilation was impaired in smokers compared with nonsmokers. Therefore, the present study extends the notion that cigarette smoking causes premature skin aging; i.e., it causes not only wrinkle formation (31) but also impairment of skin endothelial dysfunction via a similar mechanism to aging.
The effect of chronic smoking on maximal cutaneous vascular tone.
Our results showed that CVCmax induced by sodium nitroprusside administration combined with local heating was significantly attenuated in smokers (Table 2). We speculate that this attenuation is due to diminished smooth muscle function or structural limitations to vasodilation as suggested in previous studies (6, 7, 35). Chronic smoking has been reported to induce 1) an increased Rho-associated protein kinase, known to play a major role in smooth muscle contraction (33); 2) an upregulation of voltage-gated Ca2+ channels in vascular smooth muscle cells (10); 3) an elevation in endothelin-1, which activates voltage-gated Ca2+ channels in smooth muscle cell (13); and 4) smooth muscle cell proliferation (51). Furthermore, given that cigarette smoking lowers peripheral capillary density in guinea pigs (52), capillary density of the skin in smokers may have been lower than nonsmokers, resulting in lower red blood cell concentration at the laser-Doppler sites and a consequently lower CVCmax in smokers.
Whatever the mechanism, the lower CVCmax in smokers is important in terms of the interpretation of the ACh responses. To minimize the site-to-site variations in skin blood flow (27), we expressed CVCΔpeak and CVCAUC as %CVCmax; if anything, representing the data as %CVCmax would have resulted in an overestimation of the ACh-mediated cutaneous vasodilation in smokers. Had we presented absolute values, the difference between groups would have been even greater. We observed a near significant difference in CVCbaseline at the Ringer site expressed as %maximal CVC between groups (P = 0.05). It might be that impaired NO- and COX-dependent pathways in smokers upregulates EDHF-mediated vasodilation, given the existence of complex cross talk between the NO, COX, and EDHFs pathways (as discussed above). However, this difference could also be attributed to the lower CVCmax at the Ringer site in smokers.
Individual variation in drug effects.
The effects of the drugs on ACh-induced cutaneous vasodilation were largely different depending on the subjects (e.g., Keto administration almost abolished ACh-mediated cutaneous vasodilation in some subjects, but barely attenuated the vasodilation in others). This individual variation was seen in both nonsmokers and smokers. We attempted to elucidate the factor(s) underlying the individual variation with correlation analyses to relate CVCAUC and CVCΔpeak at each site with smoking years, daily numbers of cigarettes consumed, age, body weight, BMI, and arterial blood pressure. However, no significant relationships were found. Although we did not have sufficient statistical power to make a comparison between the sexes, there were no observed trends suggesting a sex difference in CVCAUC and CVCΔpeak. Perhaps the above factors and/or fitness level, smoking habit, sex, etc., interact in a complex manner, relating to the individual variation.
Limitations.
We found that the effects of Keto on CVCAUC and CVCΔpeak were significantly less in smokers than nonsmokers when expressed as %CVCmax (Fig. 2, A and B). However, the results can be interpreted differently when the data are expressed relative to the Ringer site (%change from Ringer site). CVCAUC and CVCΔpeak were similarly reduced by l-NAME in both groups when expressed as a relative value (15–34%). CVCAUC and CVCΔpeak were similarly reduced by Keto (65–76%) in both groups. However, we believe it is more appropriate to express the data as %CVCmax, meaning that our original interpretation of the data stands.
In this study, we evaluated the vasodilatory response to ACh at only one dose (137.5 μM). As such, our results cannot be simply applied to another dose of ACh. Future studies, employing a range ACh dose-response protocol (26), will be needed to address this issue.
Perspectives.
Our results indicated that impaired ACh-induced skin vasodilation in young smokers is related to diminished NO- and COX-dependent vasodilation, which resembles the effect of aging on skin endothelial function (15, 17, 29). Given that microvascular dysfunction may be a crucial step in the complications leading to cardiovascular disease (27), it is speculated that in smokers, countermeasures such as prescribing medicines that improve NO and COX pathways may ameliorate dysfunction of the microvasculature, such as in the skin, potentially reducing smoking-related cardiovascular disease and deaths, though this point needs to be tested in future studies.
GRANTS
This study was supported by the National Institutes of Health Grant HL-081671. N. Fujii is the recipient of a research fellowship for young scientists from the Japan Society for the Promotion of Science.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
N.F., M.C.R., and V.E.B. performed experiments; N.F. and M.C.R. analyzed data; N.F., M.C.R., V.E.B., and C.T.M. interpreted results of experiments; N.F. and M.C.R. prepared figures; N.F. and M.C.R. drafted manuscript; N.F., V.E.B., and C.T.M. edited and revised manuscript; N.F., M.C.R., V.E.B., and C.T.M. approved final version of manuscript; M.C.R. and C.T.M. conception and design of research.
ACKNOWLEDGMENTS
We sincerely acknowledge the volunteer subjects in this study.
REFERENCES
- 1. Alster P, Wennmalm A. Effect of nicotine on prostacyclin formation in rat aorta. Eur J Pharmacol 86: 441–446, 1983 [DOI] [PubMed] [Google Scholar]
- 2. Ambrose JA, Barua RS. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol 43: 1731–1737, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Andersson DA, Zygmunt PM, Movahed P, Andersson TL, Hogestatt ED. Effects of inhibitors of small- and intermediate-conductance calcium-activated potassium channels, inwardly-rectifying potassium channels and Na+/K+ ATPase on EDHF relaxations in the rat hepatic artery. Br J Pharmacol 129: 1490–1496, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brunt VE, Minson CT. KCa channels and epoxyeicosatrienoic acids: major contributors to thermal hyperaemia in human skin. J Physiol 590: 3523–3534, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bull HA, Pittilo RM, Woolf N, Machin SJ. The effect of nicotine on human endothelial cell release of prostaglandins and ultrastructure. Br J Exp Pathol 69: 413–421, 1988 [PMC free article] [PubMed] [Google Scholar]
- 6. Dalla Vecchia L, Palombo C, Ciardetti M, Porta A, Milani O, Kozakova M, Lucini D, Pagani M. Contrasting effects of acute and chronic cigarette smoking on skin microcirculation in young healthy subjects. J Hypertens 22: 129–135, 2004 [DOI] [PubMed] [Google Scholar]
- 7. Edvinsson ML, Andersson SE, Xu CB, Edvinsson L. Cigarette smoking leads to reduced relaxant responses of the cutaneous microcirculation. Vasc Health Risk Manag 4: 699–704, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ezzati M, Lopez AD. Estimates of global mortality attributable to smoking in 2000. Lancet 362: 847–852, 2003 [DOI] [PubMed] [Google Scholar]
- 9. Garry A, Sigaudo-Roussel D, Merzeau S, Dumont O, Saumet JL, Fromy B. Cellular mechanisms underlying cutaneous pressure-induced vasodilation: in vivo involvement of potassium channels. Am J Physiol Heart Circ Physiol 289: H174–H180, 2005 [DOI] [PubMed] [Google Scholar]
- 10. Gerzanich V, Zhang F, West GA, Simard JM. Chronic nicotine alters NO signaling of Ca2+ channels in cerebral arterioles. Circ Res 88: 359–365, 2001 [DOI] [PubMed] [Google Scholar]
- 11. Gilbert P, Tremblay J, Thorin E. Endothelium-derived endothelin-1 reduces cerebral artery sensitivity to nitric oxide by a protein kinase C-independent pathway. Stroke 32: 2351–2355, 2001 [DOI] [PubMed] [Google Scholar]
- 12. Gordon AY, Salzman P. Optimality of the Holm procedure among general step-down multiple testing procedures. Stat Probab Lett 78: 1878–1884, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goto K, Kasuya Y, Matsuki N, Takuwa Y, Kurihara H, Ishikawa T, Kimura S, Yanagisawa M, Masaki T. Endothelin activates the dihydropyridine-sensitive, voltage-dependent Ca2+ channel in vascular smooth muscle. Proc Natl Acad Sci USA 86: 3915–3918, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heitzer T, Brockhoff C, Mayer B, Warnholtz A, Mollnau H, Henne S, Meinertz T, Munzel T. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ Res 86: E36–E41, 2000 [DOI] [PubMed] [Google Scholar]
- 15. Holowatz LA, Houghton BL, Wong BJ, Wilkins BW, Harding AW, Kenney WL, Minson CT. Nitric oxide and attenuated reflex cutaneous vasodilation in aged skin. Am J Physiol Heart Circ Physiol 284: H1662–H1667, 2003 [DOI] [PubMed] [Google Scholar]
- 16. Holowatz LA, Kenney WL. Local ascorbate administration augments NO- and non-NO-dependent reflex cutaneous vasodilation in hypertensive humans. Am J Physiol Heart Circ Physiol 293: H1090–H1096, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Holowatz LA, Thompson CS, Minson CT, Kenney WL. Mechanisms of acetylcholine-mediated vasodilatation in young and aged human skin. J Physiol 563: 965–973, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ijzerman RG, Serne EH, van Weissenbruch MM, de Jongh RT, Stehouwer CD. Cigarette smoking is associated with an acute impairment of microvascular function in humans. Clin Sci (Lond) 104: 247–252, 2003 [DOI] [PubMed] [Google Scholar]
- 19. Jeremy JY, Mikhailidis DP, Dandona P. Cigarette smoke extracts, but not nicotine, inhibit prostacyclin (PGI2) synthesis in human, rabbit and rat vascular tissue. Prostaglandins Leukot Med 19: 261–270, 1985 [DOI] [PubMed] [Google Scholar]
- 20. Kellogg DL, Jr, Liu Y, Kosiba IF, O'Donnell D. Role of nitric oxide in the vascular effects of local warming of the skin in humans. J Appl Physiol 86: 1185–1190, 1999 [DOI] [PubMed] [Google Scholar]
- 21. Kellogg DL, Zhao JL, Coey U, Green JV. Acetylcholine-induced vasodilation is mediated by nitric oxide and prostaglandins in human skin. J Appl Physiol 98: 629–632, 2005 [DOI] [PubMed] [Google Scholar]
- 22. Lenasi H. The role of nitric oxide- and prostacyclin-independent vasodilatation in the human cutaneous microcirculation: effect of cytochrome P450 2C9 inhibition. Clin Physiol Funct Imaging 29: 263–270, 2009 [DOI] [PubMed] [Google Scholar]
- 23. Lenasi H, Strucl M. The effect of nitric oxide synthase and cyclooxygenase inhibition on cutaneous microvascular reactivity. Eur J Appl Physiol 103: 719–726, 2008 [DOI] [PubMed] [Google Scholar]
- 24. Lorenzo S, Minson CT. Human cutaneous reactive hyperaemia: role of BKCa channels and sensory nerves. J Physiol 585: 295–303, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCord GR, Cracowski JL, Minson CT. Prostanoids contribute to cutaneous active vasodilation in humans. Am J Physiol Regul Integr Comp Physiol 291: R596–R602, 2006 [DOI] [PubMed] [Google Scholar]
- 26. Medow MS, Glover JL, Stewart JM. Nitric oxide and prostaglandin inhibition during acetylcholine-mediated cutaneous vasodilation in humans. Microcirculation 15: 569–579, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minson CT. Thermal provocation to evaluate microvascular reactivity in human skin. J Appl Physiol 109: 1239–1246, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Minson CT, Berry LT, Joyner MJ. Nitric oxide and neurally mediated regulation of skin blood flow during local heating. J Appl Physiol 91: 1619–1626, 2001 [DOI] [PubMed] [Google Scholar]
- 29. Minson CT, Holowatz LA, Wong BJ, Kenney WL, Wilkins BW. Decreased nitric oxide- and axon reflex-mediated cutaneous vasodilation with age during local heating. J Appl Physiol 93: 1644–1649, 2002 [DOI] [PubMed] [Google Scholar]
- 30. Mori A, Suzuki S, Sakamoto K, Nakahara T, Ishii K. Role of calcium-activated potassium channels in acetylcholine-induced vasodilation of rat retinal arterioles in vivo. Naunyn Schmiedebergs Arch Pharmacol 383: 27–34, 2011 [DOI] [PubMed] [Google Scholar]
- 31. Morita A. Tobacco smoke causes premature skin aging. J Dermatol Sci 48: 169–175, 2007 [DOI] [PubMed] [Google Scholar]
- 32. Nadler JL, Velasco JS, Horton R. Cigarette smoking inhibits prostacyclin formation. Lancet 1: 1248–1250, 1983 [DOI] [PubMed] [Google Scholar]
- 33. Noma K, Goto C, Nishioka K, Hara K, Kimura M, Umemura T, Jitsuiki D, Nakagawa K, Oshima T, Chayama K, Yoshizumi M, Higashi Y. Smoking, endothelial function, and Rho-kinase in humans. Arterioscler Thromb Vasc Biol 25: 2630–2635, 2005 [DOI] [PubMed] [Google Scholar]
- 34. Orem A, Orem C, Alioglu Y, Vanizor B, Erdol C. Effect of coronary angiography on plasma endothelin-1 and nitric oxide concentrations. Angiology 52: 231–235, 2001 [DOI] [PubMed] [Google Scholar]
- 35. Pellaton C, Kubli S, Feihl F, Waeber B. Blunted vasodilatory responses in the cutaneous microcirculation of cigarette smokers. Am Heart J 144: 269–274, 2002 [DOI] [PubMed] [Google Scholar]
- 36. Peluffo G, Calcerrada P, Piacenza L, Pizzano N, Radi R. Superoxide-mediated inactivation of nitric oxide and peroxynitrite formation by tobacco smoke in vascular endothelium: studies in cultured cells and smokers. Am J Physiol Heart Circ Physiol 296: H1781–H1792, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pittilo RM, Mackie IJ, Rowles PM, Machin SJ, Woolf N. Effects of cigarette smoking on the ultrastructure of rat thoracic aorta and its ability to produce prostacyclin. Thromb Haemost 48: 173–176, 1982 [PubMed] [Google Scholar]
- 38. Pryor WA, Stone K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann NY Acad Sci 686: 12–27, 1993 [DOI] [PubMed] [Google Scholar]
- 39. Rahman MM, Laher I. Structural and functional alteration of blood vessels caused by cigarette smoking: an overview of molecular mechanisms. Curr Vasc Pharmacol 5: 276–292, 2007 [DOI] [PubMed] [Google Scholar]
- 40. Raij L, DeMaster EG, Jaimes EA. Cigarette smoke-induced endothelium dysfunction: role of superoxide anion. J Hypertens 19: 891–897, 2001 [DOI] [PubMed] [Google Scholar]
- 41. Rubanyi GM. Endothelium-derived relaxing and contracting factors. J Cell Biochem 46: 27–36, 1991 [DOI] [PubMed] [Google Scholar]
- 42. Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P. Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA 90: 7240–7244, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shi Y, Ku DD, Man RY, Vanhoutte PM. Augmented endothelium-derived hyperpolarizing factor-mediated relaxations attenuate endothelial dysfunction in femoral and mesenteric, but not in carotid arteries from type I diabetic rats. J Pharmacol Exp Ther 318: 276–281, 2006 [DOI] [PubMed] [Google Scholar]
- 44. Smith CJ, Santhanam L, Bruning RS, Stanhewicz A, Berkowitz DE, Holowatz LA. Upregulation of inducible nitric oxide synthase contributes to attenuated cutaneous vasodilation in essential hypertensive humans. Hypertension 58: 935–942, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stewart J, Kohen A, Brouder D, Rahim F, Adler S, Garrick R, Goligorsky MS. Noninvasive interrogation of microvasculature for signs of endothelial dysfunction in patients with chronic renal failure. Am J Physiol Heart Circ Physiol 287: H2687–H2696, 2004 [DOI] [PubMed] [Google Scholar]
- 46. Stewart JM, Ocon AJ, Clarke D, Taneja I, Medow MS. Defects in cutaneous angiotensin-converting enzyme 2 and angiotensin-(1–7) production in postural tachycardia syndrome. Hypertension 53: 767–774, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Takase B, Hamabe A, Satomura K, Akima T, Uehata A, Matsui T, Ohsuzu F, Ishihara M, Kurita A. Comparable prognostic value of vasodilator response to acetylcholine in brachial and coronary arteries for predicting long-term cardiovascular events in suspected coronary artery disease. Circ J 70: 49–56, 2006 [DOI] [PubMed] [Google Scholar]
- 48. Teo KK, Ounpuu S, Hawken S, Pandey MR, Valentin V, Hunt D, Diaz R, Rashed W, Freeman R, Jiang LX, Zhang XF, Yusuf S. Tobacco use and risk of myocardial infarction in 52 countries in the INTERHEART study: a case-control study. Lancet 368: 647–658, 2006 [DOI] [PubMed] [Google Scholar]
- 49. Theken KN, Schuck RN, Edin ML, Tran B, Ellis K, Bass A, Lih FB, Tomer KB, Poloyac SM, Wu MC, Hinderliter AL, Zeldin DC, Stouffer GA, Lee CR. Evaluation of cytochrome P450-derived eicosanoids in humans with stable atherosclerotic cardiovascular disease. Atherosclerosis 222: 530–536, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. World Health Organization. Global status report on noncommunicable diseases 2010. Description of the global burden of NCDs, their risk factors and determinants. Geneva, Switzerland, WHO, 2011 [Google Scholar]
- 51. Xu CB, Stavenow L, Pessah-Rasmussen H. Interactions between cultured bovine arterial endothelial and smooth muscle cells; effects of modulated low density lipoproteins on cell proliferation and prostacyclin release. Scand J Clin Lab Invest 54: 191–198, 1994 [DOI] [PubMed] [Google Scholar]
- 52. Yamato H, Sun JP, Churg A, Wright JL. Cigarette smoke-induced emphysema in guinea pigs is associated with diffusely decreased capillary density and capillary narrowing. Lab Invest 75: 211–219, 1996 [PubMed] [Google Scholar]