

Abstract
Vascular endothelial growth factor receptor-2 (VEGFR2) is a receptor tyrosine kinase that is expressed in endothelial cells and regulates angiogenic signal transduction under both physiological and pathological conditions. VEGFR2 turnover at the plasma membrane (PM) is regulated by its transport through endocytic and secretory transport pathways. Short-range cargo trafficking along actin filaments is commonly regulated by motor proteins of myosin superfamily. In the current study, performed in primary human endothelial cells, we demonstrate that unconventional myosin 1c (Myo1c; class I family member) regulates the localization of VEGFR2 at the PM. We further demonstrate that the recruitment of VEGFR2 to the PM and its colocalization with Myo1c and caveolin-1 occur in response to VEGF-A (VEGF) stimulation. In addition, VEGF-induced delivery of VEGFR2 to the cell surface requires Myo1c; surface VEGFR2 levels are reduced in the absence of Myo1c and, more importantly, are restored by the overexpression of wild-type but not mutant Myo1c. Subcellular density gradient fractionation revealed that partitioning of VEGFR2 into caveolin-1- and Myo1c-enriched membrane fractions is dependent on VEGF stimulation. Myo1c depletion resulted in increased VEGF-induced VEGFR2 transport to the lysosomes for degradation and was rescued by applying either brefeldin A, which blocks trafficking between the endoplasmic reticulum and the Golgi complex, or dynasore, an inhibitor of dynamin-mediated endocytosis. Myo1c depletion also reduced VEGF-induced VEGFR2 phosphorylation at Y1175 and phosphorylation-dependent activation of ERK1/2 and c-Src kinase, leading to reduced cell proliferation and cell migration. This is the first report demonstrating that Myo1c is an important mediator of VEGF-induced VEGFR2 delivery to the cell surface and plays a role in angiogenic signaling.
Keywords: endothelial cells, signaling, vascular endothelial growth factor receptor, Myo1c, caveolin-1
vascular endothelial growth factor receptor family members 2 (VEGFR2) and 1 (VEGFR1) are expressed primarily in the vascular endothelium. Binding of VEGF to VEGFR2 initiates signaling events that regulate diverse cellular processes such as proliferation, migration, survival, chemotaxis, and vascular permeability, which are the hallmarks of angiogenesis (44). Molecules that regulate the trafficking of intracellular vesicles and the availability of VEGFR2 at the plasma membrane (PM) may, therefore, directly regulate angiogenic events (3, 9, 16, 19, 24, 27, 42).
When bound to the ligand VEGF, VEGFR2 internalizes, traffics to endosome, and transports either to lysosome for degradation or is recycled back to the PM (3, 9, 16). Recycling has been reported to occur via both Rab4, Rab11-independent and Rab4, Rab11-dependent mechanisms (3, 16). In addition, Arf6 has been implicated in recycling VEGFR2 to the PM (19). VEGFR2 turnover at the PM is also dependent on the delivery of the newly synthesized pool of receptor via the Golgi complex (27). Caveolae, which are highly abundant in smooth muscle cells, fibroblasts, adipocytes, and endothelial cells, are heterogeneous 10–200 nm sterol- and sphingolipid-enriched domains localized in membrane rafts and play an important role in cardiovascular signaling (15, 20, 36). Ultrastructural and biochemical studies have shown that VEGFR2 localizes to caveolae, and density gradient-based fractionation studies suggest that VEGFR2 is present in caveolin-1 (Cav1)-rich membrane rafts (11, 14, 23).
Myosins are actin-based motor proteins that are involved in actin-based motility and organelle trafficking. In eukaryotes, they are required for a number of diverse functions ranging from cytokinesis to muscle contraction (41). The primary structure of an unconventional myosin (class I) consists of head (motor) domain that binds ATP, neck (regulatory) domain, followed by class-specific tail domain. (35). The neck domain contains various IQ (isoleucine-glutamine) motifs that bind calmodulin (2) and regulate the motor activity. Unlike the conventional class II myosins, class I myosins are associated with cell membranes and leading edge of migrating cells, including filopodia, lamellipodia, and membrane ruffles (5). Members of the myosin I family have also been implicated in the regulation of dynamic cellular processes such as endosomal recycling, vesicle and organelle trafficking, and calcium channel activity (18, 30, 32, 34, 43).
Myosin 1c (Myo1c) is one of nine isoforms of the human myosin I gene (17). In motile cells, Myo1c is predominantly present at the leading edge, localizing to both the cell periphery and cell surface (6). Like other unconventional myosins, Myo1c interacts with actin (7). Myo1c is involved in the transport of vesicular cargo, including the trafficking of Glut-4 glucose transporter, epithelial Na+ channel, podocyte protein Neph1, and NF-κB essential modulator (1, 7, 10, 40).
Both Myo1c and syntaxin-6 [a Golgi- and endosome-localized N-ethylmaleimide-sensitive factor-attachment protein (SNAP) receptor protein (SNARE)] are involved in the delivery of membrane raft components to the PM in fibroblasts and epithelial cells (8, 12). Although studies of endothelial cells have shown that syntaxin-6 regulates post-Golgi trafficking and delivery of VEGFR2 to the PM, thereby regulating angiogenesis (27), the role of Myo1c in VEGFR2 trafficking has not yet been investigated. The objective of this study was to determine whether Myo1c is involved in the delivery of VEGFR2 to PM and, therefore, VEGF-mediated angiogenic signaling. Our results demonstrate that Myo1c plays a role in maintaining VEGFR2 localization at the PM in response to VEGF stimulation. We further demonstrate that Myo1c and VEGFR2 colocalize in Cav1-rich membrane rafts in a VEGF-dependent manner and in Myo1c-depleted cells, VEGFR2 is degraded and the VEGF-induced angiogenic signaling associated with endothelial cell proliferation and migration is disrupted.
MATERIALS AND METHODS
Reagents.
The rabbit monoclonal antibodies (mAbs) against human VEGFR2 (55B11) and phospho-VEGFR2 (Y1175) were purchased from Cell Signaling Technology (Danvers, MA). The rabbit polyclonal Ab against Myo1c (HPA001768), as well as cycloheximide (CHX), brefeldin A (BFA), and dynasore, were purchased from Sigma (St. Louis, MO). VEGF-A165 (VEGF) was purchased from R&D Systems (Minneapolis, MN). FuGENE 6 transfection reagent, protease inhibitor, and phosphatase inhibitor cocktail tablets were obtained from Roche Diagnostics (Indianapolis, IN). The mouse mAb against Cav1, collagen type I, and endothelial cell growth supplement were obtained from BD Biosciences (San Jose, CA). The horseradish peroxidase-labeled secondary Ab used for immunoblotting was obtained from Amersham Biosciences (Piscataway, NJ). Alexa Fluor-conjugated secondary Abs were procured from Invitrogen, Molecular Probes (Eugene, OR). Vectashield mounting medium was obtained from Vector Laboratories (Burlingame, CA). SuperSignal West Femto ECL reagent was obtained from Thermo Scientific (Rockford, IL). The mouse mAb against Myo1c (used in immunofluorescence assays), green fluorescent protein (GFP)-Myo1c, and mutant GFP-Myo1c plasmid constructs were kindly provided by Dr. Deepak Nihalani and has been previously described (1, 4, 39). All other reagents were purchased from Sigma unless stated otherwise.
Cell culture.
Primary human umbilical vein endothelial cells (HUVECs) were obtained from Lonza (Walkersville, MD) and cultured on collagen-coated plates in MCDB 131 containing 7.5% fetal bovine serum, 25 ng/ml endothelial cell growth supplement, 10 ng/ml epidermal growth factor, 2 mM l-glutamine, 1 μg/ml hydrocortisone, 50 μg/ml gentamicin, and 250 ng/ml fungizone. The cells were maintained at 37°C in a humid 95% air-5% CO2 atmosphere, split at a ratio of 1:3, and used only between passages 3 and 7. Human embryonic kidney cells expressing simian virus 40 large T antigen (293T cells) (ATCC) were maintained in Dulbecco's modified minimum essential medium containing 10% fetal bovine serum. Treatment of HUVECs with VEGF was carried out at a concentration of 50 ng/ml unless otherwise indicated. For treatment with BFA, cells stably expressing the Myo1c-targeted or control small hairpin RNA (shRNA) were treated with 1 μg/ml of BFA.
Generation of stable Myo1c knockdown HUVECs.
A plasmid containing shRNA against Myo1c was purchased from Sigma (Catalog number TRCN0000122927 NM_033375.3). Lentiviral particles were generated by cotransfecting the Myo1c shRNA-expressing plasmid, envelope plasmid (pMD2.G), and packaging plasmid (pCMV-dR8.74) into HEK-293T cells using Fugene HP transfection reagent (Roche). At 48 h posttransfection, lentivirus-containing supernatant was collected and filtered through a 0.45-μM syringe filter. Early passage HUVECs were transduced with lentiviral supernatant in the presence of 8 μg/ml polybrene (Sigma). Later (24 h), the medium was replaced, and the cell population stably expressing the shRNA was enriched via puromycin (1 μg/ml) selection for 3 days. Knockdown of the Myo1c protein was confirmed by immunoblotting.
Membrane raft preparation.
Raft-enriched membrane fractions were prepared by OptiPrep density gradient centrifugation as described in the study of Katoh et al. (22). Briefly, confluent HUVECs (grown in 10-cm dishes) were scraped and pelleted after washing with phosphate-buffered saline (PBS). The pellet was lysed for 30 min on ice in lysis buffer containing 25 mM Tris·HCl (pH 7.4), 125 mM NaCl, 12.5 mM EDTA, 1% Triton X-100, and inhibitor of protease and phosphatases, and all steps were performed at 4°C on ice. The lysate was adjusted to contain 40% OptiPrep by adding four volumes of 50% OptiPrep in the same lysis buffer and placed at the bottom of an ultracentrifuge tube. Optiprep (40%)-containing lysate (0.525 ml) was overlaid with 0.75 ml of 30% Optiprep in lysis buffer and 0.225 ml lysis buffer without OptiPrep, respectively, forming a 0–40% discontinuous OptiPrep gradient when centrifuged at 55,000 rpm for 2 h in a swinging bucket, S55S rotor (Beckman Instruments, Fullerton, CA). For analysis of the resulting gradient, 13 equal volume fractions of 100 μl each were carefully collected from the top of the gradient, and proteins were resolved by SDS-PAGE and analyzed by immunoblotting.
Cell-surface biotinylation.
To measure the pool of VEGFR2 at the PM, we covalently labeled cell-surface proteins with a membrane-impermeant biotinylation reagent (NHS-SS-biotin; Pierce, Rockford, IL) using methods previously described (27). All steps were performed at 4°C. Cells were washed three times in PBS and then incubated with 0.15 mg/ml sulfo-NHS-SS-biotin in PBS for 10 min. The unreacted biotinylation reagent was quenched by washing once with a buffer containing (in mM) 25 Tris (pH 8), 137 NaCl, 5 KCl, 2.3 CaCl2, 0.5 mM MgCl2, and 1 mM Na2 HPO4. The cells were then again washed three times with PBS and resuspended in radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail. The resulting lysates were centrifuged at 14,000 g for 10 min at 4°C. A sample taken from the supernatant at this point represented total cellular VEGFR2. Streptavidin-sepharose high-performance beads (GE Healthcare) were added to the remaining supernatant (100 μl packed beads per 500 μl lysate) and left on a rotator at 4°C for 2 h. The beads were collected by centrifugation at 14,000 g for 10 s at 4°C, and the supernatant was removed. The beads were then washed three times in lysis buffer at 4°C, and protein was extracted from the beads by heating at 95°C in SDS-PAGE sample buffer.
Immunoblotting.
Cultured cells were washed twice with ice-cold PBS and lysed in radioimmunoprecipitation assay buffer supplemented with inhibitors of proteases and phosphatases for 30 min on ice and centrifuged at 13,000 g for 20 min before supernatants were collected. The protein concentration was estimated using the Bio-Rad DC Protein Assay kit (Bio-Rad). Proteins (10–25 μg) per lane were resolved by SDS-PAGE and transferred to a polyvinylidene difluoride membrane. After blocking with 5% (wt/vol) nonfat dried skimmed milk powder in TBS plus Tween-20 buffer, the membrane was incubated with the appropriate primary Abs at 4°C overnight and then with secondary Ab for 1 h at room temperature (RT). Ab binding was visualized by developing the blot with enhanced chemiluminescence reagent. The bands were visualized using ChemiDocIt (UVP) and then analyzed using ImageJ 1.42q software (http://rsbweb.nih.gov/ij/).
Immunofluorescence assay and image analysis.
Microscopy studies were performed as previously described (27). Briefly, cells grown on acid-washed glass coverslips were fixed with 4% paraformaldehyde in Dulbecco's modified PBS for 25 min at RT, after which the reaction was quenched by adding 100 mM glycine in PBS and incubating for a further 15 min at RT. The cells were then permeabilized with 0.1% Triton X-100 in PBS for 2 min at RT, blocked with PBS containing 5% glycine and 5% normal goat or donkey serum for 60 min, and incubated with primary Abs overnight at 4°C. Coverslips were incubated for 1 h in a 1:200 dilution of either Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary A and mounted using Vectashield mounting medium containing 4,6-diamidino-2-phenylindole. Fluorescence images were acquired using Zeiss LSM 700-inverted confocal microscope (Carl Zeiss) with a Plan-Apochromat 63×/1.40 oil objective or Plan-Apochromat 40×/1.3 oil objective (Zeiss) in x, y-, and z-planes. Epifluorescence images were acquired using a Leica spinning disk confocal microscope equipped with a Hamamatsu EM-CCD digital camera (Hamamatsu Photonics). The acquired images were processed using Metamorph image processing software (Molecular Device, Downingtown, PA). For a given experiment, all photomicrographs were exposed and processed identically for each fluorophore. Background fluorescence correction was done using unlabeled specimens. For double and triple Ab labeling experiments, control samples were labeled identically with the individual fluorophores and exposed identically to the dual/triple-labeled samples at each wavelength to verify that there was no crossover between emission signals for different channels at the exposure settings used.
For quantitative colocalization studies, individual cells (overlaid image of 2 or 3 wavelengths) were mapped and pasted into a new image file. The individual images recorded following excitation at two to three different wavelengths were then separated, and a lower threshold was set individually for each image to ensure that background fluorescence was reduced to zero. The thresholded (background subtracted) images were then analyzed using the quantitative colocalization function of the Metamorph software. The total pixel intensity of the thresholded protein 1 that colocalized with protein 2 or protein 3 was quantified and displayed as either a percentage of the total pixel intensity of protein 1 staining over the entire cell or that of a selected region of interest (e.g., PM or perinuclear Golgi) within a cell. This method was used for 10 cells per condition from three separate experiments.
Real-time quantitative RT-PCR analysis for VEGFR2 mRNA expression.
Endothelial cells stably expressing either scrambled shRNA (scr-shRNA) or shMyo1c were used to isolated total RNA using a Qiagen RNeasy kit (Qiagen) and quantified. cDNA was synthesized from total RNA using the iScript cDNA synthesis kit (Bio-Rad). Thermocycler conditions included a 5-min incubation at 25°C, a 30-min incubation at 42°C, and a 5-min incubation at 85°C. The cDNAs were subjected to quantitative PCR analysis with the following 5′→3′ primers (sense and antisense, respectively): VEGFR2, 5′-CACCACTCAAACGCTGACTA-3′ and 5′-CACCACTCAAACGCTGACAT-3′ (amplicon length, 74 bp); and human 18S rRNA (housekeeping gene) CCTTGGATGTGGTAGCCGTTT and AACTTTCGATGGTAGTCGCCG (amplicon length, 105 bp). Primers were designed using Primer Express software (Applied Biosystems). The assay was performed in a 96-well optical plate with a final reaction volume of 20 μl, including synthesized cDNA (20 ng), oligonucleotide primers (100 μM each), and 2× SYBR Green/ROX PCR master mix (Bio-Rad). Samples were run on an ABI PRISM Sequence Detection System (model 7000, Applied Biosystems). PCR conditions were one cycle for 10 min at 95°C, followed by 30–45 cycles (15–25 cycles for 18S rRNA) of 30 s denaturation at 95°C, 1 min annealing at 55–60°C, and 1 min extension at 72°C. Samples were checked for nonspecific products or primer/dimer amplification by melting curve analysis. The threshold cycle (Ct) values for VEGFR2 and 18S in all of the samples (analyzed in triplicate) were normalized on the basis of the abundance of the 18S transcript. Relative expression is expressed as 2−dCt, where dCt = (cycle threshold for the soluble fms-like tyrosine kinase 1) − (cycle threshold for the 18S rRNA transcript).
Cell proliferation assay.
HUVECs (2 × 104) were seeded in 96-well plates. After 24 h, they were either left untreated or were infected with syntaxin 6-cyto or syntaxin 16-cyto. After infection for 6 h, the cells were serum (0.1%) starved for 6 h and then treated with VEGF at 50 ng/ml for 24 h. Proliferation was measured using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay according to the manufacturer's recommendations (Promega). The absorbance at 490 nm was determined using a Spectra Fluor PLUS (Molecular Devices, Sunnyvale, CA). For each group, samples were prepared in triplicate. The data are presented as the fold increase over control levels.
Cell migration assay.
Harvested endothelial cells (2 × 104) were replated in triplicate onto the 0.1% collagen-coated upper chamber of a Transwell filter with 8-μm pores (6.5 mm, polycarbonate, Costar), and the chamber was placed in serum-free medium for 6 h. Serum-free medium containing VEGF (100 ng/ml) was added to the lower chamber to stimulate chemotaxis. After 4–6 h, the cells were fixed with 4% paraformaldehyde in PBS. Cells on the upper side of the filter (those that had not migrated) were removed with a cotton swab, and cells on the underside of the filter were stained with 2% crystal violet in 10% ethanol. After the cells were extensively washed in water to remove excess crystal violet, the filters were dried overnight. The number of cells that had migrated was determined by eluting the crystal violet dye from the stained cells on the underside of the filter in 250 μl of 10% acetic acid (for 15 min), followed by optical density measurement 562 nm. In parallel, cells were separately plated to transwell filters without any serum in the lower chamber (in triplicate) to control for the total number of cells that were simply attached. Relative cell migration was determined by normalizing the number of cells that had migrated to the total number of the cells adhering to transwell filters.
Statistical analysis.
All values are expressed as means ± SD. Statistical significance was determined using a two-sided Student's t-test and GraphPad Prism software (GraphPad Software, version 4.0; San Diego, CA). Unless stated otherwise, a value of P < 0.05 was considered significant.
RESULTS
VEGFR2 colocalizes with Myo1c and Cav1 at the PM and cofractionates with Cav1-enriched membrane rafts.
To establish what role, if any, Myo1c plays in VEGFR2 trafficking, we first assessed the localization of these two proteins in primary endothelial cells. Immunolocalization of the endogenous proteins revealed that VEGFR2 colocalizes with Myo1c and Cav1 at the PM (Fig. 1A). Quantitative analysis revealed ≥75% colocalization with Myo1c and ≥35% colocalization with Cav1 of VEGFR2 at the cell periphery (Fig. 1B). These results indicate that under normal conditions, VEGFR2 associates with Myo1c and Cav1 at the PM, consistent with observations made by other investigators that VEGFR2 and Myo1c are both present in membrane rafts (8, 23). We further investigated whether VEGFR2, Myo1c, and Cav1 colocalize in membrane rafts. OptiPrep gradient fractionation was performed to identify membranes enriched for the raft-associated protein Cav1. Our results show that among 13 fractions collected from the top of the gradient, fractions 3 and 4 were enriched in Cav1. Approximately 30 to 40% of total VEGFR2 cofractionated with Myo1c in Cav1-positive membrane-raft fractions (Fig. 1, C and D). These finding are consistent with VEGFR2 and Myo1c colocalization and VEGFR2 and Cav1 colocalization at the PM (Fig. 1, A and B). These results strengthen the possibility that PM pool of VEGFR2 associates with Myo1c- and Cav1-rich domains.
Fig. 1.

Vascular endothelial growth factor receptor-2 (VEGFR2) colocalizes with myosin 1c (Myo1c) and caveolin-1 (Cav1) on the endothelial cell surface. Human umbilical vein endothelial cells (HUVECs) cultured in serum-containing complete medium were used. A: representative confocal microscopy image showing colocalization (arrows) between VEGFR2 and Myo1c, and VEGFR2 and Cav1 at the plasma membrane (PM). Scale bar represent 5 μm. DAPI, 4,6-diamidino-2-phenylindole. B: quantification of overlap in fluorescence signal between cell periphery-localized VEGFR2 and Myo1c or Cav1 from images as in A. Value represents mean ± SD; n = 50 cells for each condition from 5 separate experiments (P ≤ 0.05). C: Myo1c associates with VEGFR2 in membrane raft fractions. Cell homogenates were fractionated on a discontinuous OptiPrep gradient, and fractions collected were analyzed by immunoblotting using indicated Abs. Immunoblotting with the Cav1 Ab identified raft fraction (fractions 3 and 4 at the interface of 0 and 30% OptiPrep densities). D: percentage of total VEGFR2, Myo1c, and Cav1 in each fraction, based on quantification of density of bands in each fraction obtained by OptiPrep gradient centrifugation. The percentage represents mean ± SD for n = 3.
VEGF stimulation induces the recruitment of VEGFR2 to Myo1c and Cav1-containing membrane rafts.
VEGF-A is a ligand for VEGFR2 and is known to initiate internalization and transport of the latter to the late endosomes and lysosomes for degradation (9). In addition, VEGF stimulation mobilizes VEGFR2 from internal organelles such as the Golgi complex for subsequent delivery to the PM (27). We wanted to determine whether localization of VEGFR2 along with Myo1c and Cav1 to the PM from Golgi could be modulated by stimulating cells with VEGF. Therefore, serum-starved HUVECs were pretreated with CHX (to prevent new VEGFR2 synthesis and transport to the Golgi) and then with VEGF (30 min in the presence of CHX), and the localization of VEGFR2 at Golgi and PM was monitored. Immunofluorescence analysis showed that in serum-starved endothelial cells, the majority of VEGFR2 colocalized (≥80%) with Golgi marker trans-Golgi network 46 (TGN46) (Fig. 2, A and B); however, upon VEGF stimulation, the amount of VEGFR2 at the PM was even greater (Fig. 2, A and C), and its colocalization with Myo1c and Cav1 increased (to ≥75 and ≥35%, respectively, Fig. 2C). Localization of Myo1c to the PM and the extent of its colocalization with Cav1 did not alter in response to VEGF stimulation (Fig. 2, A and C). These results suggest that VEGF alters the subcellular localization of VEGFR2 and may recruit the receptor to Cav1-enriched regions at the PM.
Fig. 2.
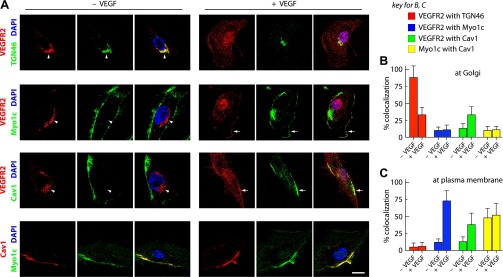
VEGF stimulates the exit of VEGFR2 from Golgi and its relocalization to Myo1c and Cav1-rich rafts at the PM. A–C: effects of VEGF treatment on VEGFR2 localization as previously described (27). Briefly, serum-starved HUVECs were treated with cycloheximide (10 μg/ml) and then cultured without or with VEGF for 30 min. A: confocal immunofluorescence imaging was used to determine VEGFR2, Myo1c, and Cav1 localization. Arrows and arrowheads show PM and Golgi locations, respectively. Scale bar represents 5 μm. B and C: quantification of the overlap in VEGFR2 signal with trans-Golgi network 46 (TGN46), Myo1c, and Cav1 at Golgi and PM. Values were calculated from images as in A using the quantitative colocalization function of Metamorph software as described in materials and methods. Myo1c-Cav1 overlap at the Golgi, and PM was also calculated. Values are expressed as percentages of colocalization of VEGFR2 with TGN46, Cav1, and Myo1c. Percentages represent means ± SD in n = 50 cells for each condition from 5 separate experiments. P ≤ 0.05.
To confirm and complement these findings, we used a density gradient fractionation approach analyzing the redistribution of VEGFR2 to Myo1c- and Cav1-containing membrane rafts upon VEGF stimulation. Cells were either serum starved or stimulated with VEGF and then processed for fractionation using OptiPrep density gradient centrifugation. The analysis of collected fractions suggests that in the absence of VEGF, ∼80 to 90% of cell-associated VEGFR2 partitioned in endosome (EEA1 enriched) and Golgi fractions (TGN46 enriched), which lack Cav1 and Myo1c (Fig. 3, A and B). However, upon stimulation with VEGF-A (30 min), ≥50% of total VEGFR2 redistributed to Myo1c- and Cav1-containing PM (syntaxin 4 enriched) fractions (Fig. 3, C and D). These results indicate that VEGFR2 is enriched in Myo1c-containing PM raft fractions in response to VEGF stimulation.
Fig. 3.
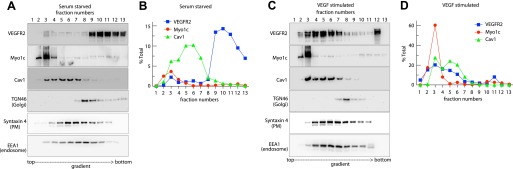
VEGF mobilizes the nonraft pool of VEGFR2 into Cav1-enriched membrane raft fractions. HUVECs cultured overnight in serum-depleted medium were either mock treated or treated with VEGF-A (50 ng/ml) for 30 min; cell homogenates were fractionated using OptiPrep gradient centrifugation and analyzed via immunoblotting using the indicated Abs. A and C: representative blots indicate the fractionation profiles of VEGFR2, Myo1c, and Cav1 with respect to markers for PM (syntaxin 4), endosomes (EEA1), Golgi (TGN46) in serum-starved cells and VEGF-stimulated cells, respectively. B and D: densitometric quantification of bands in each OptiPrep gradient fraction is represented as the percentage of total VEGFR2, Myo1c, or Cav1 in each fraction of serum-starved cells, either untreated or treated with VEGF. The percentage given is the mean ± SD for n = 3 experiments.
Myo1c depletion results in decreased cell-surface and total cellular VEGFR2 levels.
Since VEGF stimulation enhances partitioning of VEGFR2 into Myo1c-containing fractions, we investigated the possibility that Myo1c regulates the subcellular localization of VEGFR2. For this experiment, we transduced primary endothelial cells with lentivirus encoding a shRNA (shMyo1c) specific for human Myo1c. Cells stably expressing shMyo1c were selected using puromycin. In a similar fashion, we also generated cells stably expressing a scr-shRNA as a control. The shMyo1c stable cells showed 75–85% knockdown of Myo1c protein relative to controls (Fig. 4, A and B).
Fig. 4.

Depletion of Myo1c reduces cell-surface and total cellular level of VEGFR2. Puromycin-selected stable HUVECs after infection with recombinant lentiviruses expressing control scrambled small hairpin RNA (scr-shRNA) or shRNA against human Myo1c (shMyo1c). A: total cell lysates were analyzed with the indicated Abs by immunoblotting to assess the levels of Myo1c and VEGFR2. Representative blots are shown. B: Myo1c and VEGFR2 band densities from A and C were quantified; values represent relative levels of Myo1c and VEGFR2 after normalization to an arbitrary value of 100% set at the level in control shRNA cells. The percentage represents the mean ± SD for n = 3. C: biotinylation-based analysis of cell-surface VEGFR2. Serum-starved or VEGF-treated scr-shRNA or shMyo1c-expressing cells were used to label surface proteins with the biotinylation reagent sulfo-NHS-SS-biotin. Cell-surface biotinylated proteins were pulled down with streptavidin-Sepharose, and 5% of the total cell lysate and biotinylated cell-surface protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis followed by Western blot analysis with antibody against VEGFR2. D: quantification of band density for the cell-surface and total VEGFR2. Percentage is expressed as the change in VEGFR2 [relative to scr-shRNA-expressing cells without VEGF treatment (−VEGF)]. The percentage represents mean ± SD for n = 3 and P ≤ 0.03. E: stable endothelial lines expressing shRNAs were cultured in complete medium containing serum before levels of the VEGFR2 mRNA were analyzed. Quantitative RT-PCR analysis showing VEGFR2 transcript levels in shMyo1c relative to the control shRNA-expressing stable line. Data are expressed as means ± SE; n = 3. F: overexpression of green fluorescent protein (GFP)-Myo1c but not mutant GFP-Myo1c restores VEGFR2 levels in Myo1c-depleted cells in response to VEGF. scr-shRNA or shMyo1c expressing stable lines were either mock transfected or transfected with Myo1c-GFP or mutant Myo1c-GFP [(Myo1c 690-1028), lacking both ATP and actin-binding domains, dominant negative form (1)]. After 36 h of transfection, cells were serum starved for 16 h, followed by VEGF treatment (30 min). Immunolocalization of VEGFR2 and Myo1c was performed by epifluorescence microscopy. Fluorescence images of representative field are shown. Scale bar represents 5 μm. G: quantitation of relative levels of VEGFR2 at the PM. VEGFR2 antibody staining along the cell edges and total cell associated was quantified by Metamorph image analysis. Values represent relative changes in the levels of VEGFR2 at the cell periphery (calculated as %total cell-associated VEGFR2). Results are expressed as means ± SE; n = 70 cells for each condition from 3 separate experiments and P ≤ 0.05.
We used these Myo1c knockdown and control cell lines to examine the role of Myo1c in localizing VEGFR2 at the cell surface and its effect on total cellular level of VEGFR2. Cell-surface biotinylation revealed a ≥70% reduction at the cell surface and a ≥75% reduction in the total pool of VEGFR2 in Myo1c-depleted cells relative to controls following stimulation with VEGF (Fig. 4, C and D). The intracellular localization of VEGFR2 was significantly altered in the shMyo1c-expressing stable line compared with control, as the VEGFR2 levels at PM and its perinuclear accumulation were reduced (Fig. 4F). To understand the mechanism by which intracellular VEGFR2 levels are reduced in Myo1c-depleted cells, we first analyzed the expression of VEGFR2 mRNA in scr-shRNA and shMyo1c-expressing stable lines by quantitative real-time RT-PCR. Expression levels were similar in shMyo1c and scr-shRNA stable lines (Fig. 4E), suggesting that the decrease in the levels of VEGFR2 in the shMyo1c stable line relative to controls was not due to reduced mRNA expression.
To distinguish between the possibilities that Myo1c is required for VEGF-induced delivery or maintenance of VEGFR2 at the cell surface, we overexpressed GFP-tagged wild-type murine Myo1c (GFP-Myo1c) and mutant GFP-Myo1c (dominant negative form that lacks ATP and actin-binding domains) in shMyo1c stable cells. GFP-Myo1c overexpression in these cells rescued cell-surface VEGFR2, resulting in a fivefold increase relative to that in Myo1c-depleted cells in response to VEGF stimulation (Fig. 4, F and G). However, overexpression of mutant GFP-Myo1c failed to rescue VEGFR2 at the cell surface in Myo1c-depleted cells. Our data also show that overexpression of mutant GFP-Myo1c reduces PM and total VEGFR2 levels in control (scr-shRNA), an effect similar to that observed upon Myo1c depletion (Fig. 4, F and G). These results suggest that functional Myo1c is essential for maintaining normal cellular levels of VEGFR2.
VEGFR2 level in Myo1c-depleted cells is restored by treatment with either BFA, dynasore, or chloroquine.
To determine the mechanism by which loss of Myo1c alters cellular distribution of VEGFR2 in response to VEGF, we inhibited endoplasmic reticulum (ER) -to-Golgi transport by subjecting cells to treatment with BFA for 2 h (that results in resorption of the Golgi complex into ER, Fig. 5A) (21, 27). We then assessed the intracellular distribution of VEGFR2 in the presence or absence of VEGF in cell lines stably expressing either scr-shRNA or shMyo1c by immunofluorescence imaging. Myo1c-depleted cells showed a decrease in intracellular VEGFR2 levels only in response to VEGF stimulation (Fig. 5, B and C). In response to VEGF, BFA-treated shMyo1c-expressing cells showed increased intracellular accumulation of VEGFR2 (relative to that in mock-treated cells) within punctate vesicular structures (Fig. 5, B and C). As expected, in the control group, BFA treatment resulted in the accumulation of VEGFR2 in perinuclear area and corresponding loss of prominent Golgi-like perinuclear staining (Fig. 5B). Quantitation revealed 75% increase in intracellular accumulation of VEGFR2 in BFA-treated, VEGF-exposed shMyo1c stable cells (Fig. 5C). The PM level of VEGFR2 was reduced in control and Myo1c-depleted cells after BFA treatment most likely because of a block in membrane trafficking between ER and Golgi complex (Fig. 5, A, B, and D).
Fig. 5.

Myo1c depletion targets post-Golgi and PM pool of VEGFR2 for degradation in response to VEGF stimulation. A: schematic diagram showing the direction of vesicular cargo transport along endocytic and secretory pathways, as well as the known intracellular sites of action of brefeldin A (BFA), dynasore, and proposed site where Myo1c may be involved in facilitating VEGFR2 delivery to the PM. ER, endoplasmic reticulum. B: stable cells were serum starved for 6 h. Cells were then either mock pretreated or pretreated with BFA (1 μg/ml, 2 h) or dynasore (80 μM in DMSO, 2 h) before being cultured in the absence or presence of VEGF (50 ng/ml, 30 min) in medium containing respective inhibitors. Cells were then fixed, permeabilized, and labeled with Abs specific for VEGFR2 and Myo1c, followed by incubation with the appropriate fluorescently tagged secondary Ab. Representative images obtained by epifluorescence microscopy show localization of VEGFR2. Scale bar represents 5 μm. C: quantification of intracellular retention of VEGFR2 upon BFA and dynasore treatments as in B. Total cell-associated fluorescence was quantified by image analysis. Values represent relative changes in the levels of VEGFR2 normalized to an arbitrary value of 100% for mock-treated control (scr-shRNA, -VEGF). Results are expressed as means ± SE; n = 70 cells for each condition from 3 separate experiments and P ≤ 0.03. D: in a similar study as in B, confocal imaging was performed to quantitatively access VEGFR2 levels at the PM. Fluorescence signal of VEGFR2 at cell periphery from the confocal images was quantified by Metamorph image analysis. Value represents mean ± SD; n = 30 cells for each condition from 3 separate experiments and P ≤ 0.05. E: Stable scr-shRNA or shMyo1c cells were serum starved for 6 h before treatment for an additional 6 h with or without chloroquine (100 μM). Pretreated samples were then incubated in the presence or absence of VEGF in medium with or without chloroquine. F: quantification of band density for total VEGFR2. Values are expressed as the percent changes in VEGFR2 [relative to scr-shRNA (−VEGF)]. The percentage represents mean ± SD for n = 3 and P ≤ 0.05.
We next investigated whether Myo1c depletion affects the targeting of endocytic VEGFR2 pool (PM-localized) for degradation. We used dynasore, a small-molecule reversible inhibitor of dynamin 1 and 2 to inhibit clathrin-dependent and -independent endocytic pathways (Fig. 5A) (26, 29). We expected that if Myo1c has a role in regulating VEGF-induced trafficking of endocytic VEGFR2, then dynasore treatment should trap VEGFR2 at the PM and prevent its degradation in Myo1c-depleted cells. Indeed, dynasore treatment increased the PM expression of VEGFR2 by ≥200% in controls and Myo1c-depleted cells in response to VEGF (Fig. 5, B and D). In addition, dynasore treatment also rescued intracellular upregulation of VEGFR2 levels in Myo1c-depleted cells exposed to VEGF; these receptors were trapped in the Golgi complex and at the PM (Fig. 5B). Quantitation of the images revealed a ≥75% VEGF-mediated increase in intracellular accumulation of VEGFR2 in dynasore-treated shMyo1c stable cells (Fig. 5C). Golgi accumulation of VEGFR2 upon dynasore treatment suggests that dynamin GTPase is required for VEGFR2 to exit the Golgi complex. These results indicate that blocking membrane transport from ER to Golgi complex or anterograde transport from Golgi prevents Myo1c depletion-induced VEGFR2 reduction following stimulation with VEGF. We previously showed that blocking syntaxin 6-regulated post-Golgi trafficking of VEGFR2 targets this receptor to the lysosomes for degradation (27). We next investigated whether the decreased VEGFR2 level in Myo1c-depleted cells is due to a block in VEGF-induced VEGFR2 delivery to the PM. We found that treatment with chloroquine [an inhibitor of lysosomal hydrolases that effectively blocks VEGFR2 degradation in lysosomes (27, 38)] rescued intracellular levels of VEGFR2 in Myo1c-depleted cells treated with VEGF (Fig. 5, E and F). Thus VEGF appears to stimulate delivery of the post-Golgi and endocytic pools of VEGFR2 to the PM via a Myo1c-dependent mechanism, and in the absence of Myo1c, VEGFR2 appears to move from PM to lysosomes where it is processed for degradation.
Myo1c is required for VEGF-induced endothelial cell proliferation and migration.
The functional significance of Myo1c depletion was first assessed in response to VEGF-induced endothelial cell proliferation. Endothelial cells stably expressing shMyo1c showed a ≥3-fold reduction in VEGF-induced cell proliferation relative to wild-type HUVECs and scr-shRNA-expressing cells (Fig. 6A). The effect of Myo1c depletion on endothelial cell migration was also determined. Specifically, directed cell migration in the presence or absence of VEGF was analyzed using the Boyden chamber assay. The stable endothelial line expressing shMyo1c showed a ≥2-fold reduction in migration to the lower chamber in response to VEGF relative to HUVECs and scr-shRNA-expressing cells (Fig. 6B). These results suggest that Myo1c is required for maintaining VEGFR2 at the cell surface where it can associate with VEGF and transmit cell proliferation and migration signals. VEGF-induced phosphorylation of VEGFR2 has been shown to regulate endothelial cell migration via Src activation and cell proliferation via mitogen-activated protein kinase/extracellular signal-regulated kinase-1/2 (ERK1/2) activation (28, 37). We found that in Myo1c knockdown cells, levels of phosphorylated VEGFR2 (pY1175), c-Src (pY419, active), and Erk1/2 (active) kinases were reduced (Fig. 6, C and D). These experiments indicate that Myo1c participates in the regulation of dynamic endothelial cell processes such as proliferation and migration by regulating the signaling events downstream of VEGFR2.
Fig. 6.

Depletion of Myo1c blocks VEGFR2 signaling associated with VEGF-induced cell proliferation and migration. A: stably selected endothelial cells expressing shRNAs were serum starved and then cultured in the presence or absence of VEGF. Cell proliferation was determined using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay. Values are expressed as fold changes relative to that in control shRNA expressing cells. B: directional migration of stable endothelial cells toward VEGF (100 ng/ml) by Boyden chamber assay in the presence or absence of VEGF in the lower well. The number of migrating cells assessed by crystal violet dye extraction was normalized to the number in control (−VEGF) cells. Data in A and B are means ± SD from 4 independent experiments. P ≤ 0.001 in A, and P ≤ 0.005 in B. C: stable endothelial cells expressing control shRNA or shMyo1c were cultured in serum-depleted medium and treated with VEGF-A (50 ng/ml) for the indicated times; total cell lysate was immunoblotted for VEGFR2, VEGFR2 phosphorylated at Y1175 (pVEGFR2), c-Src, c-Src phosphorylated at Y419 (c-Src pY419), ERK1/2, phosphorylated form of ERK1/2. Representative blots are shown (n = 3). D: band densities were quantified, and values represent relative levels of protein phosphorylation (at various time points of VEGF treatment) after normalization to arbitrary value of 100% for cells expressing a control scr-shRNA (n = 4).
DISCUSSION
VEGFR2 is expressed at the endothelial cell surface and regulates angiogenesis in response to VEGF. Binding of VEGF to VEGFR2 at the cell surface initiates signal transduction and alters receptor localization and its distribution in distinct domains within the PM. Membrane rafts located at the cell surface (caveolae) are enriched for Cav1. Previous biochemical and ultrastructural studies have demonstrated that VEGFR2 is present in both raft and nonraft regions of cell membranes. The motor protein Myo1c is a member of unconventional myosin family and has been shown to localize at the leading edge of motile cells (13, 33). Our analysis reveals that a significant fraction of total Myo1c colocalizes with Cav1 at the cell surface in primary human endothelial cells. We show that exposure of cells to VEGF-A increases the recruitment of VEGFR2 to Myo1c- and Cav1-rich cell-surface membrane rafts. Importantly, Myo1c depletion drastically reduces total cellular VEGFR2 concentration in response to VEGF treatment, but does not alter VEGFR2 mRNA levels. Given the importance of VEGFR2 in angiogenesis our study represents a key advancement in the study of angiogenic receptors regulation.
Cell-surface VEGFR2 exists in a dynamic state, and its levels are maintained by internalization, recycling, secretory transport, and delivery of receptor to the PM (3, 9, 16, 19, 24, 27, 42). In response to VEGF stimulation, endothelial cells mobilize VEGFR2 from the Golgi complex for subsequent transport to the PM (27), which most likely involves intermediate vesicular compartments. In both adipocytes and epithelial cells, Myo1c is involved in PM delivery of transmembrane cargo proteins such as NF-κB essential modulator, Glut-4, and Neph1 (1, 7, 33). We observed that in human endothelial cells, Myo1c and VEGFR2 colocalize at the PM and cofractionate with PM-rich domains by density gradient analysis. We further demonstrate that VEGFR2 colocalizes with Myo1c at PM in a VEGF-dependent fashion. In epithelial cells, Myo1c localizes to membrane rafts (8), which is in agreement with our data showing colocalization of Myo1c with Cav1 at the PM of endothelial cells. Previous subcellular fractionation of aortic endothelial cells had shown that VEGFR2 is partitioned into Cav1-enriched membrane raft fractions but that VEGFR2 is mobilized out of raft membranes following activation by VEGF (23). In contrast, our data suggest that VEGFR2 is redistributed from the nonraft to raft fractions in response to VEGF. Our results are supported by immunolocalization studies demonstrating that VEGFR2 is recruited to PM and colocalizes with Myo1c and Cav1 in response to VEGF. We also observed that a fraction of VEGFR2 in the perinuclear region colocalizes with TGN46 (indicating that it is present in the Golgi) and that VEGF-stimulation leads to reduced colocalization at these locations and increased colocalization with Myo1c and Cav1 at PM. Our results and previous studies by other investigators have prompted us to hypothesize that VEGF mobilizes inactive intracellular VEGFR2 from Golgi for delivery to the PM that is mediated through a Myo1c-dependent mechanism for subsequent receptor activation and signaling (11, 23).
Previous studies indicating the involvement of Myo1c in translocation of Glut-4 and Neph1 to the PM (1, 7) and ours demonstrating restoration of cell-surface VEGFR2 levels in Myo1c-depleted cells in which Myo1c is overexpressed support our hypothesize that Myo1c is required for the delivery of VEGFR2 to PM in endothelial cells. When cells are depleted of Myo1c, the PM pool of VEGFR2 is reduced in a VEGF-dependent manner as is the total intracellular pool of VEGFR2. Since our data indicate that Myo1c depletion altered neither expression of the VEGFR2 mRNA nor trafficking of the protein between the ER and Golgi but it did affect the delivery of receptor to the PM, we postulate that loss of Myo1c may lead to a transient accumulation of VEGFR2 destined for PM delivery in vesicles at the interface between actin cytoskeleton and PM [site at which Myo1c is thought to contribute to exocytic delivery of cargo to the PM (30)] and that it may eventually be rerouted for subsequent degradation in lysosomes. Notably, in a previous study we observed that blocking post-Golgi VEGFR2 trafficking by inhibiting syntaxin 6 function reroutes receptor from PM to lysosomes. Since Myo1c is known to regulate short-range transport events, it is likely that VEGF stimulation causes a pool of VEGFR2 to be recycled back to the PM shortly after endocytosis in a Myo1c-dependent manner. This is supported by the finding that blocking endocytosis prevented Myo1c depletion-induced VEGFR2 degradation. We previously showed that syntaxin 6 plays a role in the post-Golgi trafficking of VEGFR2 for subsequent transport via intermediate vesicular compartments along the secretory pathway before being delivered to the PM (27). Alternatively, Myo1c may play a role in the delivery of VEGF-responsive secretory post-Golgi pool of VEGFR2 to the PM. Therefore, our study indicates that Myo1c plays a critical role in rapid VEGF-induced VEGFR2 recruitment to the PM from endocytic and secretory pools and thus participates in the regulation of endothelial cell dynamics.
VEGF binding to VEGFR2 leads to phosphorylation of VEGFR2 and activation of MAPKs including ERK1/ERK2 to promote endothelial cell proliferation. VEGF-induced activation of VEGFR2 also stimulates Src phosphorylation in endothelial cells (31). Here we report for the first time that loss of Myo1c can antagonize both VEGF-induced endothelial cell proliferation and migration. Reduced proliferation in Myo1c-depleted cells is most likely due to decreased total and phosphorylated (Y1175) VEGFR2, which accounts for the reduced ERK1/ERK2 phosphorylation (activation). VEGF also induces the formation of a complex by VEGFR2, VEGFR-associated protein (also known as TSAD), and c-Src; this interaction has been suggested to regulate Src activation and endothelial cell migration (28). Reduced Src activation in Myo1c-depleted cells may be due to the absence of active VEGFR2. Myo1c has been shown to regulate the delivery of membrane-raft contents at the PM (8) and c-Src activation has been shown to require membrane rafts (25). Hence, Myo1c may regulate not only VEGFR2 trafficking but also c-Src activation. Given the limited information available on the role of unconventional myosins in VEGFR2 trafficking and angiogenesis, our study provides significant advancement in this field. This is the first study that demonstrates the role of Myo1c in regulating VEGF-induced angiogenic signal transduction by regulating VEGFR2 localization at the endothelial cell surface.
GRANTS
This work was supported in part by National Institutes of Health Grants R01-HL-089599 (to A. Choudhury) and R01-DK-087956 (to D. Nihalani).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.T., J.-J.J., and S.M.I. performed experiments; A.T., J.-J.J., S.M.I., and A.C. analyzed data; A.T., J.-J.J., S.M.I., and A.C. interpreted results of experiments; A.T., J.-J.J., and S.M.I. prepared figures; A.T., J.-J.J., S.M.I., and A.C. drafted manuscript; A.T., J.-J.J., S.M.I., D.N., and A.C. edited and revised manuscript; A.T., J.-J.J., S.M.I., D.N., and A.C. approved final version of manuscript; A.C. conception and design of research.
ACKNOWLEDGMENTS
We thank Dr. Christine Blaumueller for editorial contributions.
REFERENCES
- 1. Arif E, Wagner MC, Johnstone DB, Wong HN, George B, Pruthi PA, Lazzara MJ, Nihalani D. Motor protein Myo1c is a podocyte protein that facilitates the transport of slit diaphragm protein Neph1 to the podocyte membrane. Mol Cell Biol 31: 2134–2150, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bahler M, Rhoads A. Calmodulin signaling via the IQ motif. FEBS Lett 513: 107–113, 2002 [DOI] [PubMed] [Google Scholar]
- 3. Ballmer-Hofer K, Andersson AE, Ratcliffe LE, Berger P. Neuropilin-1 promotes VEGFR-2 trafficking through Rab11 vesicles thereby specifying signal output. Blood 118: 816–826, 2011 [DOI] [PubMed] [Google Scholar]
- 4. Barletta GM, Kovari IA, Verma RK, Kerjaschki D, Holzman LB. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J Biol Chem 278: 19266–19271, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Barylko B, Jung G, Albanesi JP. Structure, function, and regulation of myosin 1C. Acta Biochim Pol 52: 373–380, 2005 [PubMed] [Google Scholar]
- 6. Barylko B, Wagner MC, Reizes O, Albanesi JP. Purification and characterization of a mammalian myosin I. Proc Natl Acad Sci USA 89: 490–494, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bose A, Guilherme A, Robida SI, Nicoloro SM, Zhou QL, Jiang ZY, Pomerleau DP, Czech MP. Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature 420: 821–824, 2002 [DOI] [PubMed] [Google Scholar]
- 8. Brandstaetter H, Kendrick-Jones J, Buss F. Myo1c regulates lipid raft recycling to control cell spreading, migration and Salmonella invasion. J Cell Sci 125: 1991–2003, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bruns AF, Herbert SP, Odell AF, Jopling HM, Hooper NM, Zachary IC, Walker JH, Ponnambalam S. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 11: 161–174, 2010 [DOI] [PubMed] [Google Scholar]
- 10. Chen XW, Leto D, Chiang SH, Wang Q, Saltiel AR. Activation of RalA is required for insulin-stimulated Glut4 trafficking to the plasma membrane via the exocyst and the motor protein Myo1c. Dev Cell 13: 391–404, 2007 [DOI] [PubMed] [Google Scholar]
- 11. Cho CH, Lee CS, Chang M, Jang IH, Kim SJ, Hwang I, Ryu SH, Lee CO, Koh GY. Localization of VEGFR-2 and PLD2 in endothelial caveolae is involved in VEGF-induced phosphorylation of MEK and ERK. Am J Physiol Heart Circ Physiol 286: H1881–H1888, 2004 [DOI] [PubMed] [Google Scholar]
- 12. Choudhury A, Marks DL, Proctor KM, Gould GW, Pagano RE. Regulation of caveolar endocytosis by syntaxin 6-dependent delivery of membrane components to the cell surface. Nat Cell Biol 8: 317–328, 2006 [DOI] [PubMed] [Google Scholar]
- 13. Fan Y, Eswarappa SM, Hitomi M, Fox PL. Myo1c facilitates G-actin transport to the leading edge of migrating endothelial cells. J Cell Biol 198: 47–55, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Ultrastructural localization of platelet endothelial cell adhesion molecule (PECAM-1, CD31) in vascular endothelium. J Histochem Cytochem 52: 87–101, 2004 [DOI] [PubMed] [Google Scholar]
- 15. Frank PG, Woodman SE, Park DS, Lisanti MP. Caveolin, caveolae, and endothelial cell function. Arterioscler Thromb Vasc Biol 23: 1161–1168, 2003 [DOI] [PubMed] [Google Scholar]
- 16. Gampel A, Moss L, Jones MC, Brunton V, Norman JC, Mellor H. VEGF regulates the mobilization of VEGFR2/KDR from an intracellular endothelial storage compartment. Blood 108: 2624–2631, 2006 [DOI] [PubMed] [Google Scholar]
- 17. Gillespie PG, Cyr JL. Myosin-1c, the hair cell's adaptation motor. Annu Rev Physiol 66: 521–545, 2004 [DOI] [PubMed] [Google Scholar]
- 18. Holt JR, Gillespie SK, Provance DW, Shah K, Shokat KM, Corey DP, Mercer JA, Gillespie PG. A chemical-genetic strategy implicates myosin-1c in adaptation by hair cells. Cell 108: 371–381, 2002 [DOI] [PubMed] [Google Scholar]
- 19. Ikeda S, Ushio-Fukai M, Zuo L, Tojo T, Dikalov S, Patrushev NA, Alexander RW. Novel role of ARF6 in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res 96: 467–475, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Insel PA, Patel HH. Membrane rafts and caveolae in cardiovascular signaling. Curr Opin Nephrol Hypertens 18: 50–56, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jung JJ, Tiwari A, Inamdar SM, Thomas CP, Goel A, Choudhury A. Secretion of soluble vascular endothelial growth factor receptor 1 (sVEGFR1/sFlt1) requires Arf1, Arf6, and Rab11 GTPases. PLoS One 7: e44572, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katoh SY, Kamimoto T, Yamakawa D, Takakura N. Lipid rafts serve as signaling platforms for Tie2 receptor tyrosine kinase in vascular endothelial cells. Exp Cell Res 315: 2818–2823, 2009 [DOI] [PubMed] [Google Scholar]
- 23. Labrecque L, Royal I, Surprenant DS, Patterson C, Gingras D, Beliveau R. Regulation of vascular endothelial growth factor receptor-2 activity by caveolin-1 and plasma membrane cholesterol. Mol Biol Cell 14: 334–347, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev Cell 18: 713–724, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu S, Ouyang M, Seong J, Zhang J, Chien S, Wang Y. The spatiotemporal pattern of Src activation at lipid rafts revealed by diffusion-corrected FRET imaging. PLoS Comput Biol 4: e1000127, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10: 839–850, 2006 [DOI] [PubMed] [Google Scholar]
- 27. Manickam V, Tiwari A, Jung JJ, Bhattacharya R, Goel A, Mukhopadhyay D, Choudhury A. Regulation of vascular endothelial growth factor receptor 2 trafficking and angiogenesis by Golgi localized t-SNARE syntaxin 6. Blood 117: 1425–1435, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsumoto T, Bohman S, Dixelius J, Berge T, Dimberg A, Magnusson P, Wang L, Wikner C, Qi JH, Wernstedt C, Wu J, Bruheim S, Mugishima H, Mukhopadhyay D, Spurkland A, Claesson-Welsh L. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J 24: 2342–2353, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 8: 603–612, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McConnell RE, Tyska MJ. Leveraging the membrane-cytoskeleton interface with myosin-1. Trends Cell Biol 20: 418–426, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meyer RD, Dayanir V, Majnoun F, Rahimi N. The presence of a single tyrosine residue at the carboxyl domain of vascular endothelial growth factor receptor-2/FLK-1 regulates its autophosphorylation and activation of signaling molecules. J Biol Chem 277: 27081–27087, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Montes de Oca G, Lezama RA, Mondragon R, Castillo AM, Meza I. Myosin I interactions with actin filaments and trans-Golgi-derived vesicles in MDCK cell monolayers. Arch Med Res 28: 321–328, 1997 [PubMed] [Google Scholar]
- 33. Nakamori Y, Emoto M, Fukuda N, Taguchi A, Okuya S, Tajiri M, Miyagishi M, Taira K, Wada Y, Tanizawa Y. Myosin motor Myo1c and its receptor NEMO/IKK-gamma promote TNF-alpha-induced serine307 phosphorylation of IRS-1. J Cell Biol 173: 665–671, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neuhaus EM, Soldati T. A myosin I is involved in membrane recycling from early endosomes. J Cell Biol 150: 1013–1026, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oliver TN, Berg JS, Cheney RE. Tails of unconventional myosins. Cell Mol Life Sci 56: 243–257, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol 8: 185–194, 2007 [DOI] [PubMed] [Google Scholar]
- 37. Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci USA 102: 1076–1081, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith RM, Jarett L. Ultrastructural basis for chloroquine-induced increase in intracellular insulin in adipocytes: alteration of lysosomal function. Proc Natl Acad Sci USA 79: 7302–7306, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wagner MC, Barylko B, Albanesi JP. Tissue distribution and subcellular localization of mammalian myosin I. J Cell Biol 119: 163–170, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner MC, Blazer-Yost BL, Boyd-White J, Srirangam A, Pennington J, Bennett S. Expression of the unconventional myosin Myo1c alters sodium transport in M1 collecting duct cells. Am J Physiol Cell Physiol 289: C120–C129, 2005 [DOI] [PubMed] [Google Scholar]
- 41. Woolner S, Bement WM. Unconventional myosins acting unconventionally. Trends Cell Biol 19: 245–252, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu C, Agrawal S, Vasanji A, Drazba J, Sarkaria S, Xie J, Welch CM, Liu M, Anand-Apte B, Horowitz A. Rab13-dependent trafficking of RhoA is required for directional migration and angiogenesis. J Biol Chem 286: 23511–23520, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wu X, Jung G, Hammer JA., 3rd Functions of unconventional myosins. Curr Opin Cell Biol 12: 42–51, 2000 [DOI] [PubMed] [Google Scholar]
- 44. Yang S, Toy K, Ingle G, Zlot C, Williams PM, Fuh G, Li B, de Vos A, Gerritsen ME. Vascular endothelial growth factor-induced genes in human umbilical vein endothelial cells: relative roles of KDR and Flt-1 receptors. Arterioscler Thromb Vasc Biol 22: 1797–1803, 2002 [DOI] [PubMed] [Google Scholar]