

Abstract
This study investigates the impact of pressure overload on vascular changes after myocardial infarction (MI) in rats. To evaluate the effect of pressure overload, MI was induced in three groups: 1) left coronary artery ligation for 1 mo (MI-1m), 2) ischemia 30 min/reperfusion for 1 mo (I/R-1m), and 3) ischemia-reperfusion (I/R) was performed after pressure overload induced by aortic banding for 2 mo; 1 mo post-I/R, aortic constriction was released (Ab+I/R+DeAb). Heart function was assessed by echocardiography and in vivo hemodynamics. Resin casting and three-dimensional imaging with microcomputed tomography were used to characterize changes in coronary vasculature. TTC (triphenyltetrazohum chloride) staining and Masson's Trichrome were conducted in parallel experiments. In normal rats, MI induced by I/R and permanent occlusion was transmural or subendocardial. Occluded arterial branches vanished in MI-1m rats. A short residual tail was retained, distal to the occluded site in the ischemic area in I/R-1m hearts. Vascular pathological changes in transmural MI mostly occurred in ischemic areas and remote vasculature remained normal. In pressure overloaded rats, I/R injury induced a sub-MI in which ischemia was transmural, but myocardium in the involved area had survived. The ischemic arterial branches were preserved even though the capillaries were significantly diminished and the pathological changes were extended to remote areas, characterized by fibrosis, atrial thrombus, and pulmonary edema in the Ab+I/R+DeAb group. Pressure overload could increase vascular tolerance to I/R injury, but also trigger severe global ventricular fibrosis and results in atrial thrombus and pulmonary edema.
Keywords: myocardial infarction, congestive heart failure, left coronary artery, fibrosis, hypertension
cardiac ischemia usually can result from arterial atherosclerosis, which is characterized by neointimal formation and vessel narrowing. Atherosclerosis is mainly associated with chronic high blood pressure, in addition to metabolic disorders and genetic defects (22). Experimental atherosclerosis can be induced from inside (endothelial injury) and outside (cuff placement) in common carotid and femoral arteries (25, 39). In most animal models, coronary arterial ischemia is induced by temporal or permanent mechanical occlusion (9, 32). The degree of ischemia and injury area depends directly on arterial branch (level) and occlusion length (2, 24, 36). Occlusion of blood supply more than 30 min results in myocardial infarction (MI) distal from the ligation site in small animals. The onset of irreversible injury begins 20 to 30 min after occlusion. Later, restoration of blood supply could minimize the damage, but the extent of injury is often increased in acute studies (6). Multiple factors contribute to reperfusion injury including free radicals (19), leukocytes and inflammatory mediators (17), platelet activation, pro-apoptotic signaling cascade (18), endothelium damage, and vasoconstriction (33).
Many patients with ischemic heart disease have also a history of hypertension, increasing the risk of MI (13, 30). However, reduced diastolic blood pressure has also been associated with an increased incidence of MI. This might be due to reduced myocardial perfusion, which could increase thrombogenic tendency (15). However, the impact of left ventricular (LV) pressure on ischemia-reperfusion (I/R), especially on ischemic arterial branches and capillary, is unclear.
The myocardium is composed of three integrated components: myocytes, extracellular matrix, and capillary microcirculation that serves the contractile unit assembly. Heart failure is not simply a decrease of contractility but also a multiple organ dysfunction (28). After MI, remote area undergoes remodeling: ventricular dilation, hypertrophy, and collagen scar formation (35).
Myocyte hypertrophy, fibrosis, ventricular compliance, and coronary patency play distinct roles in different postinfarction phases (5, 7, 31, 34). MI in human patients is much more complicated than those in small animals because most MI patients have a long history of hypertension and atherosclerosis (1, 16, 29), although it is unclear how hypertension affects ischemic remodeling.
The objective of the present study is to investigate the impact of pressure overload on vascular pathological changes in transmural and sub-MI in congestive heart failure in rats. The ischemic vasculature was casted and characterized with digital imaging and microcomputed tomography to study the vascular tolerance to I/R injury and coronary vascular mechanisms associated with fibrosis, ventricular stiffness, and atrial and pulmonary pathology.
METHODS
All procedures were followed by the recommendations of the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and were approved by the Mount Sinai School of Medicine Animal Care and Use Committee.
Animal protocol.
MI was induced in all three groups studied: 1) left coronary artery ligation (LCA) ligation 1 mo (MI-1m); 2) ischemia 30 min/reperfusion 1 mo (I/R-1m) using male Sprague-Dawley rats of 350∼400 g. Subendocardial infarction was induced by loose ligation of LCA 30 min and reperfusion 24 h or 1 mo as special control; and 3) pressure overloaded MI was induced by aortic banding 2 mo plus I/R 1 mo followed by aortic debanding (Ab+I/R+DeAb) as previously described (9). Briefly, aortic banding (Ab) was performed in male Sprague-Dawley rats (180–200 g) by constricting the ascending aorta with a 4-0 suture against a polyethylene-50 tube through the right thoracotomy at the second intercostal space. Two months after Ab, rats underwent an LCA ligation for 30 min followed by reperfusion to induce I/R injury. One mo after I/R, the rats underwent a third thoracotomy at the upright side of the sternum. The roots of 2∼4 ribs were cut along the right side of sternum to expose aorta. The aortic banding suture was cut with microdissecting scissors and separated with microdissecting forceps. For all surgical procedures, anesthesia was induced by intraperitoneal administration of pentobarbital (60 mg/kg). Animals underwent intratracheal intubation and mechanical ventilation (9).
Echocardiography.
The animals were sedated by intraperitoneal injection of ketamine (40 mg/kg). Echocardiograms were performed as previously described (8, 9). LV ejection fraction and fractional shortening were obtained in M-mode.
Coronary artery resin casting.
Hearts were cast at the time of euthanization using Batson's No. 17 Plastic Replica and Corrosion Kit (cat. No. 07349; Polysciences). The manufacturer's protocol was followed (2, 24). Briefly, under ketamine anesthesia (90 mg/kg ip), the chest was opened, the right atrium was cut open, and red latex (30 ml) was infused into the LV after blood was flushed out with 60 ml of PBS (pH 7.4; RT) containing heparin (0.2 ml, 1.000 USP units/ml) with a 14-gauge catheter. Thirty minutes later, the heart and lungs were removed and washed in PBS. Thereafter, the heart was corroded in a 50-ml tube with 10 ml of Maceration Solution (No. 07359) at 50°C for 2 to 3 h and then room temperature for 24 to 48 h. After digestion, the cast was carefully washed in water and photographed. LCA diameter, length, and branch patterns were measured and recorded. LCA length loss was defined as loss segment (from dead end to apex)/[loss segment + residual LCA segment (from aortic root to dead end)} × 100.
Three-dimensional imaging of casted coronary artery.
CT images were obtained using a dual-head micro-SPECT/CT (Triumph SPECT/CT; Gamma Medica-Ideas, Northridge, CA) with an X-ray tube operating at 80 kVp and 0.6 mA, capturing 1,024 views in 360 degrees rotation at 3.5 zoom and with a pixel size of 70 μm. Images were reconstructed to obtain three-dimensional data sets. LCA diameter, length, and branch patterns were measured and recorded again.
TTC stain.
To distinguish sub-MI and subendocardial infarction, TTC (triphenyltetrazohum chloride) stain was conducted on acute I/R injury (I/R-24 h) hearts as previously described (8).
In vivo hemodynamics.
Left ventricular pressure and ejection fraction were acquired and analyzed as previously described (9).
Histology.
Hearts were harvested after the final hemodynamic measurements. The hearts were perfused with 30 ml of cold PBS with 0.1 ml of 1% heparin. The LV was cut from the root of the pulmonary artery to the ventricular apex and photographed. The samples were embedded in optimum cutting temperature and stored at −80°C. Masson's Trichrome was performed as previous described (10). Paraffined samples were used for immunohistochemistry of CD31 (ABCAM, ab28364, dilution 1:100). The staining procedures and confocal imaging have been described in detail elsewhere (11).
Statistics.
Variables are expressed as means ± SE. One-way ANOVA followed by Tukey's Multiple Comparison Test was performed to compare experimental groups. Correlation data was analyzed by regression, using GraphPad Prism software. P values <0.05 were considered statistically significant.
RESULTS
Patterns of LCA.
We used digital photo and three-dimensional CT imaging to characterize the left coronary arteries. Our data showed that every rat had a different and distinct coronary anatomy. Four patterns of LCA were identified and classified (Fig. 1). The MI size mainly depends on the length and size of injured branch, but not on the patterns (data not showed). Variation in MI size is inevitable due to the anatomic disparity. Only 24% of LCA are visible in vivo in rats (n = 46).
Fig. 1.
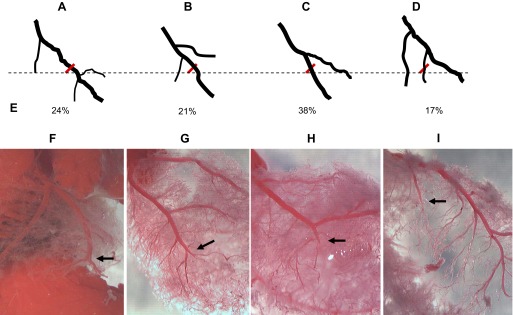
Patterns of ligation of left coronary artery (LCA). A–D: illustration of 4 main types of LCA ligation. E: incidence of different type, total animal number = 29. F–I: different position of ligation (arrow) on LCA branch leads to variation of myocardial infarction size.
Changes of vasculature in ischemic and remote myocardium.
The fate of an artery after occlusion depends on the ischemic length, ligated pressure, and reperfusion pressure. Ischemic segments disappeared, leaving a sharp end at the sutured site after permanent ligation. However, after reperfusion a short residual tail was retained, distal to the occluded site in I/R-injured LCA. High reperfusion pressure could significantly increase the tolerance of artery to ischemic, preserving the ischemic LCA branches, although capillary density was significantly decreased in pressure overloaded rats (aortic banding 2 mo plus I/R 1 mo followed by aortic debanding 1 mo, or Ab+I/R+DeAb; Fig. 2).
Fig. 2.

Vascular changes in ischemic area. A: normal LCA. B: LCA branch snags after ligation I mo (MI-1m). C: short bare branch without capillary in ischemia 30 min/reperfusion 1 mo (I/R-1m) heart. D: bare LCA branches with diminished capillaries distal to ligated suture in aortic banding plus ischemia/reperfusion plus de-banding (Ab+I/R+DeAb) rat. Arrow: ligated location.
Vascular responses were mostly limited within an ischemic region in MI-1m and I/R-1m animals, whereas remote areas remained mostly unaltered. In Ab+I/R+DeAb hearts, increased fibrosis in remote area was also observed (Fig. 3). Fibrotic-like component in remote myocardium of Ab+I/R+DeAb hearts had a cotton wool appearance, equally distributed from endocardium to epicardium and from vein to arteries, and it could not be digested by NaOH (Fig. 4). Micro CT three-dimensional images showed a better view of these global pathological changes (Supplement videos S1–S4).
Fig. 3.
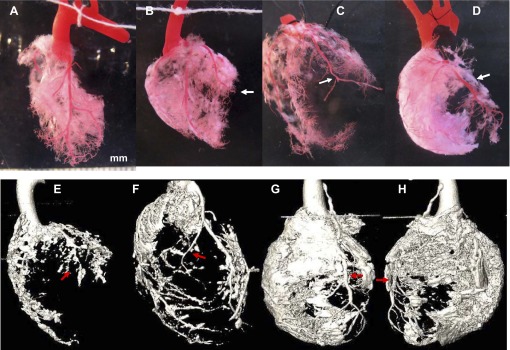
Fibrosis in remote/nonischemic myocardium. Top row: regular photos. A: control. B: LCA ligation 1 mo (MI-1m). C: ischemia/reperfusion injury 1 mo (I/R-1m) with residential vessel tail distal to ligated site. D: Ab+I/R+DeAb. Bottom row: micro CT images. E: MI-1m. F: I/R-1m. G: anterior view of Ab+I/R+DeAb. H: posterior view of Ab+I/R+DeAb. Arrow: ligated site.
Fig. 4.

Characteristics of capillary in ischemic and remote myocardium. A: capillaries of LCA in apex in control. ED, endocardium; EP, epicardium. B: capillaries in middle anterior wall in control rat. C: fibrous component and bare branches (BB) in aortic banding plus ischemia/reperfusion plus de-banding (Ab+I/R+DeAb) rat. IBZ, ischemic border zone. Arrow indicates narrowing segment. D: increase of fibrotic-like component in remote area in Ab+I/R+DeAb rat. V, vein; A, artery.
Cardiac dysfunction versus LCA ischemic injured length and remote area.
Aortic reperfusion pressure was significantly increased after aortic constriction as demonstrated in Fig. 5A. Echocardiography data showed cardiac dysfunction in MI-1m, I/R-1m, and Ab+I/R+DeAb rats (Table 1, and Fig. 5B). LV function (fractional shortening) inversely correlated with LCA injured length (loss length/LCA length in MI-1m and I/R-1m, ischemic length/LCA length in Ab+I/R+DeAb; Fig. 5C). Cast LCA weight-to-body weight ratio showed that global fibrosis was significantly increased in Ab+I/R+DeAb group (Fig. 5D).
Fig. 5.

Cardiac hemodynamics and coronary arterial injury. A: pressure-volume loop shows normal and overloaded pressure induced by aortic constriction. B: left ventricle (LV) fractional shortening of echocardiography in different groups. **P < 0.01 compared with control. C: correlation between LV fractional shortening and LCA injured length. Injured length = lost branch/LCA in MI-1m, I/R-1m and ischemic bare branch/LCA in Ab+I/R+DeAb hearts (R2 = 0.62, P < 0.01). D: ratio of casted coronary artery (CA) weight and body weight (BW) in different groups. **P < 0.01 compared with control, MI-1m; and P < 0.05 compared with I/R-1m.
Table 1.
Echocardiography on different groups
| Control | 1 mo LCA Ligation | 30 min Ischemia and 1 mo Reperfusion | 2 mo Aortic Banding plus 1 mo I/R, Followed by 1 mo Aortic Debanding | |
|---|---|---|---|---|
| N | 10 | 12 | 9 | 8 |
| Body weight, g | 455 ± 19 | 508 ± 25 | 485 ± 17 | 530 ± 12 |
| Heart rate, beats/min | 392 ± 15 | 378 ± 24 | 381 ± 14 | 380 ± 30 |
| Wall thickness, cm | ||||
| Diastolic anterior | 0.21 ± 0.01 | 0.16 ± 0.01** | 0.19 ± 0.01 | 0.21 ± 0.01 |
| Diastolic posterior | 0.23 ± 0.04 | 0.19 ± 0.02 | 0.24 ± 0.01 | 0.22 ± 0.01 |
| Systolic anterior | 0.35 ± 0.02 | 0.26 ± 0.02 | 0.30 ± 0.03 | 0.35 ± 0.02 |
| Systolic posterior | 0.40 ± 0.02 | 0.29 ± 0.02 | 0.36 ± 0.02 | 0.37 ± 0.01 |
| Volume, ml | ||||
| End diastolic (Teich) | 0.60 ± 0.03 | 1.19 ± 0.10** | 1.05 ± 0.13* | 1.20 ± 0.12** |
| End systolic | 0.02 ± 0.003 | 0.33 ± 0.07** | 0.25 ± 0.06* | 0.22 ± 0.05 |
| Ejection fraction, % | 96 ± 0.7 | 74 ± 3.45** | 78 ± 3.52** | 82 ± 3.49* |
Values are means ± SE. LCA, left coronary artery; I/R, ischemic-reperfusion.
P < 0.05;
P < 0.01 compared with control.
Cardiac histological changes.
We observed three types of myocardial infarction post-LCA ischemia with or without reperfusion: transmural MI, sub-MI, and subendocardial infarction. Transmural MI was observed in both I/R-1m and MI-1m groups. In transmural MI, LV wall became thinner as it could clearly be shown by echocardiogram; myocytes were lost and were mostly replaced by scar tissue (fibrin and connective adipose tissue). Furthermore, scattered isolated myocyte islands could also be seen, but it was apparent that this structural component on LV wall could not contract to form a force (Fig. 6, A–D). Sub-MI was observed in Ab+I/R+DeAb rats. In sub-MI, massive ischemic myocardium was preserved. MI could not be definitely diagnosed easily by echocardiogram. However, bright field images showed severe fibrosis in the ischemic myocardium. The apex was also thin, but there was some muscular sheet that seemed being able to contract (Fig. 6, E–H). Immunohistochemistry and fluorescent imaging data (Fig. 7, A–C) showed that small vessels in the infarcted area in MI-1m heart could probably be small veins, instead of small arteries or capillaries. Subendocardial infarction is not a common model in small animals. It could be induced by loosely ligation of LCA in rats. Ischemic necrosis in transmural MI involves the whole ventricular wall in I/R-24h heart (Fig. 7D). In contrast, subendocardial infarction was nontransmural and constituted an ischemic necrotic area, limited to one third or one half of the inner ventricular wall (Fig. 7, E and F). Nevertheless, fibrosis was not evident in nonischemic areas in subendocardial infarct heart (Fig. 7G).
Fig. 6.
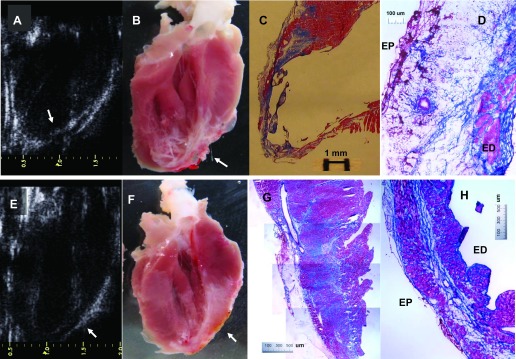
Transmural and submyocardial infarction. A and B: echocardiography and regular photo on transmural myocardial infarction (arrow). C and D: Masson's staining; ventricular wall is thinner and mostly replaced by scar tissue. E–H: submyocardial infarction (arrow). Left ventricular wall is thinner and fibrosis significantly increased, but muscle cells are mostly survived. ED, endocardium; EP, epicardium.
Fig. 7.
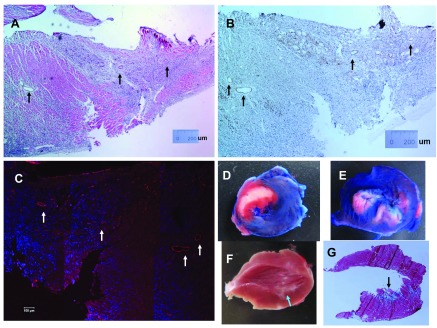
Small vessels in transmural myocardial infarction and subendocardial infarction. A: hematoxylin-eosin stain. B: small vessels in transmural myocardial infarction stained by CD31-DAB. C: fluorescent imaging of CD31 in MI-1m heart. Arrows indicate small vessels. D and E: triphenyltetrazolium chloride staining on transmural myocardial infarction (D) and subendocardial infarction (E) induced by ischemia 30 and reperfusion 24 h. Blue, normal area; white, infarct area; red, ischemic area. F and G: regular photo (F) and Masson's staining (G) on subendocardial infarction induced by ischemia 30 and reperfusion 1 mo. Arrows indicate subendocardial infarction area.
Evidence of congestive heart failure in the animal models.
Figure 8A shows MI and atrial thrombus in a heart after Ab+I/R+DeAb. Figure 8, B and C, showed thrombus in the opened atrium and fibrosis in atrial section. Figure 8, D and E, shows pulmonary edema: bronchiolar and alveolar wall thickness was increased; exudation, fibrin, and cellular infiltration were significantly increased in lung compared with control (Fig. 8F). Ventricular interstitial fibrosis, neointimal formation, and arterial narrowing in remote myocardium were also observed in the Ab+I/R+DeAb animals. Multiple organs were affected beyond the regional anatomical changes of coronary artery in the development of heart failure (Fig. 8, G and H).
Fig. 8.
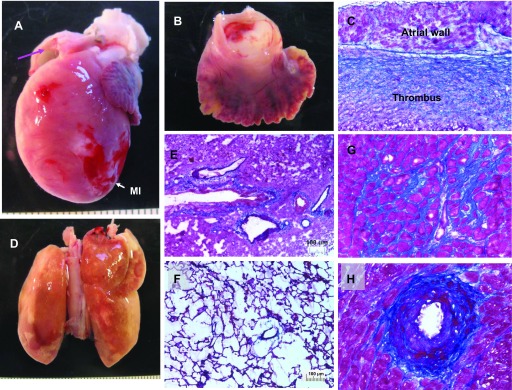
Macro perspective of congestive heart failure in one rat. A: heart with myocardial infarction (MI). B: atrial thrombus. C: Masson's staining on atrial section. Thrombus is in the left atrium. Blue indicates fibrosis. D: pulmonary edema. Lung became yellow. E: Masson's staining on the lung section. Bronchiole and alveolar wall thickness were increased. Exudation, fibrin, and cellular infiltration were significantly increased compared with lung from control (F). G: interstitial fibrosis in remote area. H: neointima and arterial narrowing in remote myocardium.
Comprehensive comparison of three types of MI.
Our data (Table 2) showed that the cardiovascular injury after arterial occlusion depends on several processes including the ischemic length, interruption degree, size of the involved vessels, and reperfusion pressure. Pressure overload hearts (Ab+I/R+DeAb) had higher resistance to I/R injury, but more damage was observed in remote areas and in other organs.
Table 2.
Comparison of transmural MI, sub-MI, and subendocardial infarction in rats
| Transmural MI | Sub-MI | Subendocardial MI | |
|---|---|---|---|
| Ischemic time, min | ≥30 | =30 | ≤30 |
| LCA casted, N | 18 | 8 | 3 |
| Ligated LCA | Major branch | Major branch | Small branch |
| Ischemic LCA | Not preserved | Preserved | Not preserved |
| TTC on I/R-24 h, N | 6 | 0 | 3 |
| Ischemic area | Transmural | Transmural | Nontransmural |
| Hemodynamic and histological study, N | 10 | 9 | 3 |
| LVPs at I/R, mmHg | 118 ± 4 | 227 ± 14** | 112 ± 12 |
| Ejection fraction, % | Decreased (41 ± 3) | Decreased (53 ± 4) | Normal (68 ± 7) |
| Ischemic myocardium | Infarction cross LV wall | Fibrosis cross ischemic LV wall | Inner 1/3 or 1/2 of LV wall |
| LV wall thinning | Severe (+++) | Moderate (++) | Little or no (+/−) |
| Fibrosis in non-ischemic area | Little or no (+/−) | Severe (+++) | No |
| Neointima in remote myocardium, incidence | 0 | 6/9 | 0 |
| Atrial thrombus | 0 | 4/9 | 0 |
| Pulmonary edema | 0 | 4/9 | 0 |
Values are means ± SE. MI, myocardial infarction; TTC, triphenyltetrazohum chloride staining; LVPs, left ventricular (LV) pressure, systolic.
P < 0.01 compared with transmural MI.
DISCUSSION
In the present study, we explored the impact of pressure overload on vascular pathological changes in transmural and sub-MI. We described vascular characteristics of transmural MI and sub-MI in different models of heart failure. Our data showed that LCA arterial branches were lost in transmural MI after silk suture ligation (≥30 min) in normal LV pressure rats. However, ischemic arterial branches were preserved, whereas capillaries diminished post-I/R injury in pressure overload rats (Ab+I/R+DeAb). High reperfusion pressure was able to increase tolerance of LCA to ischemia. Nevertheless, pressure overload also prompted cardiac adaptation, which resulted in severe fibrosis in the remote vasculature, leading to heart failure. The Ab+I/R+DeAb rats had not only low cardiac function but also atrial thrombus and pulmonary edema.
MI could be classified as transmural and nontransmural, which was traditionally labeled as subendocardial (27). Whether MI is transmural or subendocardial depends on the severity and length of the ischemia (26). The gravity of ischemic damage also depends on the size of the vessels involved and LV pressure (7, 21). Sub-MI was induced by I/R injury in pressure-overloaded rats. The morphological characteristics of sub-MI were that the ischemic area was transmural, but the majority of involved myocytes had survived; fibrosis was significantly increased in both ischemic area and remote myocardium (9). The leading cause of sub-MI was that the involved arterial branches were preserved in the transmural ischemia even when capillary density was significantly decreased.
The loss of myocytes and decrease in contractility is a dominant mechanism in the pathogenesis of congestive heart failure (23). The role of vasculature changes in CHF has not been fully investigated, especially in three-dimensional studies. There have been many studies inducing angiogenesis with gene or stem cell therapies to rescue the function of the ischemic heart (3, 14, 37). One of the obstacles to judge the efficacy of stem cells therapy on angiogenesis is the uncertain vessel characteristics in the infarct or border myocardium. Histological assessment of angiogenesis with CD31 or CD34 immunostaining could not specifically identify capillary, small artery, and vein based on diaminobenzidine and fluorescent imaging (4, 14, 20). According to current cast data, small vessels in infarcted area could probably be small veins (Fig. 7), which are usually running on the surface of the heart. Nevertheless, it is unknown the reason why most survived small vessels are located in the middle of the infarcted ventricular wall (Figs. 6 and 7).
It has been very difficult to get consistent MI size in a rat model by LCA ligation due to the inherent variability in anatomic structure (31), even though the LAC ligature position could be determined by anterior cardiac vein and adjusted by multiple loosely preocclusion in some cases. The dead end branch in transmural MI made this model almost irreversible. Scar tissue in the infarcted area had very limited approaches to regain nutrition or blood supply. But it appears that therapies that aim to rescue ischemic/infarcted myocardium rely on the preservation or regeneration of a functional vasculature (12). It is unknown in which condition the damaged vessel could be regenerated and whether there are any difference in angiogenesis of arteries and capillaries between transmural MI and sub-MI.
Ischemia 30 min and reperfusion usually leads to MI, but the MI size is smaller than that resulting from permanent LCA occlusion (9). Our current data showed that most arterial branches were lost post-I/R, leaving a residual short tail distal to the ligated site. In our study, the length of residual vessel tail depended on pressure from outside and inside the vessel wall: on the one hand, if the aortic pressure was low (induced by bleeding), a dead end vessel could be seen in I/R-1m rat (1/9); on the other hand, if the occlusion on LCA was not firm enough, a residual vessel tail was left in MI-1m heart (1 of 12). I/R also could induce transmural MI if ischemic time was longer than 45 min even in pressure overload rats (data not shown). Not every Ab+I/R+DeAb heart automatically resulted in sub-MI; some of them (1 of 8) had transmural MI probably due to lower LV reperfusion pressure caused by surgical bleeding. Atrial thrombus, pulmonary edema, and neointima in coronary artery were not observed in I/R and permanent LCA ligation rats in the present study. It has been known that inflammation and oxidative stress could induce extra injury after reperfusion in accurate ischemic experiment (6), but no significant pathologic changes were observed in remote myocardium in chronic I/R-1m group in current study. Cardiac function was significantly decreased in Ab+I/R+DeAb rats, even though the arterial branches were preserved. The defined mechanism is unknown. Inflammatory cytokines, both in circulation and in failing heart, neurohormonal activation, and innate immune system activation secondary to cardiac stress, might contribute to the development and progression of CHF by promoting myocardial fibrosis, matrix metalloproteinase activation, provoking apoptosis, and contractile dysfunction (38).
The chemical properties of fiber-like structures in Ab+I/R+DeAb hearts are unclear. Collagen fibers in infarcted area were digested, but the fiber-like components in remote area could not be digested by sodium hydroxide. In pressure-overload hearts, fibrosis largely occurred in internal and middle myocardial layers, but very little in the epicardium (9). However, in the present data, the increased fiber-like component was almost universally distributed across the LV wall in Ab+I/R+DeAb hearts (Figs. 3 and 4). More histochemical works need to be done to identify the chemical properties of the fiber-like structure in remote area in Ab+I/R+DeAb rats because this compound might play an important role in the development of congestive heart failure.
In summary, occlusion of LCA (≥30 min) induces transmural MI or subendocardial infarction in normal rats. The pathological changes in transmural MI are mostly local. Cardiac dysfunction is associated with the size of regional injury. Pressure overload (mimicking hypertension) could increase vascular tolerance to I/R injury but also trigger severe cardiac global fibrosis, atrial thrombus, and pulmonary edema. Macroperspective of cardiovascular pathophysiology in the present study, from coronary microvessels to pulmonary circulation, probably offers a better view of congestive heart failure in consideration of angiogenesis for MI therapies.
GRANTS
This work is supported by National Institute of Health Grants R01-HL-093183, HL-088434, P20-HL-100396, Program of Excellence in Nanotechnology Contract No. HHSN268201000045C, and P50-HL-112324.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.C. and R.J.H. conception and design of research; J.C., A.P., E.Y.-G., L.L., and H.J.d.H. performed experiments; J.C., L.L., and H.J.d.H. analyzed data; J.C. interpreted results of experiments; J.C., A.P., and E.Y.-G. prepared figures; J.C. and H.J.d.H. drafted manuscript; J.C., E.Y.-G., J.N., and R.J.H. edited and revised manuscript; R.J.H. approved final version of manuscript.
Supplementary Material
REFERENCES
- 1. Ahmed A, Pitt B. A history of systemic hypertension and incident heart failure hospitalization in patients with acute myocardial infarction and left ventricular systolic dysfunction. Am J Cardiol 103: 1374–1380, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahn D, Cheng L, Moon C, Spurgeon H, Lakatta EG, Talan MI. Induction of myocardial infarcts of a predictable size and location by branch pattern probability-assisted coronary ligation in C57BL/6 mice. Am J Physiol Heart Circ Physiol 286: H1201–H1207, 2004 [DOI] [PubMed] [Google Scholar]
- 3. Anversa P, Leri A, Kajstura J. Cardiac regeneration. J Am Coll Cardiol 47: 1769–1776, 2006 [DOI] [PubMed] [Google Scholar]
- 4. Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125: 1234–1245, 2012 [DOI] [PubMed] [Google Scholar]
- 5. Bozkurt B, Mann DL, Deswal A. Biomarkers of inflammation in heart failure. Heart Fail Rev 15: 331–341, 2010 [DOI] [PubMed] [Google Scholar]
- 6. Buja LM. Myocardial ischemia and reperfusion injury. Cardiovasc Pathol 14: 170–175, 2005 [DOI] [PubMed] [Google Scholar]
- 7. Burke AP, Virmani R. Pathophysiology of acute myocardial infarction. Med Clin North Am 91: 553–572, 2007 [DOI] [PubMed] [Google Scholar]
- 8. Chen J, Chemaly E, Liang L, Kho C, Lee A, Park J, Altman P, Schecter AD, Hajjar RJ, Tarzami ST. Effects of CXCR4 gene transfer on cardiac function after ischemia-reperfusion injury. Am J Pathol 176: 1705–1715, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen J, Chemaly ER, Liang LF, Larocca TJ, Yaniz-Galende E, Hajjar RJ. A new model of congestive heart failure in rats. Am J Physiol Heart Circ Physiol 301: H994–H1003, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen J, Lee SK, Abd-Elgaliel WR, Liang L, Galende EY, Hajjar RJ, Tung CH. Assessment of cardiovascular fibrosis using novel fluorescent probes. PLos One 6: e19097, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J, Tung CH, Allport JR, Chen S, Weissleder R, Huang PL. Near-infrared fluorescent imaging of matrix metalloproteinase activity after myocardial infarction. Circulation 111: 1800–1805, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chien KR. Lost and found: cardiac stem cell therapy revisited. J Clin Invest 116: 1838–1840, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cushman WC, Ford CE, Cutler JA, Margolis KL, Davis BR, Grimm RH, Black HR, Hamilton BP, Holland J, Nwachuku C, Papademetriou V, Probstfield J, Wright JT, Jr, Alderman MH, Weiss RJ, Piller L, Bettencourt J, Walsh SM. Success and predictors of blood pressure control in diverse North American settings: the antihypertensive and lipid-lowering treatment to prevent heart attack trial (ALLHAT). J Clin Hypertens (Greenwich) 4: 393–404, 2002 [DOI] [PubMed] [Google Scholar]
- 14. Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P, Verma S, Weisel RD, Keating A, Li RK. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J Clin Invest 116: 1865–1877, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Green KG, Heady A, Oliver MF. Blood pressure, cigarette smoking and heart attack in the WHO co-operative trial of clofibrate. Int J Epidemiol 18: 355–360, 1989 [DOI] [PubMed] [Google Scholar]
- 16. Houser SR, Margulies KB, Murphy AM, Spinale FG, Francis GS, Prabhu SD, Rockman HA, Kass DA, Molkentin JD, Sussman MA, Koch WJ. Animal models of heart failure: a scientific statement from the American Heart Association. Circ Res 111: 131–150, 2012 [DOI] [PubMed] [Google Scholar]
- 17. Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res 43: 860–878, 1999 [DOI] [PubMed] [Google Scholar]
- 18. Jugdutt BI, Idikio HA. Apoptosis and oncosis in acute coronary syndromes: assessment and implications. Mol Cell Biochem 270: 177–200, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Kako KJ. Free radical effects on membrane protein in myocardial ischemia/reperfusion injury. J Mol Cell Cardiol 19: 209–211, 1987 [DOI] [PubMed] [Google Scholar]
- 20. Kawamoto A, Iwasaki H, Kusano K, Murayama T, Oyamada A, Silver M, Hulbert C, Gavin M, Hanley A, Ma H, Kearney M, Zak V, Asahara T, Losordo DW. CD34-positive cells exhibit increased potency and safety for therapeutic neovascularization after myocardial infarction compared with total mononuclear cells. Circulation 114: 2163–2169, 2006 [DOI] [PubMed] [Google Scholar]
- 21. Koike MK, Frimm Cde C, Curi M. Low coronary driving pressure early in the course of myocardial infarction is associated with subendocardial remodelling and left ventricular dysfunction. Int J Exp Pathol 88: 279–290, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lusis AJ. Atherosclerosis. Nature 407: 233–241, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McMurray JJ, Pfeffer MA. Heart failure. Lancet 365: 1877–1889, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM. Myocardial ischemia and reperfusion: a murine model. Am J Physiol Heart Circ Physiol 269: H2147–H2154, 1995 [DOI] [PubMed] [Google Scholar]
- 25. Moroi M, Zhang L, Yasuda T, Virmani R, Gold HK, Fishman MC, Huang PL. Interaction of genetic deficiency of endothelial nitric oxide, gender, and pregnancy in vascular response to injury in mice. J Clin Invest 101: 1225–1232, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ng AS, Johan A, Quek SS, Koh TH, Tan AM. Subendocardial versus transmural myocardial infarction: clinical comparison and review. Singapore Med J 24: 324–332, 1983 [PubMed] [Google Scholar]
- 27. Nicod P, Corbett JR, Sanford CF, Mukharji J, Dehmer GJ, Croft CH, Rude RE, Lewis S, Willerson JT. Comparison of the influence of acute transmural and nontransmural myocardial infarction on ventricular function. Am Heart J 107: 28–34, 1984 [DOI] [PubMed] [Google Scholar]
- 28. Patel KC, Leyva F, Frenneaux MP. Heart failure is not a diagnosis. Int J Clin Pract 62: 526–528, 2008 [DOI] [PubMed] [Google Scholar]
- 29. Pedrinelli R, Ballo P, Fiorentini C, Denti S, Galderisi M, Ganau A, Germano G, Innelli P, Paini A, Perlini S, Salvetti M, Zaca V. Hypertension and acute myocardial infarction: an overview. J Cardiovasc Med 13: 194–202, 2012 [DOI] [PubMed] [Google Scholar]
- 30. Pell AC, Dunn FG. The interface of hypertension and ischemic heart disease. Curr Opin Cardiol 10: 473–479, 1995 [DOI] [PubMed] [Google Scholar]
- 31. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81: 1161–1172, 1990 [DOI] [PubMed] [Google Scholar]
- 32. Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E. Myocardial infarct size and ventricular function in rats. Circ Res 44: 503–512, 1979 [DOI] [PubMed] [Google Scholar]
- 33. Seccombe JF, Schaff HV. Coronary artery endothelial function after myocardial ischemia and reperfusion. Ann Thorac Surg 60: 778–788, 1995 [DOI] [PubMed] [Google Scholar]
- 34. Struthers AD. Pathophysiology of heart failure following myocardial infarction. Heart 91, Suppl 2: ii14–ii16, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation 101: 2981–2988, 2000 [DOI] [PubMed] [Google Scholar]
- 36. Wang QD, Swardh A, Sjoquist PO. Relationship between ischaemic time and ischaemia/reperfusion injury in isolated Langendorff-perfused mouse hearts. Acta Physiol Scand 171: 123–128, 2001 [DOI] [PubMed] [Google Scholar]
- 37. Yaniz-Galende E, Chen J, Chemaly ER, Liang L, Hulot JS, McCollum L, Arias T, Fuster V, Zsebo K, Hajjar RJ. Stem cell factor gene transfer promotes cardiac repair after myocardial infarction via in situ recruitment and expansion of c-kit+ cells. Circ Res 111: 1434–1445, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yndestad A, Damas JK, Oie E, Ueland T, Gullestad L, Aukrust P. Role of inflammation in the progression of heart failure. Curr Cardiol Rep 9: 236–241, 2007 [DOI] [PubMed] [Google Scholar]
- 39. Zhou M, Sutliff RL, Paul RJ, Lorenz JN, Hoying JB, Haudenschild CC, Yin M, Coffin JD, Kong L, Kranias EG, Luo W, Boivin GP, Duffy JJ, Pawlowski SA, Doetschman T. Fibroblast growth factor 2 control of vascular tone. Nat Med 4: 201–207, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.