

Abstract
Channelrhodopsin-2 (ChR2) is a light-activated nonselective cation channel that is found in the eyespot of the unicellular green alga Chlamydomonas reinhardtii. Despite the wide employment of this protein to control the membrane potential of excitable membranes, the molecular determinants that define the unique ion conductance properties of this protein are not well understood. To elucidate the cation permeability pathway of ion conductance, we performed cysteine scanning mutagenesis of transmembrane domain three followed by labeling with methanethiosulfonate derivatives. An analysis of our experimental results as modeled onto the crystal structure of the C1C2 chimera demonstrate that the ion permeation pathway includes residues on one face of transmembrane domain three at the extracellular side of the channel that face the center of ChR2. Furthermore, we examined the role of a residue at the extracellular side of transmembrane domain three in ion conductance. We show that ion conductance is mediated, in part, by hydrogen bonding at the extracellular side of transmembrane domain three. These results provide a starting point for examining the cation permeability pathway for ChR2.
Introduction
Channelrhodopsin-2 (ChR2) is a light-gated nonselective channel, which can conduct mono- and divalent cations (1). In the unicellular green alga Chlamydomonas reinhardtii, it has been proposed that ChR2 serves as a sensory photoreceptor, controlling phototaxis signaling (2). ChR2 is a retinylidene protein. Retinylidene proteins (commonly called rhodopsins with the chromophore retinal or opsins without retinal) use retinal as a chromophore for light reception and are present in many species from bacteria to algae and animals. Type 1 and type 2 retinylidene proteins are differentiated by three features: a), apoprotein sequences; b), near-atomic to atomic crystallographic data showing the orientation of their seven helices; and c), the isomeric configuration and ring/chain conformation of the retinal (3). ChR2 is a type 1 rhodopsin that also includes proteins from eukaryotic microbes (3). All type 2 rhodopsins are reported in higher eukaryotes. Despite differences, both types of proteins have similar overall structural features: seven transmembrane (TM) α-helices and the light-isomerizable chromophore retinal. The chromophore is covalently attached to a lysine residue (K257 in ChR2) located in TM7 through a protonated Schiff base (4). In type 1 rhodopsins, light isomerizes all-trans-retinal to 13-cis-retinal. Photoisomerization of the chromophore triggers conformational changes of the protein that induces ion translocation (5). In the absence of light, 13-cis-retinal isomerizes back to all-trans-retinal.
Our understanding of ChR2 structure has been greatly enhanced following cryo-electron microscopy experiments and molecular dynamics (MD) simulations, as well as through comparison with the crystal structure of bacteriorhodopsin (BR) (6–8). The 6 Å projection map of ChR2, obtained by cryo-electron microscopy of two-dimensional crystals revealed that the protein is a dimer, and suggested that the conductance pathway of ChR2 is located at the dimer interface on the twofold axis, lined by TM helices 3 and 4 (6). However, the localization of the pore at the dimer interface was called into question in MD simulation studies (7). The dimer interface was shown to be densely packed and unlikely to be the ion conductance pathway. The recently published x-ray crystal structure of the channelrhodopsin-1 and channelrhodopsin-2 chimera (C1C2) resolved in the dark state at 2.3 Å resolution clarified the architecture of the protein (Fig. 1) (9). Similar to the previous structure, it was shown that channelrhodopsin is a dimer with multiple tight interactions at the N-domain, extracellular loop 1 (ECL1), TM3, and TM4. Cysteine residues in the N-domain form disulfide bonds between protomers. The intracellular end of TM7 is shifted toward the central axis of the monomer and projects into the intracellular space. The extracellular ends of TM1 and TM2 are tilted outward when compared with BR.
Figure 1.
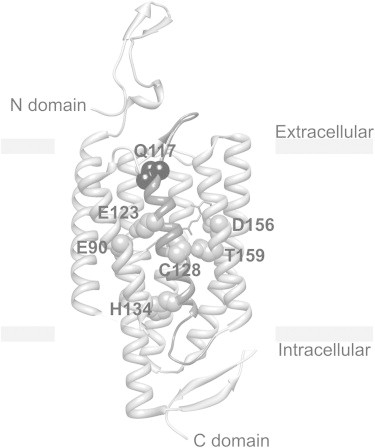
Structure of ChR2 monomer based on the crystal structure of a C1C2 chimera (PDB ID 3UG9). TM3 with Q117 residue on the top is highlighted (black bead). Retinal is highlighted (gray). E90 and H134 are key residues crucial for the gating and selectivity mechanism of the channel. Closed to the Schiff base residue E123 accelerates channel kinetics. Fast kinetic E123 mutants have been used to designate an engineered opsin gene ChETA. The T159C mutation resulted in a dramatic increase in stationary photocurrent. Modifications at the C128 position extend the lifetime of the open state by a modification of the hydrogen bond between C128 and D156.
Functionally important residues of ChR2 have most often been identified by sequence alignment with other opsin proteins. Homologous residues that are critical for BR function may also be essential for ChR2 gating, ion selectivity, and rectification of the channel. For example, mutation of E123, a homolog to D85 in BR, which serves as an acceptor of the proton from the Schiff base, alters the photocycle kinetics of ChR2 (10,11). The double mutant E123T and T159C exhibited rapid kinetics and large amplitudes and thereby increased reliability of high-frequency stimulation (12). Likewise, it was established that residue H134, which is a homolog to proton donor D96 in BR, together with E90, contributes to the selectivity filter of ChR2 (10,13–15). E90 is located in TM2 and described as critical for pore function (15). An analysis of recent experiments suggests that TM2 contributes to ChR2 selectivity and ion conductance (9). In contrast, most residues that alter the kinetics of ChR2, are located in TM3. TM3 contains several amino acids that interact with the all-trans-retinal Schiff base chromophore (16). This includes C128 (T90 in BR) (Fig. 2). Recently, it has been shown that modifications at C128 extend the lifetime of the open state by affecting the hydrogen bond between C128 and D156, indicating the same possible function as T90 in BR (16–19). However, this point is not entirely resolved as the distance between these residues in the C1C2 chimera is too great to support a hydrogen bond (9).
Figure 2.

Amino acid sequence alignment of the predicted third transmembrane domain of the ChR2 from Chlamydomonas reinhardtii with the sequences of ChR1, BR from the Halobacterium sp., and Halorhodopsin from the Natronomonas pharaonis. The GenBank accession numbers of the protein sequences are AAM15777 (ChR2), AAL08946 (ChR1), AAG19772 (BR), and AAA72222, respectively. Conserved amino acid sequences across all proteins are indicated by boxes. Sequence alignment obtained using ClustalW2 (EMBL-EBI).
Although some functionally important residues of ChR2 have been identified, the molecular determinants that contribute to ion channel function are largely unknown. Considering that residues that comprise TM3 have been implicated in ion conductance, our objective was to systemically examine the contribution of residues within this TM domain to ion flux. Therefore, we performed cysteine-scanning mutagenesis along TM3, followed by 2-aminoethylmethanethiosulfonate (MTSEA) and 2-sulfonatoethyl methanethiosulfonate (MTSES) labeling. An analysis of our experimental results has determined that MTS labeling of residues along one face of TM3 contribute to ion channel conductance. Furthermore, we have identified an extracellular surface-exposed residue that modulates channel permeability for cations. We propose that this functional effect is due to the destabilization of helix-helix interactions at the extracellular side of ChR2.
Materials and Methods
Expression of ChR2 mutants in Xenopus oocytes
The cDNA of ChR2 (residues 1–309) with a hemagglutinin tag (YPYDVPDYA) on the C-terminus was subcloned into the vector pTLN as described (20–22). The ChR2 cysteine double replacement mutant (ChR2C34A/C36A) was created using the polymerase chain reaction-based QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer’s instructions. The ChR2C34A/C36A mutant was used as a template for further incorporation of single cysteines. All mutants were verified by sequencing of the entire gene.
Oocytes were obtained after partial ovarectomy from Xenopus laevis females (20). Freshly isolated oocytes were exposed to Ca2+-free Ringer’s buffer containing 3 mg/ml collagenase for 2.5–3 h at room temperature. Defolliculated oocytes were washed extensively and kept in Ringer’s buffer (5 mM MOPS/NaOH, 110 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, pH = 7.6) until injection. mRNA of the desired construct was prepared using the SP6 mMessage mMachine kit (Life Technologies, Grand Island, NY), and quantified with a Nanodrop 2000c (Thermo Scientific, Wilmington, DE). Verification of mRNA synthesis was performed after running an aliquot of mRNA on an agarose gel. Oocytes were injected with 50 ng ChR2 mRNA. After mRNA injection, oocytes were kept in the dark in Ringer’s buffer supplemented with 1 mg/ml gentamycin and 1 μM all-trans-retinal at 17°C for at least 3 days before two-electrode voltage-clamp experiments.
Two-electrode voltage-clamp measurements, chemical labeling with MTS
Glass microelectrodes, filled with 3 M KCl and with tip resistances between 0.5 and 2.5 MΩ were used for all experiments. Bath electrodes were connected to the experimental chamber via agar bridges filled with 3% agar in 3 M KCl. For photocurrent measurements, oocytes expressing each wild-type (WT) or a mutant ChR2 was clamped to −40 mV. A 75-W xenon arc lamp (Sciencetech, Port Huron, MI) was used as a light source. The Sciencetech power supply (model SCI 200) delivered power for the lamp. Illumination was controlled by a computer-triggered shutter Uniblitz VCM-D1 (Vincent Associates, Rochester, NY). The light was applied to the oocytes using a light-guiding fiber with a diameter of 2 mm. The light intensity was ∼4 × 1021 photons s−1 m−2 at the surface of the oocyte and calculated as a derivative of the total power of light falling onto a surface deviated by energy of a single photon. The total power of light was measured with a single pixel power detector PM100D (Thorlabs GmbH, Dachau, Germany) at the end of the light-guiding fiber. The voltage pulses were configured by the pCLAMP10 program (Molecular Devices, Sunnyvale, CA) between −120 and +60 mV in increments of 20 mV. A 10 s recovery between voltage pulses was sufficient to allow recovery from desensitization of the stationary state photocurrent properties (see Fig. S1 in the Supporting Material) (23). Photocurrent measurements were carried out in Tris buffer (5 mM Tris/HCl, 115 mM alkali-metal chloride salt, 2 mM BaCl2, 1 mM MgCl2, pH = 9.0). Each oocyte was equilibrated in buffer for ∼2 min before beginning the recording of sequences. Chemical modification of cysteine residues was achieved by incubating the oocyte in Ringer’s buffer containing 1 mM of either MTSEA or MTSES (Toronto Research Chemicals, Ontario, Canada) for 10 min in the measuring chamber according to our established methodology (24). The MTS reagents were weighed out before each experiment, kept on ice, and diluted in buffer just before use. Photocurrents were measured before and after cysteine-specific labeling. Photocurrents before and after chemical modification were measured using Ringer’s buffer as the extracellular solution.
Data analysis and structural modeling
All analyses were performed using Clampfit 10.2 (Molecular Devices) and SigmaPlot 11.0 (Systat Software, San Jose, CA). Numerical values were given as mean ± SE and calculated from at least five measurements obtained with oocytes from at least three frogs. Statistical t-tests were used to compare two groups of data. Time constants τon and τin characterize the monoexponential transition from flash-to-peak (τon) and from peak-to-stationary currents (τin), respectively (12). A biexponential fit was used to analyze current kinetics after the light was switched off (τoff) (12,19,25). Reversal potentials (Vrev) were determined manually from current-voltage (I/V) plots. The apparent permeability ratios (PX/PNa) of monovalent cations relative to Na+ were calculated with the Goldman Hodgkin Katz equation from the measured changes of reversal potentials, where PX and PNa are the permeabilities of ion X+ (X+: Li+, K+, Rb+, or Cs+) and Na+, respectively (26). Structural models were created using visual molecular dynamics and Chimera software with PDB entry 3UG9 (9,27). Incorporation and evaluation of glutamine residue at position 117 as well as calculation of H-bonds were carried out with the Rotamers tool of the Chimera software (28).
Results
Introduction of single cysteines into ChR2 by site-directed mutagenesis
Following replacement of two extracellular cysteine residues (C34 and C36) with alanine, mRNA of the ChR2C34A/C36A mutant was injected into X. laevis oocytes. Functional expression was examined by measuring stationary currents upon application of light in two-electrode voltage clamp experiments. No significant change in kinetic parameters for the ChR2C34A/C36A mutant was observed when compared to the WT protein (Fig. 3 A). The ChR2C34A/C36A mutant was used as a template for cysteine scanning mutagenesis and for all subsequent MTS labeling experiments.
Figure 3.
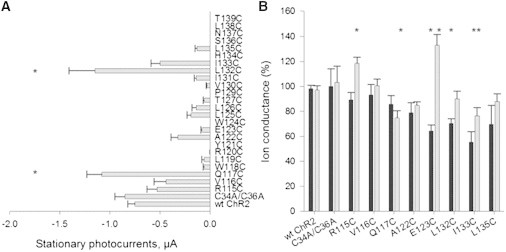
Effect of cysteine substitution and thiol-reactive reagents treatment on functional activity of the protein. (A) Mean stationary photocurrents of WT ChR2, ChR2C34A/C36A, and cysteine mutants of the channel recorded at a Vm of −120 mV in Ringer’s buffer (5 mM MOPS/NaOH, 110 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, pH = 7.6), and measured as in μA. The currents shown are the average of stationary currents from 7 to 61 cells. The error bars represent SE. ∗, p ≤ 0.05. (B) Analysis of cysteine-substituted mutants after MTSEA and MTSES treatment via two-electrode voltage clamp. Each oocyte was incubated in Ringer’s buffer in presence of 1 mM sulfhydryl-specific reagents MTSEA (in black) or MTSES (in gray) at room temperature for 10 min in the measuring chamber. Currents were recorded at −120 mV before and after incubation with MTS reagents and are expressed as the percent of the stationary currents at −120 mV before treatment. The data are means ± SE from 5 to 35 oocytes. ∗p ≤ 0.002.
Cysteine scanning mutagenesis was performed on residues R115 to T139, which comprise the third transmembrane domain (Fig. 2). Stationary currents were highly variable. The largest increase in stationary current when compared to WT ChR2 (photocurrents at −120 mV: −0.75 ± 0.07 μA, n = 61) was observed with the Q117C (−1.08 ± 0.15 μA, n = 17) and L132C (−1.15 ± 0.26 μA, n = 13) mutants (Fig. 3 A). L132 is a highly conserved amino acid in ChR2, ChR1, BR, and halorhodopsin (Fig. 2). The L132C mutant was already described as CatCh (calcium-translocating channelrhodopsin), which has a higher Ca2+ permeability, light sensitivity, and faster response kinetics when expressed in hippocampal neurons than the WT ChR2 (29). In contrast, Q117 is variable among homologous proteins (Fig. 2).
Cysteine replacement at residues R115, V116, or I133 did not have a significant effect on the stationary current of the channel when compared to the ChR2 WT. The remaining constructs had reduced photocurrents.
Mapping the effects of MTS on cysteine mutants in TM3
To assess the functional significance of residues along transmembrane domain three, cysteine mutants were labeled with the sulfhydryl-specific reagents MTSEA and MTSES. MTSES introduces a net negative charge, whereas MTSEA introduces a positive charge following site-specific labeling of cysteine residues (30). We compared stationary photocurrents generated by the oocytes expressing cysteine mutants with WT ChR2 after hyperpolarization to −120 mV in Ringer’s buffer. Photocurrents were obtained from the same oocyte before and after 10 min incubation with 1 mM MTSEA or 1 mM MTSES. Ion conductance was calculated as the percent of stationary currents at −120 mV before exposure to MTS. Neither the WT ChR2, nor the ChR2C34A/C36A constructs were affected by either MTS reagent (Fig. 3 B). However, the ChR2C34A/C36A construct was used for all subsequent experiments to minimize indirect effects of MTSEA or MTSES labeling at these positions. Five mutants showed statistically significant changes in ion conductance after treatment with MTS reagents. The negatively charged modifier (MTSES) enhanced currents of R115C and E123C and impaired the stationary current of Q117C and I133C mutants. Addition of the positively charged MTSEA reagent reduced photocurrents of E123C, L132C, and I133C.
The effect of sulfhydryl modifications on the photocurrents of mutants was further examined as changes in reversal potentials (ΔVrev, see Table 1). Small changes in Vrev for untreated oocytes were observed between different batches of oocytes that can be directly attributed to changes in the intracellular components of oocytes. Therefore, changes in the reversal potential before and after MTS labeling were directly compared on a single day. I/V plots recorded from wt-expressing oocytes before and after MTS treatment were indistinguishable. This is consistent with data in Fig. 3 B, showing that MTS reagents did not alter stationary currents of WT. MTSES labeling had negligible effects on the reversal potential of the ChR2 mutant proteins (data not shown). In contrast, MTSEA labeling resulted in either no change or depolarization of reversal potentials following labeling at positions R115, Q117, E123, and I133. The positive shift in reversal potential indicates that fewer cations are conducted when compared to the untreated mutant proteins.
Table 1.
Reversal potential values (mV) calculated from oocytes expressing WT ChR2 and cysteine mutants
| Protein | MTSEA | MTSEA |
|---|---|---|
| − | + | |
| WT ChR2 | −4.53 ± 0.45 (35) | −3.5 ± 0.04 |
| R115C | −3.68 ± 0.33 (9) | 11.76 ± 0.84∗ |
| V116C | −5.76 ± 1.11 (5) | −1.74 ± 0.95 |
| Q117C | -4.22 ± 0.81 (5) | 2.93 ± 0.49∗ |
| A122C | 0.73 ± 2.44 (5) | 11.85 ± 2.07 |
| E123C | −12.24 ± 1.23 (8) | −5.79 ± 1.16∗ |
| L132C | n/d | n/d |
| I133C | 0.95 ± 0.74 (8) | 5.21 ± 0.93∗ |
| L135C | 7.58 ± 2.36 (5) | 9.94 ± 2.30 |
Reversal potentials were determined in Ringer’s solution (pH = 7.6) before (−) and after (+) treatment with MTS reagents at −120 mV. Mean values ± SE of the mean (number of experiments). n/d (not determined), as L132C mutant (CatCh) has an elevated Ca2+ permeability (29). Asterisks indicate p ≤ 0.005 when compared to untreated oocytes.
Replacement of Q117 on the extracellular side of TM3 alters channel permeability
Upon the observation of the large photocurrents by the Q117C ChR2 mutant as well as the change in ion conductance upon labeling with MTSEA, we hypothesized that this residue at the extracellular side of the third transmembrane domain of ChR2 could contribute to cation selectivity. Therefore, we performed a sequence alignment of ChR2 with ChR1 and BR and determined that Q117 corresponds to valine and tyrosine, respectively, in these homologous proteins. We subsequently replaced glutamine with these amino acids at this position. In addition, we replaced Q117 with charged amino acids (glutamic acid and arginine) to gain further insight into the role of this residue in ion conductance. Four ChR2 constructs with point mutants were expressed in X. laevis oocytes and characterized via two-electrode voltage clamp. Light-induced currents were recorded in the presence of 115 mM monovalent cation X+ (X+: Li+, Na+, K+, Rb+, and Cs+) at pH 9, and the reversal potential of each cation was determined (Table 2). Although valine substitution had minimal effects on Vrev, replacement of glutamine by negatively charge glutamic acid resulted in a hyperpolarizing shift of reversal potentials of all monovalent ions. Q117R and Q117V exchanges did not markedly alter reversal potentials, with the exception of K+ Vrev (Q117R: Vrev (K+) = −28.6 ± 0.4 mV; Q117V: Vrev (K+) = −20.9 ± 2.4 mV) when compared to WT ChR2 (Vrev (K+) = −14.8 ± 2.1 mV).
Table 2.
Reversal potentials of Q117 mutants
| Ion X+ | WT ChR2 | Q117E | Q117R | Q117V | Q117Y |
|---|---|---|---|---|---|
| Li+ | −8.6 ± 0.5 (9) | −11.7 ± 0.7 (6) | −10.3 ± 0.7 (6) | −8.9 ± 0.5 (7) | −6.2 ± 0.7 (10) |
| Na+ | −9.4 ± 0.5 (11) | −25.8 ± 2.0 (19) | −10.5 ± 0.3 (7) | −10.0 ± 0.6 (7) | −9.4 ± 0.7 (13) |
| K+ | −14.8 ± 2.1 (14) | −27.8 ± 1.0 (8) | −28.6 ± 0.4 (5) | −22.5 ± 1.7 (13) | −12.2 ± 1.2 (21) |
| Rb+ | −29.3 ± 0.8 (5) | −35.8 ± 3.6 (6) | −27.7 ± 0.3 (5) | −29.6 ± 0.7 (5) | −24.7 ± 0.3 (10) |
| Cs+ | −28.4 ± 2.6 (5) | −35.3 ± 4.2 (6) | −29.9 ± 0.2 (5) | −29.7 ± 0.6 (8) | −27.6 ± 2.0 (8) |
Reversal potentials were determined by current-voltage relationships and 115 mM XCl in the external solution (pH = 9.0) on oocytes expressing WT ChR2, Q117E, Q117R, Q117V, and Q117Y. Mean values ± SE (n).
In addition, ΔVrev were used to estimate permeability ratios (PX/PNa) of each monovalent cation for WT ChR2 and mutants. Ratios were determined as changes in reversal potentials of photocurrents when replacing 115 mM Na+ by another monovalent cation X+ (Table 3). Values >1 indicates a higher permeability for cation X+ over Na+, whereas values <1 indicates reduced permeability for cation X+. We observed the same dependency as previously described for WT ChR2 (1). Furthermore, the conductance of WT ChR2 and mutants for all monovalent cations decreased with increasing atomic radius. The most significant effects were observed for the Q117E mutant where each cation had reduced permeability when compared to the WT ChR2.
Table 3.
Permeability ratios PX/PNa of monovalent cations for Q117 mutants
| Ion X+ | WT ChR2 | C34A/C36A | Q117E | Q117R | Q117V | Q117Y |
|---|---|---|---|---|---|---|
| Li+ | 1.03 (9) | 1.07 (5) | 0.91 (6) | 0.96 (6) | 1.02 (7) | 1.13 (10) |
| Na+ | 1.00 (11) | 0.97 (8) | 0.52 (19) | 0.95 (7) | 0.98 (7) | 1.00 (13) |
| K+ | 0.81 (14) | 0.77 (7) | 0.48 (8) | 0.47 (5) | 0.59 (13) | 0.89 (21) |
| Rb+ | 0.45 (5) | 0.50 (5) | 0.35 (6) | 0.48 (5) | 0.45 (5) | 0.54 (10) |
| Cs+ | 0.47 (5) | 0.45 (5) | 0.36 (6) | 0.44 (5) | 0.45 (8) | 0.49 (8) |
Permeability ratios determined as described in Materials and Methods. [X+] = 115 mM, pH = 9. Mean values ± SE of the mean (number of experiments).
The Q117Y mutant generated >fivefold larger photocurrents when compared to the WT (data not shown). Large photocurrents are often accompanied by slower channel kinetics (13). We analyzed kinetics (flash-to-peak (τon), peak-to-stationary currents (τin), and switched-off (τoff) time constants) to determine whether mutations of Q117 alter the kinetics of the channel photocycle (Table 4). Current traces taken in 115 mM Na+ solution at −120 mV were fit as described in the Materials and Methods. WT ChR2 had τon of 5.51 ± 0.27 ms, τin of 136 ± 7.26 ms, and τoff 6.75 ± 0.60 ms (n = 7). The Q117E and Q117V constructs had similar on-, in-, and off-kinetics rates. In contrast, the Q117R and Q117Y were slower in one or more steps of the photocycle compared with those of WT ChR2. Next, we analyzed steady-state/peak current ratios (S/P) that indicate inactivation of the channel during light exposure. S/P ratios were calculated on photocurrents evoked at −120 mV (115 mM Na+, pH = 9) (Table 4). Larger S/P ratio, when compared with the WT, indicates either less inactivation for the mutant construct or a higher conductance in O2 for cations. Only the Q117Y mutant showed a significant difference in signal inactivation with S/P = 0.30 ± 0.02 when compared to the WT ChR2 (S/P = 0.44 ± 0.02).
Table 4.
Kinetics and steady-state/peak current ratios of WT ChR2 and Q117 mutants
| τon, ms | τin, ms | τoff, ms | S/P | |
|---|---|---|---|---|
| WT ChR2 | 5.51 ± 0.27 (7) | 136.06 ± 7.26 | 6.75 ± 0.60 | 0.44 ± 0.02 |
| Q117E | 4.83 ± 0.42 (12) | 112.63 ± 10.19 | 6.04 ± 0.45 | 0.46 ± 0.3 |
| Q117R | 8.57 ± 0.26 (7)∗ | 162.83 ± 5.43 | 8.68 ± 0.36 | 0.33 ± 0.02 |
| Q117V | 7.69 ± 0.65 (7) | 180.03 ± 12.84 | 8.80 ± 0.59 | 0.63 ± 0.05 |
| Q117Y | 7.45 ± 0.27 (12)∗ | 151.21 ± 8.70 | 9.28 ± 0.36∗ | 0.30 ± 0.02∗ |
Kinetics values and steady-state/peak current ratios (S/P) were determined by fitting photocurrents taken in 115 mM Na+ solution at −120 mV. Time constants τon, τin, and τoff correspond to flash-to-peak, peak-to-stationary current, and switched-off regions of a current trace, respectively. on- and in-rates were determined by monoexponential fitting of current traces, whereas off-rates were determined by biexponential fit. Mean values ± SE of the mean (number of experiments). t-test with level of significance p ≤ 0.005 (indicated by asterisks) was used to compare Q117 mutants with WT ChR2.
Discussion and Conclusions
ChR2 is a nonselective cation channel, which is functionally distinct from other opsin proteins as it can act as an ion channel, whereas other opsin proteins are ion pumps. Although this protein has become a valuable tool in controlling cellular membrane potential with light, the cation permeation pathway for ion conductance is not well defined. To examine this open question, we performed cysteine scanning mutagenesis on transmembrane domain three.
To reduce nonspecific MTS labeling of ChR2, we first made two mutations of cysteine residues located on the extracellular side, C34 and C36. It has recently been shown that these residues form disulfide bonds as the protein is dimeric (9). Mutagenesis of these residues and subsequent loss of disulfide bonding had no functional effect on ChR2. Subsequent to this experiment, 25 residues in transmembrane domain three, from R115 to T139, were replaced by cysteine and screened for functional changes in X. laevis oocytes. An analysis of our experiments showed that stationary photocurrents were highly variable; but that nine mutants (R120C, Y121C, W124C, P129C, H134C, S136C, N137C, L138C, and T139C) had highly reduced photocurrents compared to WT ChR2 or were inactive. As can be seen from our sequence alignment of ChR2 with ChR1, BR, and halorhodopsin (Fig. 2), R120, Y121, W124, P129, and L138 are highly conserved amino acids. Therefore, mutagenesis of these residues disrupts the structural and/or functional role of residues at these positions (31). Furthermore, H134, S136, N137, and T139 are conserved between ChR1 and ChR2 suggesting a structural/functional role for these residues as well. Replacement of histidine at position 134 has been shown to be involved in H+ selectivity and release (32). An analysis of our experimental results showed there is a significant increase of stationary photocurrents at two positions (Q117C and L132C). The L132C mutant was previously described as CatCh and has been shown to modulate the relative permeability to Ca2+ and Na+ (29).
To further explore the functional significance of residues that comprise transmembrane domain three in ion channel conductance, functionally active cysteine mutants were labeled with methanethiosulfonate derivates. MTS labeling has previously been shown to be an effective method to examine the ion conductance pathway of membrane proteins (30). An analysis of our experimental results showed that labeling with MTSEA, a positively charged reagent, inhibited cation conductance, whereas negatively charged MTSES labeling induced ion conductance. These results could simply be due to charge-charge attraction or repulsion between the MTS reagent and cations. Labeling with MTS reagents at five positions along transmembrane domain three (R115C, Q117C, E123C, L132C, and I133C) had a significant effect on ion conductance. Each of these residues faces transmembrane domains six and seven and residues, which comprise each of these transmembrane domains and could participate in the ion conductance pathway. Labeling of R115, located at the extracellular pore entrance of ChR2, with MTSES significantly increased stationary current and could mark the extracellular entrance of the ion channel pore. Residue E123 has already been shown to be important for proton pumping and also appears to play a role in ion channel function as MTSEA labeling decreased ion conductance, whereas MTSES labeling resulted in a large enhancement of ion conductance (∼40%). Labeling at two positions (L132C and I133C) occurred deep within ChR2 on the intracellular side of the retinal cofactor. It has previously been demonstrated that the MTS reagents would fit inside a cylinder ∼0.6 nm in diameter, which suggests that the pore is conformationally flexible enough to enable labeling with these bulky reagents deep within the protein (33).
To further understand the molecular determinants of these residues along the cation permeation pathway, we modeled each residue, which demonstrated a significant change in ion conductance upon MTS labeling on the structure of C1C2 (Fig. 4). Viewed from the extracellular side of the channel, the cation permeation pathway progresses along the pathway of three residues (R115, Q117, and E123), which align from the extracellular space into the pore of ChR2, on top of each other. Interestingly, L132 and I133 are located more distal from these positions on the other side of the retinal moiety. Nevertheless, R115, Q117, and E123 could represent the entrance pathway for cation conductance of ChR2. It should be noted that an absence of functional change upon application of MTS reagents can be due to either no labeling or no effect upon labeling at these positions.
Figure 4.
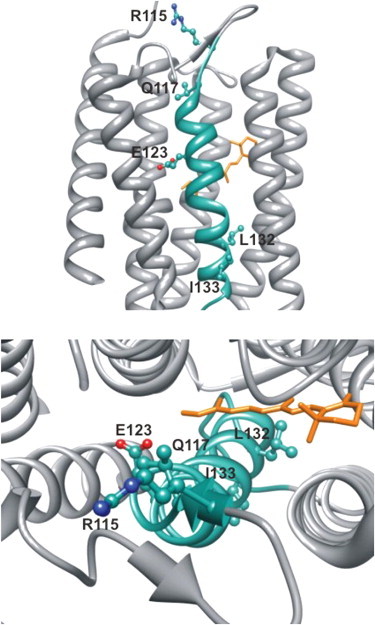
A model of residues that show changes in cation permeability upon cysteine labeling and labeling with MTSEA or MTSES. Views include along the membrane (top) and into the center of the C1C2 chimera (PDB ID 3UG9) from the extracellular side of the protein (bottom).
During the course of our experiments, we observed that replacement of Q117 with cysteine resulted in significantly greater stationary current when compared to the WT ChR2. To further examine the mechanistic role of this residue, glutamine was replaced with charged residues (glutamic acid and arginine) as well as tyrosine and valine, which are the homologous residues in BR and ChR1, respectively. The most significant effects on cation permeability were observed for the Q117E mutation. To understand the cause of this change in permeability, we modeled changes in the side chain at this position (V156 in ChR1 (28)) using the crystal structure of the channelrhodopsin chimera C1C2 (PDB ID 3UG9). Replacement of valine with glutamine at this position using the Richardson rotamer library (Fig. 5) indicates that glutamine can form a hydrogen bond with E140 (E101 in ChR2) on transmembrane domain two. Replacement of glutamine with glutamate at position 117 in ChR2 disrupts H-bonding with E101 as glutamate is the only tested residue that has an unprotonated carboxylic acid side chain under our experimental conditions. In contrast, the Q117Y, Q117V, and Q117R mutations preserve the hydrogen bond between the transmembrane domains. Furthermore, we observe that although the permeability for all ions is reduced with the Q117E mutant (Table 3), the decrease in permeability is most dramatic for Na+ and K+. Considering that it is well established that ions pass through ChR2 in a dehydrated state, we speculate that the extracellular pore entrance could be involved in cation dehydration before ion conductance and that small changes in H-bonding at the entrance of the pore have a variable effect on dehydrating cations as they are conducted by ChR2 (1,21). This suggests that H-bonding at the extracellular side of ChR2 is an important determinant of ion conductance where disruption of interhelical H-bonding results in a destabilization of helix-helix assembly and a subsequent decrease in ion permeability.
Figure 5.
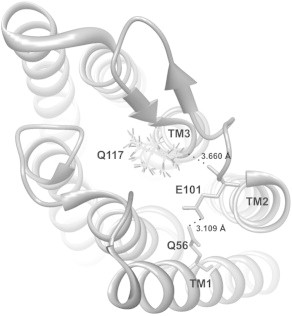
A structure of ChR2 based on the crystal structure of a channelrhodopsin C1C2 chimera (PDB ID 3UG9) with glutamine rotamers at position 117 (V156 in ChR1). Insertion of a glutamine residue was carried out with the Chimera program using the Richardson rotamer library. Hydrogen bonds between Q117 in TM3 and E101 in TM2, as well as between E101 and Q56 in TM1 are shown (dotted lines). Nitrogen atom of glutamine side chain is hydrogen bonded to the backbone carbonyl oxygen of E101 (E140 in ChR1).
In summary, an analysis of our experimental results provides, to our knowledge, new insights into the permeation pathway of ion conductance for ChR2: 1), whereas highly conserved residues within TM3 are required for the structure and/or function of ChR2, residues located on one face at the extracellular end of TM3 toward TM6 and TM7 contribute to the cation permeability pathway; 2), the pore of ChR2 is conformationally flexible as MTS reagents can penetrate the protein deep within the channel; and 3), ion conductance is mediated, in part, by H-bonding at the extracellular side of transmembrane domain three.
Acknowledgments
We thank Sagar Antala for help in the laboratory and Ryan Richards for critically reading the manuscript.
The research described within this manuscript was supported by the Worcester Polytechnic Institute Research Foundation.
Supporting Material
References
- 1.Nagel G., Szellas T., Bamberg E. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sineshchekov O., Jung K., Spudich J. Two rhodopsins mediate phototaxis and photophobic responses to low- and high-intensity light in Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 2002;99:8689–8694. doi: 10.1073/pnas.122243399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spudich J.L., Yang C.S., Spudich E.N. Retinylidene proteins: structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 2000;16:365–392. doi: 10.1146/annurev.cellbio.16.1.365. [DOI] [PubMed] [Google Scholar]
- 4.Terakita A. The opsins. Genome Biol. 2005;6:213. doi: 10.1186/gb-2005-6-3-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang F., Vierock J., Deisseroth K. The microbial opsin family of optogenetic tools. Cell. 2011;147:1446–1457. doi: 10.1016/j.cell.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller M., Bamann C., Kühlbrandt W. Projection structure of channelrhodopsin-2 at 6 Å resolution by electron crystallography. J. Mol. Biol. 2011;414:86–95. doi: 10.1016/j.jmb.2011.09.049. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe H.C., Welke K., Elstner M. Structural model of channelrhodopsin. J. Biol. Chem. 2012;287:7456–7466. doi: 10.1074/jbc.M111.320309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsui Y., Sakai K., Kouyama T. Specific damage induced by X-ray radiation and structural changes in the primary photoreaction of bacteriorhodopsin. J. Mol. Biol. 2002;324:469–481. doi: 10.1016/s0022-2836(02)01110-5. [DOI] [PubMed] [Google Scholar]
- 9.Kato H.E., Zhang F., Nureki O. Crystal structure of the channelrhodopsin light-gated cation channel. Nature. 2012;482:369–374. doi: 10.1038/nature10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tittor J., Oesterhelt D., Bamberg E. Bacteriorhodopsin mutants D85N, D85T and D85,96N as proton pumps. Biophys. Chem. 1995;56:153–157. doi: 10.1016/0301-4622(95)00027-u. [DOI] [PubMed] [Google Scholar]
- 11.Gunaydin L.A., Yizhar O., Hegemann P. Ultrafast optogenetic control. Nat. Neurosci. 2010;13:387–392. doi: 10.1038/nn.2495. [DOI] [PubMed] [Google Scholar]
- 12.Berndt A., Schoenenberger P., Oertner T.G. High-efficiency channelrhodopsins for fast neuronal stimulation at low light levels. Proc. Natl. Acad. Sci. USA. 2011;108:7595–7600. doi: 10.1073/pnas.1017210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gradmann D., Berndt A., Hegemann P. Rectification of the channelrhodopsin early conductance. Biophys. J. 2011;101:1057–1068. doi: 10.1016/j.bpj.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyden E.S., Zhang F., Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhauer K., Kuhne J., Gerwert K. In channelrhodopsin-2 Glu-90 is crucial for ion selectivity and is deprotonated during the photocycle. J. Biol. Chem. 2012;287:6904–6911. doi: 10.1074/jbc.M111.327700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berndt A., Yizhar O., Deisseroth K. Bi-stable neural state switches. Nat. Neurosci. 2009;12:229–234. doi: 10.1038/nn.2247. [DOI] [PubMed] [Google Scholar]
- 17.Radu I., Bamann C., Heberle J. Conformational changes of channelrhodopsin-2. J. Am. Chem. Soc. 2009;131:7313–7319. doi: 10.1021/ja8084274. [DOI] [PubMed] [Google Scholar]
- 18.Bamann C., Gueta R., Bamberg E. Structural guidance of the photocycle of channelrhodopsin-2 by an interhelical hydrogen bond. Biochemistry. 2010;49:267–278. doi: 10.1021/bi901634p. [DOI] [PubMed] [Google Scholar]
- 19.Stehfest K., Ritter E., Hegemann P. The branched photocycle of the slow-cycling channelrhodopsin-2 mutant C128T. J. Mol. Biol. 2010;398:690–702. doi: 10.1016/j.jmb.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 20.Richards R., Dempski R.E. Examining the conformational dynamics of membrane proteins in situ with site-directed fluorescence labeling. J. Vis. Exp. 2011;51 doi: 10.3791/2627. pii: 2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Richards R., Dempski R.E. Re-introduction of transmembrane serine residues reduce the minimum pore diameter of channelrhodopsin-2. PLoS ONE. 2012;7:e50018. doi: 10.1371/journal.pone.0050018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lorenz C., Pusch M., Jentsch T.J. Heteromultimeric CLC chloride channels with novel properties. Proc. Natl. Acad. Sci. USA. 1996;93:13362–13366. doi: 10.1073/pnas.93.23.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berndt A., Prigge M., Hegemann P. Two open states with progressive proton selectivities in the branched channelrhodopsin-2 photocycle. Biophys. J. 2010;98:753–761. doi: 10.1016/j.bpj.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dempski R.E., Lustig J., Bamberg E. Structural arrangement and conformational dynamics of the gamma subunit of the Na+/K+-ATPase. Biochemistry. 2008;47:257–266. doi: 10.1021/bi701799b. [DOI] [PubMed] [Google Scholar]
- 25.Sugiyama Y., Wang H., Yawo H. Photocurrent attenuation by a single polar-to-nonpolar point mutation of channelrhodopsin-2. Photochem. Photobiol. Sci. 2009;8:328–336. doi: 10.1039/b815762f. [DOI] [PubMed] [Google Scholar]
- 26.Hille B. Sinauer; Sunderland, MA: 2001. Ion Channels of Excitable Membranes. [Google Scholar]
- 27.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 28.Lovell S.C., Word J.M., Richardson D.C. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 29.Kleinlogel S., Feldbauer K., Bamberg E. Ultra light-sensitive and fast neuronal activation with the Ca²+-permeable channelrhodopsin CatCh. Nat. Neurosci. 2011;14:513–518. doi: 10.1038/nn.2776. [DOI] [PubMed] [Google Scholar]
- 30.Stauffer D.A., Karlin A. Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry. 1994;33:6840–6849. doi: 10.1021/bi00188a013. [DOI] [PubMed] [Google Scholar]
- 31.Plazzo A.P., De Franceschi N., Mongillo M. Bioinformatic and mutational analysis of channelrhodopsin-2 protein cation-conducting pathway. J. Biol. Chem. 2012;287:4818–4825. doi: 10.1074/jbc.M111.326207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagel G., Brauner M., Gottschalk A. Light activation of channelrhodopsin-2 in excitable cells of Caenorhabditis elegans triggers rapid behavioral responses. Curr. Biol. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 33.Akabas M.H., Stauffer D.A., Karlin A. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 1992;258:307–310. doi: 10.1126/science.1384130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.