

Abstract
Ataxin-1 is a human protein responsible for spinocerebellar ataxia type 1, a hereditary disease associated with protein aggregation and misfolding. Essential for ataxin-1 aggregation is the anomalous expansion of a polyglutamine tract near the protein N-terminus, but the sequence-wise distant AXH domain modulates and contributes to the process. The AXH domain is also involved in the nonpathologic functions of the protein, including a variety of intermolecular interactions with other cellular partners. The domain forms a globular dimer in solution and displays a dimer of dimers arrangement in the crystal asymmetric unit. Here, we have characterized the domain further by studying its behavior in the crystal and in solution. We solved two new structures of the domain crystallized under different conditions that confirm an inherent plasticity of the AXH fold. In solution, the domain is present as a complex equilibrium mixture of monomeric, dimeric, and higher molecular weight species. This behavior, together with the tendency of the AXH fold to be trapped in local conformations, and the multiplicity of protomer interfaces, makes the AXH domain an unusual example of a chameleon protein whose properties bear potential relevance for the aggregation properties of ataxin-1 and thus for disease.
Introduction
Ataxin-1 is the protein responsible for spinocerebellar ataxia type-1 (SCA1), a genetically inherited human neurodegenerative disorder that shares a common mechanism with eight other so-called polyglutamine diseases (1,2). They are all caused by expansion of an unstable trinucleotide CAG repeat in the coding region of the respective gene. The mutation leads to lengthening of a polyglutamine tract in the gene products. When the number of glutamines exceeds a threshold, which is variable in each member of the disease family but is mostly at ∼35–42 repeats, a progressive motor deterioration is observed (3). Degeneration is associated with the formation of toxic aggregates and with the death of specific neural cells in the cerebellum, by a mechanism that resembles those observed in other neurodegenerative pathologies caused by protein misfolding such as Alzheimer’s, Parkinson’s, and prion diseases (4,5).
It is now established that polyglutamine expansion is essential for disease onset (2) but other regions play a role in modulating the aggregation process and influencing the development of the disease (2,6–11). One such region is the AXH domain of ataxin-1, so far the only structured region identified along the ∼820-residue protein sequence (12). The AXH domain is an important element of ataxin-1, responsible for both the normal and the aberrant functions of ataxin-1; it has transcriptional repression activity and is essential for interaction with a wide variety of proteins, including the SMRT/SMRTER and Capicua proteins (reviewed in de Chiara and Pastore (13)). AXH also participates in ataxin-1 self-assembly (14). We demonstrated that, although determined by polyglutamine expansion, in-cell ataxin-1 aggregation is noticeably reduced by deletion of the AXH domain or by replacement with the homologous sequence from the transcription factor HBP1, which has no known tendency to aggregate (9). In the same study, we also showed that the isolated domain spontaneously misfolds and forms amyloid fibers in vitro without the need for chemical denaturants, temperature increase, or other destabilizing conditions.
The AXH domain is an unusual chameleon protein (15) that constitutes the first known example in which the existence of alternative structures is not induced by mutations, ligand binding, or different protein contexts (16,17). The crystal structure of the isolated AXH domain contains four AXH molecules in the asymmetric unit arranged as two globular dimers, with the protomer assuming the oligonucleotide/oligosaccharide binding-fold (18). Each protomer adopts different conformations leading to asymmetric dimer interfaces. Differences that are even more striking are observed between the structures of the ataxin-1 AXH protomers and the homologous HBP1 AXH domain (15,18). Despite sharing the same secondary structure elements, the N-terminus adopts different topologies (15). A detailed study by analytical ultracentrifugation showed that the dimer is the predominant species in solution for the ataxin-1 domain whereas the HBP1 domain is a monomer (12).
These findings have raised the intriguing question of whether the adoption of different topologies and the presence of higher-order oligomers are intrinsic features of the AXH motif of ataxin-1 or merely an artifact of crystallization.
To gain fresh insights into these issues, we have investigated the chameleon properties of the ataxin-1 AXH domain and its behavior in solution by a combination of liquid state NMR, small angle x-ray scattering (SAXS), and x-ray crystallography. We show that the chameleon behavior of the domain is not confined to local structural asymmetries but also contributes to a complex equilibrium among monomer, dimer, and higher molecular-weight species. These results may be of relevance for interactions with cellular partners and for SCA1 pathogenesis (9).
Materials and Methods
Protein expression and purification
In this study we have mainly used a recombinant fragment of ataxin-1 spanning residues 567–689 of human ataxin-1, hereinafter referred to as wt-AXH (for consistency with all previous literature, numbering refers to the UniProtKB/Swiss-Prot entry No. P54253 +1). This construct differs slightly from that previously used for crystal structure determination (residues 563–694) (18). Wt-AXH and its mutants were produced as previously described (12). In short, they were overexpressed in the Escherichia coli host strain BL21 (DE3)pLysS in Luria Broth medium using a kanamycin-resistant pETM30 vector with tobacco etch virus-cleavable N-terminal His6-GST tags. 15N single- or 15N,13C double-labeled samples were expressed in minimal (M9) medium containing 15N-ammonium sulfate and/or 13C-glucose as the sole source of nitrogen and carbon. The cells were grown at 37°C until an optical density of 0.6 at 600 nm was reached, before inducing protein expression with Isopropyl β-D-1-thiogalactopyranoside (IPTG, 0.25 mM) for 3 h at 37°C, and harvested. A standard purification protocol was performed using a Ni-NTA agarose column (Qiagen, Germantown, MD). The proteins were further purified by FPLC size-exclusion chromatography using a prepacked HiLoad 16/60 Superdex 75 prep grade column (Pharmacia, Uppsala, Sweden). Protein identity was checked by electrospray ionization mass spectrometry.
Crystallization and data collection
Wt-AXH samples at 20 mg/mL concentration were crystallized in 0.1 M MES, pH 6.5, 10% PEG 20,000 for crystal X1 and 0.4 M potassium sodium tartrate for crystal X2. These were considerably different from the original recipe (i.e., 0.1 M sodium citrate, pH 5.8, 0.1 M ammonium sulfate, 5 mM DTT, 16% PEG 4000). A quantity of 30% glycerol was present for cryoprotection. Datasets for the two crystals were recorded at beamlines I02 and I04 at the Diamond Light Source (Oxfordshire, UK). Diffraction data were indexed and integrated using the software MOSFLM (19). Both crystals belong to the space group P212121. The data were scaled and merged using the software SCALA (20), and further processed because of the presence of severe diffraction anisotropy (21). The final statistics are summarized in Table 1. Summary of the anisotropic correction is included in Table S1 in the Supporting Material.
Table 1.
Data processing and refinement statistics
| Categories | Structure X1 | Structure X2 |
|---|---|---|
| Cell dimensions, Å | a = 47.54, b = 82.70, c = 137.11 | a = 41.47, b = 80.83, c = 139.96 |
| Resolution, Å | 70.9–2.45 | 40.4–2.50 |
| Unique reflections | 16,989 | 14,350 |
| Completeness, % | 82.7 (85.4/32.5) | 84.4 (91.7/35.5) |
| Mean I/σI | 14.8 (5.8/4.5) | 12.5 (4.4/3.0) |
| Rmergea | 0.097 (0.33/0.70) | 0.060 (0.25/0.61) |
| Wilson B, Å2 | 56 | 51 |
| Data truncation along d1, d2, d3, Å | 2.90, 2.45, 2.45 | 2.90, 2.50, 2.50 |
| R-factor, Rfreeb | 22.0, 26.7 | 23.9, 28.8 |
| Model | ||
| No. of protein atoms | 3845 | 3835 |
| No. of solvent atoms | 74 | 46 |
| Overall B factor Å2 | 53.4 | 59.5 |
| RMS bonds | 0.008 | 0.006 |
| RMS angles | 0.72 | 0.76 |
| Coordinate errorc | 0.78 | 0.88 |
| Ramachandran analysisd | ||
| Most favored | 96.6 | 98.2 |
| Disallowed | 0 | 0 |
Values in parentheses are for the highest resolution shells (two values: one for along d1, and the other for along d2 and d3). For comparison, the cell dimensions of the original crystal structure was a = 48.78, b = 82.44, and c = 137.78.
The Rmerge values reported are for the raw data before anisotropy correction.
Free R set test size is 5%.
Estimated by maximum likelihood.
Checked with the software MOLPROBITY.
Structure solution and refinement
Both new crystal structures were solved by molecular replacement with the software PHASER (22), using the C:D dimer of the AXH domain (PDB:1OA8) as the search model. The solutions were unambiguous with translation search z-scores well over 40. For both structures, refinement was performed using the software PHENIX (23), with manual model rebuilding carried out in the software COOT (24). Disordered atoms were modeled with reference to corresponding atoms from the original AXH domain structure. The Rfree routine was used to monitor the progress of refinement. The TLS method was introduced in later stages of the refinement, with groups chosen using the TLSMD server (25). Noncrystallographic symmetry was not imposed. Water molecules were identified automatically with PHENIX, and checked in COOT. The geometry of the final models was assessed using the software MOLPROBITY (26). The results of refinement are summarized in Table 1. The atomic coordinates and structure factors of the two crystal structures have been deposited in the Protein Data Bank (PDB), Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org) and are designated PDB:4AQP (X1) and PDB:4APT (X2).
NMR measurements
NMR measurements were performed on an Avance spectrometer (Bruker BioSpin, Billerica, MA) equipped with a cryo-probe and operating at 700 MHz proton frequency. 1H-15N- Heteronuclear single quantum coherencies (HSQCs) of wild-type AXH domain and mutant A567G were acquired at 300 K in 20 mM Tris-HCl buffer (pH 7.0), 2 mM TCEP. HSQCs for wt-AXH and mutant A567G-AXH were acquired at different protein concentrations (0.40, 0.22, 0.11, 0.055, 0.027, and 0.012 mM) obtained by stepwise dilution of a concentrated stock solution. Partial spectral assignment of wt-AXH was obtained using standard procedures and based on the (15N1H)-TOCSY- and NOESY-HSQC (with 50- and 100-ms mixing times, respectively), HNCA, and HN(CO)CA, CBCANH, and CBCA(CO)NH experiments recorded at 308 K and 600 MHz. Also benefiting from the backbone assignment for the sequence- and structurally- conserved C-terminal region of the homologous HBP1 AXH domain (15), we could confidently assign the backbone of residues G578-L594, E597-S606, L609-E620, G626-E636, A639-E644, F651-V653, G655-S660, E664-L672, S675-V682, S685, and T687-K689, mostly corresponding to secondary structure regions. Further assignment was prevented by the challenging spectral complexity.
Small angle x-ray scattering measurements
Synchrotron radiation x-ray scattering data were collected on the European Molecular Biology Laboratory (EMBL) X33 beamline on the storage ring DORIS III (DESY, Hamburg, Germany) (27) and the EMBL P12 beamline of the storage ring PETRA-III. Samples were measured at 10°C in a concentration range 0.5–18.6 mg/mL. The data were recorded using a 1 M/2 M PILATUS detector (DECTRIS, Baden, Switzerland) for the X33 and P12 beamlines, respectively, at a sample-detector distance of 2.7 m and a wavelength of λ = 0.15 nm, covering the range of momentum transfer 0.12 < s < 6.0 nm−1 (s = 4π sinθ/λ, where 2θ is the scattering angle). No measurable radiation damage was detected by comparison of eight successive time frames with 15 s exposures (X33)/20 successive frames with 1 s exposures (P12). The data were averaged after normalization to the intensity of the transmitted beam and the scattering of the buffer was subtracted using the program PRIMUS (28).
The radius of gyration Rg and forward scattering I(0), the maximum particle dimension Dmax, and the distance distribution function p(r) were evaluated using the program GNOM (29). The molecular masses of the samples were calculated by comparison with reference bovine serum albumin samples. The scattering patterns from the structure models were calculated using the program CRYSOL (30). The best fits in terms of mixtures of monomers, dimers, and tetramers for the AXH domain (as well as extended octamers and hexadecamers) were computed using the program OLIGOMER (28).
Results
The search for alternative structures of wt-AXH
Based on the observation that the homologous AXH domains of ataxin-1 and HBP1 adopt different topologies (9), we hypothesized that the overall molecular topology may be dependent on the crystallization conditions. To test whether the protein could be trapped into alternative conformations and understand further the extent of structural asymmetry, we searched for different crystallization conditions (18). Two new crystal structures, X1 and X2, were solved to medium resolutions. They belong to the same space group and have similar unit cell dimensions as the original structure (X0) (Table 1). The new structures preserve the overall arrangement in the asymmetric unit, having four chains assembled into pairs of globular dimers (A:B and C:D) which stack onto each other along the longest cell dimension. Although the resolution limits are similar, structure X1 is better defined than X2. When sister protomers are compared (i.e., A–C and B–D), the ordered backbone structures of all chains in X1 and X2 are similar to those of X0 within the limit of experimental error. The root-mean-square (RMS) deviations in Cα atomic positions between the individual chains of X1 and the corresponding chains of X0 are 0.4–0.5 Å; those between X2 and X0 are 0.5–0.6 Å. Comparison of the 12 independent protomers within the three crystal structures allows us to appreciate differences even while having statistics rarely available (Fig. 1). Thus, the overall conformation of the ataxin-1 AXH domain remained invariant at the applied crystallization conditions.
Figure 1.
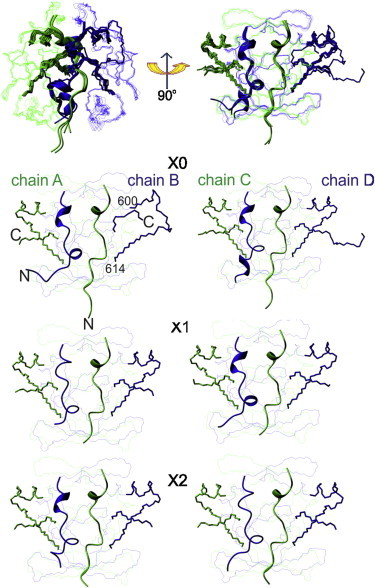
Comparison between the crystal structures of the AXH domain of Ataxin-1. (Top) Overlay of the six dimers from the three structures solved under different crystallization conditions and the same bundle rotated by 90°. (Green and blue) Alternated protomers. (Bottom) Individual comparison of the dimers with the chameleon sequence (shown as stick representations).
Within the framework of the robust overall topology, many local structural variations are observed. The main differences involve the first ∼20 N-terminal amino acids that adopt different conformations and confer a well-defined intrinsic asymmetry to the dimerization interface. Chains A and C consistently form a helical turn between residues 573–575, as supported by the clear electron density (see Fig. S1 in the Supporting Material). Chains B and D differ from A and C and form regular secondary structures in only some of the dimers (helical turns between residues 564–566 or 568–570 and 574–576). At the C-terminal region, which is also involved in the dimer interface, the electron densities vary among protomers, from being well defined to being completely missing—suggesting an intrinsically higher flexibility. In X0, residues 600–614 of chain B are disordered compared with the other protomers that contain a well-formed helix (α1) from residues 597–605.
The emerging picture is that, while the overall conformation is well preserved at the tertiary and quaternary levels, the AXH domain exhibits extensive degrees of local structural plasticity.
NMR spectra of AXH reveal the presence of more than one species in solution
To understand whether the structural asymmetry observed in the crystal is retained in solution or averaged-out by molecular motions, we recorded a NMR 15N-1H HSQC spectrum of wt-AXH that constitutes the fingerprint of a protein. The spectrum is that typical of a protein folded into a stable three-dimensional structure but contains many more resonances than expected (approximately double the number) (Fig. 2 A). The resonances of glutamine and asparagine side chains, for instance, are many more than the expected eight (circled in figure). The resonances also have inhomogeneous intensities, suggesting the presence of unequal populations of different species. Attempts to reduce the spectral complexity by changing pH, buffer composition, and temperature failed (data not shown).
Figure 2.
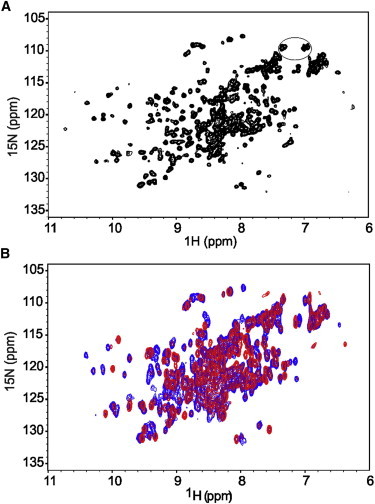
The NMR spectrum of wt-AXH indicates sample heterogeneity. (A) 15N-1H HSQC spectrum of wt-AXH showing spectral complexity recorded on a 0.40 mM protein solution at 27°C on an Avance spectrometer (Bruker) operating at 700 MHz. (B) Concentration dependence of 15N-1H HSQC spectra of wt-AXH at 0.40 mM (blue) and 0.012 mM (red). Note that the spectra in panel B were collected at lower resolution than the one in panel A (64 vs. 128 increments) to reduce the recording time of the diluted sample. This explains their overall broader appearance. The resonances of glutamines/asparagines are circled.
To exclude the possibility that the complexity of the HSQC spectrum could be related to the choice of the domain boundaries, we checked the spectrum of a previously characterized longer construct in which both the N- and C-termini had been elongated (long-AXH, residues 556–700) (12). The spectrum of this construct also contains more resonances than expected (see Fig. S2 A).
The complexity of the HSQC spectrum of wt-AXH has at least three nonmutually exclusive explanations: proline cis/trans isomerization; an asymmetric arrangement of the protomers; and/or by different aggregation states. Initially, the first hypothesis seemed the simplest because four of the eight prolines of wt-AXH are near the N-terminus, which is in the dimeric interface and is where the main structural differences between protomers occur (18). However, partial assignment of the wt-AXH spectrum did not support this hypothesis: cis/trans isomerization usually leads to resonance doubling for residues sequentially close to prolines, whereas we observed doubling for residues all over the sequence (see below). Furthermore, none of the prolines is in a cis configuration in the crystal structures.
We investigated whether the globular dimers interact with each other, forming higher-order oligomers, and also whether they contribute to the observed conformational heterogeneity by recording HSQC spectra of wt-AXH as a function of protein concentration. Formation of higher molecular weight species would be consistent with previous analytical ultracentrifugation data, which suggested the presence of higher-order oligomers in addition to the predominant dimeric species (12,18). Stepwise dilutions from 0.40 to 0.012 mM led to disappearance of some of the peaks and to variation of the relative intensities of others (Fig. 2 B). This supports the hypothesis that higher level organization contributes to conformational heterogeneity and that this is a cause of the spectral complexity.
Identification of potential protein-protein interfaces in solution
To analyze dimerization and possible organization of dimers into higher order assemblies in wt-AXH, we used the PISA server (31) to identify potential intermolecular contact interfaces within the crystal structures. The dimerization interface (interface 1) within protomer pairs A:B and C:D in all the three X0-2 structures buries ∼1500–1700 Å2 of accessible surface area. As mentioned above, this interface is dominated by the N-terminus and, to a minor extent, by the C-terminus of the domain (Fig. 3 A). In addition, two possible crystallographic interfaces between dimers are identified in the crystal structure. These are as follows:
Figure 3.
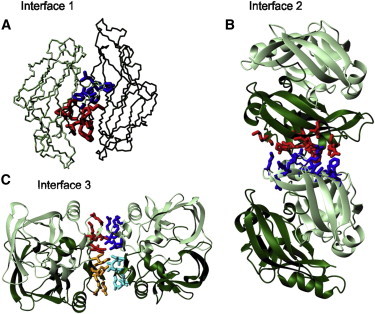
Cartoon representations of the structural interfaces. (A) Monomer-monomer interface 1 which comprises the first ∼19 residues of protomers A and B of structure X1. (B) Interface 2 between dimers using the same orientation shown in Fig. 2. (Red and blue) Residues from protomers B and C, respectively. (C) Interface 3 between dimers. (Different shades of red and blue) Side chains from different protomers.
The first (interface 2), that buries ∼600–700 Å2, involves residues F654(B)-F654(C), I600(B)-F654(C), K595(B)-E604(C), I600(B)-I600(C), F654(B)-I600(C), and F654(B)-I611(C) (Fig. 3 B). The contacts between protomers are mainly mediated by hydrophobic residues with the only exception being the salt bridge between residues K595(B) and E604(C).
The second (interface 3), that buries 520–540 Å2 in total, is highly symmetric with the two globular dimers related by twofold rotation, and forms an overall tetramer with pseudo-222 symmetry. This interface involves the β1/β2-hairpin from each protomer packing against each other through the exposed hydrophobic side chains of I580 and L588 and a hydrogen bond between Q582 and K590 (Fig. 3 C).
The populations of the species can be modified by mutations
Next, we investigated the structural consequences of three sets of mutations that were each designed to disrupt specific intermolecular interfaces. The first mutation, A567G, affects a centrally located residue in the dimerization interface and the first residue of the natural sequence (interface 1) (Fig. 4 A, left). The second set of mutations was designed to probe the potential dimer-dimer interaction (interface 2), with single (F654K) and double (I600K/F654K) mutations (Fig. 4 A, middle). In the third set of mutations, we introduced single and double alanine mutations of I580 and L588 (Fig. 4 A, right). They could affect the monomer-dimer and the dimer-tetramer equilibrium.
Figure 4.
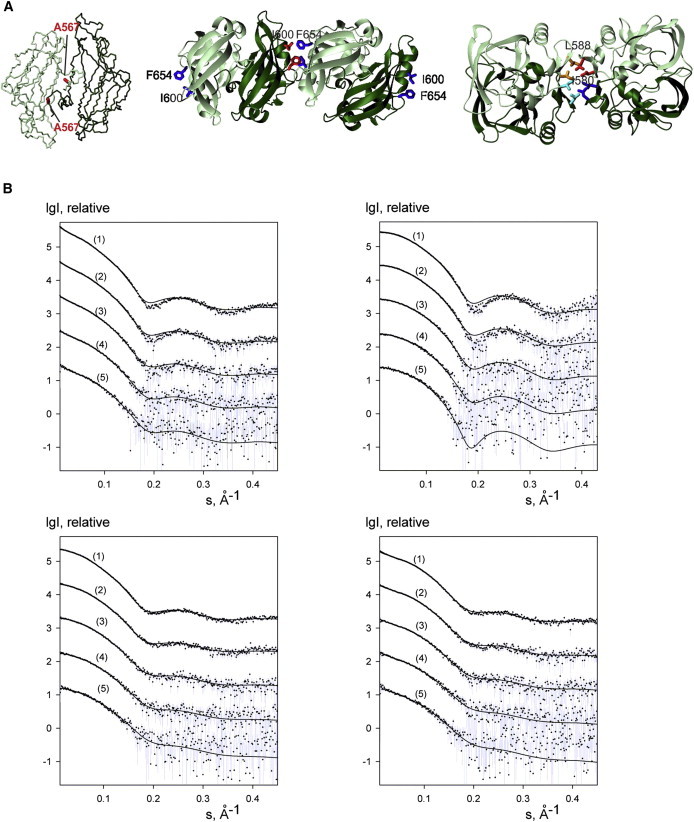
SAXS data for wt-AXH and its mutants. (A) Mapping the mutations on the structure. The side chains of the mutated residues are indicated. The orientations are the same used in Fig. 3, A, B, and C. (B) (Top panel) wt-AXH (left) and wt-AXH freshly prepared (right). (Bottom panel) A567G-AXH (left) and I580A-AXH (right). (Curves 1–5) Solute concentrations 1.35, 0.67, 0.33, 0.17, and 0.08 mM, respectively, in 20 mM Tris-HCl pH 7, 2 mM TCEP. (Dots with error bars) Experimental data. (Solid lines) Best fits calculated by the software OLIGOMER (Konarev et al. (28)) for mixtures of monomers, dimers, and tetramers (in the case of wt-AXH and I580A-AXH, higher-order oligomers, i.e., extended octamers and hexadecamers, were taken into account). The intensities are displayed as a function of the momentum transfer and successive curves are displaced down by one logarithmic unit for clarity.
To characterize the populations of the species in solution, we used SAXS measurements on wt-AXH and the two single mutants I580A-AXH and A567G-AXH in the protein concentration range 0.040–1.35 mM. The data collection parameters and overall structural parameters derived from SAXS data are summarized in Table S2 and Table S3. We observed an increase of the scattered intensity at low angles for wt-AXH and I580A-AXH, but not for A567G-AXH, suggesting the presence of a high molecular mass species. To understand whether this could be related to sample freezing, we collected datasets on a new freshly produced batch of wt-AXH, transporting it in two aliquots (one frozen, the second kept on ice). No increase of the scattered intensity at low angles was revealed. We fitted the data using a multiple equilibria model in which we assumed the presence of monomeric, dimeric, and tetrameric (dimer of dimer) species (as well as extended octamers and hexadecamers) and calculated their relative populations. While we observed no effect of freezing we obtained in all cases fits of reasonably good quality (Table 2 and Fig. 4 B).
Table 2.
SAXS data fitting results from the software OLIGOMER for the estimate of tetramer, dimer, and monomer percentages of wt-AXH, A567G-AXH, and I580A-AXH at different sample concentrations
| Concentration (mM) | Monomer % | Dimer % | Tetramer % | High order oligomer, % | χ2 | |
|---|---|---|---|---|---|---|
| WT-AXH | 1.35 | 21 | 39 | 26 | 14 | 1.56 |
| 0.67 | 27 | 42 | 20 | 11 | 1.28 | |
| 0.33 | 32 | 45 | 15 | 8 | 1.22 | |
| 0.17 | 38 | 45 | 10 | 7 | 0.90 | |
| 0.08 | 41 | 46 | 7 | 6 | 0.91 | |
| 0.04 | 43 | 45 | 7 | 5 | 0.98 | |
| WT-AXH (fresh) | 1.35 | 28 | 55 | 17 | none | 1.76 |
| 0.67 | 32 | 55 | 13 | none | 1.38 | |
| 0.33 | 30 | 63 | 7 | none | 1.23 | |
| 0.17 | 27 | 73 | 0 | none | 1.15 | |
| 0.08 | 25 | 75 | 0 | none | 1.33 | |
| 0.04 | 30 | 70 | 0 | none | 1.12 | |
| A567G-AXH | 1.35 | 37 | 42 | 21 | none | 1.53 |
| 0.67 | 46 | 40 | 14 | none | 1.14 | |
| 0.33 | 56 | 33 | 11 | none | 1.03 | |
| 0.17 | 66 | 26 | 8 | none | 1.00 | |
| 0.08 | 75 | 16 | 9 | none | 0.98 | |
| 0.04 | 81 | 11 | 8 | none | 0.98 | |
| I580A-AXH | 1.35 | 55 | 39 | 3 | 3 | 1.51 |
| 0.67 | 61 | 34 | 3 | 2 | 1.16 | |
| 0.33 | 68 | 26 | 4 | 2 | 1.01 | |
| 0.17 | 75 | 18 | 4 | 3 | 0.92 | |
| 0.08 | 80 | 14 | 4 | 2 | 0.91 | |
| 0.04 | 80 | 15 | 3 | 2 | 0.90 |
In the case of wt-AXH and I580A-AXH, the presence of higher oligomers (i.e., extended octamers and hexadecamers) are necessary to fit the low-angle region. The label Wt-AXH (fresh) corresponds to the freshly produced batch of WT-AXH sample.
The data are thus compatible with the presence of a multiple equilibrium measurement among monomer, dimer, and tetramer with an appreciable fraction of the monomeric species. The contribution of higher molecular weight species becomes increasingly relevant for wt-AXH and I580A-AXH samples. The discrepancy at higher angles (∼0.2 nm−1) could be explained by the fact that the scattering patterns from the components were calculated with the software CRYSOL using default parameters for excluded volume and solvation shell density. Small modifications of the tertiary structure can also not be excluded.
For I580A-AXH and A567G-AXH, we observed an appreciable increase in the population of the monomeric species in agreement with a role of both these mutations in destabilizing the dimer. We did not, however, attempt a more quantitative analysis of the data (i.e., an estimate of the dissociation constants) because the contribution of the higher molecular weight species and the lack of clearer knowledge of the species present could bias the analysis. There is also a potential influence of interparticle interactions at high concentrations and a high noise level of the data at low concentrations; both effects prevent us from getting better fits and worsen the accuracy of the species populations.
Nevertheless, together with the NMR data, these results provide solid evidence of a complex equilibrium among multiple species in solution where high molecular weight species have an important role.
The role of multiple equilibria
We then monitored the effects of the mutations by NMR. The spectrum of A567G-AXH shares common resonances with that of wt-AXH, suggesting that the mutation does not lead to drastic structural changes. It contains fewer resonances than the wild-type but it still has more resonances than expected (Fig. 5, left). The single F654K and double I600K/F654K mutants expressed well, but purified samples led to NMR spectra that are very similar to that of wt-AXH (see Fig. S2 B), thereby ruling out interface 2 as an interaction surface.
Figure 5.
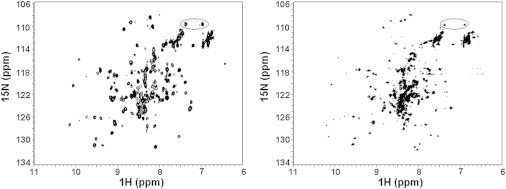
The effect of mutations on the spectral complexity. 15N-1H HSQC spectra of A567G-AXH (left) and of I580A-AXH (right). The spectra were acquired at 0.4 mM protein concentrations and 27°C on an Avance spectrometer (Bruker) operating at 700 MHz. The resonances of asparagines/glutamines are circled.
The I580A/L588A double mutant is probably misfolded or partially unfolded as the protein was mostly in inclusion bodies and the soluble fraction was already prone to degradation after cell lysis. The L588A-AXH single mutant is folded but unstable toward degradation, indicating that L588 is an important residue for maintaining protein integrity. In striking contrast, the spectrum of the I580A-AXH mutant is simpler and contains approximately the number of resonances expected for a monomer or a symmetric dimer (Fig. 5, right). Just as an example, the glutamine/asparagine side-chain resonances at 7.55–6.90 1H-ppm and 109.8 15N-ppm that showed multiplicity in the spectra of the wt-AXH are now reduced to a unique pair of peaks (circled in figure). While definitely much improved, the spectrum still contains minor resonances. These results indicate that I580 plays an important role in mediating the AXH self-association and confirms the role of chemical equilibria in the spectral complexity of wt-AXH that can be altered by destabilizing the dimer interface.
Rationalizing the spectral complexity
Finally, we evaluated the relative importance of the state of aggregation versus the conformational plasticity of the domain to help us further understand the complexity of the species in solution and because it will significantly help future studies of the aggregation pathway of AXH. We can identify two independent sets of major resonances in the spectrum of wt-AXH, which can be sequence-specifically assigned with reasonable confidence. We observe, for instance, 14 resonances that correspond to the seven glycines distributed along the sequence (Fig. 6 A). It is striking that the two sets of peaks have approximately the same linewidth, making it unlikely that they represent the monomer and the dimer species in a slow exchange regime. The same is observed for all the other assigned resonances from all over the sequence.
Figure 6.
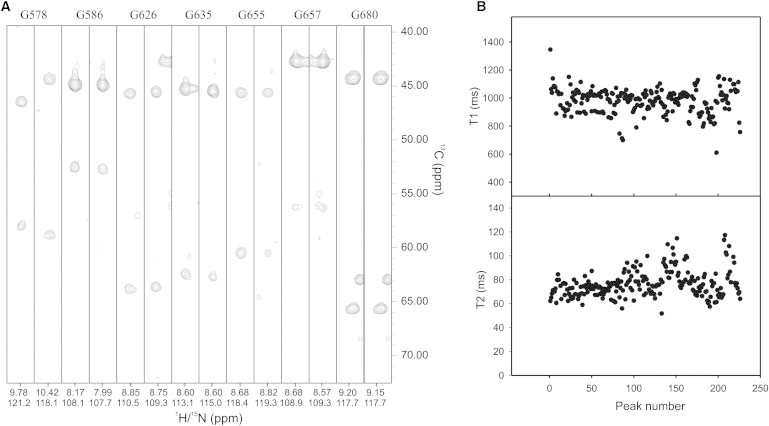
Further analysis of the spectral complexity. (A) Representative strips from a HNCA spectrum showing at least duplication of the glycine resonances. (B) T1 and T2 values for all peaked resonances. The spectra were recorded on a 0.4 mM protein solution at 27°C on a Varian-INOVA spectrometer operating at 600 MHz.
Even more revealing are the relaxation parameters: Plots of the T1 and T2 values of all nonoverlapping main intensity resonances (225) are relatively flat within the variation normally observed for compact globular proteins (Fig. 6 B). The estimated correlation time of wt-AXH at 27°C for a 0.4 mM sample concentration is ∼12 ns. A calibration plot recorded for proteins of different sizes at the same temperature predicts a correlation time of ∼8 ns for a protein of the molecular weight of the wt-AXH monomer at the same temperature. The value we observed is therefore in agreement with a dimer in the presence of an appreciable population of the monomer in a fast exchange regime. The contribution of the tetramer is likely to be in the broader resonances of lower intensities, as is expected for the molecular weight of this species (55 kDa). Accordingly, the correlation time at the same protein concentration estimated for the A567G-AXH mutant is 10 ns, in agreement with an increased contribution of the monomer population.
We can thus conclude that wt-AXH is present in solution as a mixture of species of different sizes in mutual equilibrium; however, some of the species also experience a conformational heterogeneity.
Discussion
This project was initially formulated with the goal of solving the solution structure of the AXH domain of ataxin-1 when no other structural information was available. The unexpected complexity of the spectrum hindered the work for a long time, although we could not understand its exact cause. It was only after the crystal structure was solved that it appeared clear that the AXH domain has an inherent plasticity so that a part of the protein can be trapped in different conformations. The solution structure of the homologous AXH domain from HBP1 supported this view because, although having similar secondary structure elements, it has a distinctly different fold (15). The exact role of these chameleon properties nevertheless remained elusive. Although described, asymmetric homo-dimeric interfaces are rarely observed in solution where molecular motions often average-out local differences (32,33). The scenario was further complicated by the presence of multiple states of oligomerization, as suggested by analytical ultracentrifugation data (12,18). It is only by an extensive analysis that we can now rationalize all our observations.
To explore the role of asymmetric assembly in the dimer, we searched for new structural solutions and obtained two new crystal structures, which we could then compare with the previous ones. Although the structures were crystallized under different conditions, they all belong to the same space group and are very similar in terms of unit cell dimensions, crystal packing, asymmetric unit content, and quaternary and monomeric backbone structures. The availability of 12 protomers from the three independent structures gave us an unusually rich collection of statistics. It confirms an inherent tendency of the AXH domain to show local variations in conformations and/or dynamics that can be frozen in the crystal environment. The N-terminus of the wt-AXH protomers adopts different conformations, especially around the dimer interfaces.
The crystal structures also suggested another potentially important source of complexity: dimers are further assembled as dimers of dimers. Accordingly, SAXS analysis is compatible with a model in which the isolated AXH domain is present not only as a dimer but also as a complex mixture of monomeric, dimeric, tetrameric, and higher molecular weight populations in equilibrium. This is not the only cause of the spectral complexity. We have now solid evidence suggesting that the structure of the dimeric species is not unique, as supported by the NMR analysis described above. This very complex mixture of species explains the objective difficulties encountered in interpreting the NMR and SAXS data. Our results lead us to conclude that the AXH domain is a unique example of a chameleon protein able to adopt multiple aggregation states as well as different local or global conformations.
While finally clarifying the original observations, this study has two potentially important implications that could be of direct relevance for the cellular properties of the AXH domain and to our understanding of the polyglutamine expansion disease SCA1:
The first potential implication is that our data suggest a mechanism by which AXH could recognize other proteins. Both the crystal structure and NMR measurements of wt-AXH clearly indicate that the predominant species in solution is a dimer, but in copresence with an appreciable population of monomer. This monomer must be unstable because its sickle-cell shape would leave the hydrophobic core exposed. Because the dimer is in equilibrium with the monomeric species, dimerization could easily be disrupted by a different partner (since our identification, the AXH domain of ataxin-1 has been implicated in several nonaberrant protein-protein interactions with several important protein partners). Dimerization and protein interactions could thus be competing pathways and the competition would then be a possible way to regulate protein functions.
The second potential implication concerns the tendency of ataxin-1 AXH to aggregate. Although several isolated domains aggregate in the absence of the protein context, we have previously demonstrated by domain deletion and swapping experiments in cells that ataxin-1 AXH (but not the homologous AXH domain from HBP1) plays a role in modulating the aggregation properties of polyglutamine tract (9). The AXH domain has also been proven to be essential for the self-association of full-length ataxin-1 (14). It is thus tempting to speculate that the monomer/dimer/tetramer equilibrium is an important part of the aggregation pathway of ataxin-1. Further studies will be needed to verify this hypothesis and provide a clearer understanding of the role of the AXH domain in the SCA1 pathology.
Acknowledgments
We thank Clelia Giannini for her contribution in the early stages of this work, John E. McCormick for preparation, Katrin Rittinger, Steve Smerdon, and Phil Walker for help and advice on crystallization, and Laura Masino for critical reading of the manuscript.
This work was supported by the Medical Research Council, UK (grant No. U117584256). M.R. was supported by an EPSRC Studentship.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Contributor Information
Yu Wai Chen, Email: yu-wai.chen@kcl.ac.uk.
Annalisa Pastore, Email: apastor@nimr.mrc.ac.uk.
Supporting Material
References
- 1.Orr H.T., Chung M.Y., Zoghbi H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 2.Zoghbi H.Y., Orr H.T. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J. Biol. Chem. 2008;284:7425–7429. doi: 10.1074/jbc.R800041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matilla-Dueñas A., Goold R., Giunti P. Clinical, genetic, molecular, and pathophysiological insights into spinocerebellar ataxia type 1. Cerebellum. 2008;7:106–114. doi: 10.1007/s12311-008-0009-0. [DOI] [PubMed] [Google Scholar]
- 4.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 5.Soto C., Estrada L.D. Protein misfolding and neurodegeneration. Arch. Neurol. 2008;65:184–189. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- 6.Klement I.A., Skinner P.J., Orr H.T. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 7.Emamian E.S., Kaytor M.D., Orr H.T. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 8.Masino L., Nicastro G., Pastore A. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J. Mol. Biol. 2004;344:1021–1035. doi: 10.1016/j.jmb.2004.09.065. [DOI] [PubMed] [Google Scholar]
- 9.de Chiara C., Menon R.P., Pastore A. Polyglutamine is not all: the functional role of the AXH domain in the ataxin-1 protein. J. Mol. Biol. 2005;354:883–893. doi: 10.1016/j.jmb.2005.09.083. [DOI] [PubMed] [Google Scholar]
- 10.Tsuda H., Jafar-Nejad H., Zoghbi H.Y. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 11.Saunders H.M., Bottomley S.P. Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng. Des. Sel. 2009;22:447–451. doi: 10.1093/protein/gzp033. [DOI] [PubMed] [Google Scholar]
- 12.de Chiara C., Giannini C., Pastore A. The AXH module: an independently folded domain common to ataxin-1 and HBP1. FEBS Lett. 2003;551:107–112. doi: 10.1016/s0014-5793(03)00818-4. [DOI] [PubMed] [Google Scholar]
- 13.de Chiara C., Pastore A. Polyglutamine Diseases and Neurodegeneration: The Example of Ataxin-1. In: Brnjas-Kraljević J., Pifat-Mrzljak G., editors. Supramolecular Structure and Function, Vol. 10. Springer; New York: 2011. pp. 87–99. [Google Scholar]
- 14.Burright E.N., Davidson J.D., Orr H.T. Identification of a self-association region within the SCA1 gene product, ataxin-1. Hum. Mol. Genet. 1997;6:513–518. doi: 10.1093/hmg/6.4.513. [DOI] [PubMed] [Google Scholar]
- 15.de Chiara C., Menon R.P., Pastore A. The AXH domain adopts alternative folds the solution structure of HBP1 AXH. Structure. 2005;13:743–753. doi: 10.1016/j.str.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Andreeva A., Murzin A.G. Evolution of protein fold in the presence of functional constraints. Curr. Opin. Struct. Biol. 2006;16:399–408. doi: 10.1016/j.sbi.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Yeates T.O. Protein structure: evolutionary bridges to new folds. Curr. Biol. 2007;17:R48–R50. doi: 10.1016/j.cub.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y.W., Allen M.D., Bycroft M. The structure of the AXH domain of spinocerebellar ataxin-1. J. Biol. Chem. 2004;279:3758–3765. doi: 10.1074/jbc.M309817200. [DOI] [PubMed] [Google Scholar]
- 19.Powell H.R. The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 1999;55:1690–1695. doi: 10.1107/s0907444999009506. [DOI] [PubMed] [Google Scholar]
- 20.Evans P.E. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 21.Strong M., Sawaya M.R., Eisenberg D. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA. 2006;103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCoy A.J., Grosse-Kunstleve R.W., Read R.J. PHASER crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams P.D., Afonine P.V., Zwart P.H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emsley P., Lohkamp B., Cowtan K. Features and development of COOT. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Painter J., Merritt E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 26.Davis I.W., Leaver-Fay A., Richardson D.C. MOLPROBITY: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35(Web Server issue):W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roessle M.W., Klaering R., Svergun D. Upgrade of the small-angle x-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Cryst. 2007;40:s190–s194. [Google Scholar]
- 28.Konarev P.V., Volkov V.V., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Cryst. 2003;36:1277–1282. [Google Scholar]
- 29.Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 30.Svergun D., Barberato C., Koch M.H. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 31.Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 32.Nooren I.M., George A.V., Boelens R. NMR structure determination of the tetramerization domain of the Mnt repressor: an asymmetric α-helical assembly in slow exchange. J. Biomol. NMR. 1999;15:39–53. doi: 10.1023/A:1008312309535. [DOI] [PubMed] [Google Scholar]
- 33.Nooren I.M., Kaptein R., Boelens R. The tetramerization domain of the Mnt repressor consists of two right-handed coiled coils. Nat. Struct. Biol. 1999;6:755–759. doi: 10.1038/11531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.