

Abstract
This study aims to identify the potential mechanisms by which perivascular adipose tissue (PVAT) reduces tone in small arteries. Small mesenteric arteries from wild-type and large-conductance Ca2+-activated K+ (BKCa) channel knockout mice were mounted on a wire myograph in the presence and absence of PVAT, and contractile responses to norepinephrine were assessed. Electrophysiology studies were performed in isolated vessels to measure changes in membrane potential produced by adiponectin. Contractile responses from wild-type mouse small arteries were significantly reduced in the presence of PVAT. This was not observed in the presence of a BKCa channel inhibitor or with nitric oxide synthase (NOS) inhibition or in BKCa or adiponectin knockout mice. Solution transfer experiments demonstrated the presence of an anticontractile factor released from PVAT. Adiponectin-induced vasorelaxation and hyperpolarization in wild-type arteries were not evident in the absence of or after inhibition of BKCa channels. PVAT from BKCa or adiponectin knockout mice failed to elicit an anticontractile response in wild-type arteries. PVAT releases adiponectin, which is an anticontractile factor. Its effect on vascular tone is mediated by activation of BKCa channels on vascular smooth muscle cells and adipocytes and by endothelial mechanisms.
Keywords: BKCa channels, perivascular adipose tissue, arteries, adipocyte, adiponectin
the release of adipokines from perivascular fat [perivascular adipose tissue (PVAT)] has been linked to reduced tone in a variety of vascular beds from a number of species including humans (11–13, 15). The possible health implications of PVAT are highlighted by the fact that such anticontractile activity is lost in obesity and disease states such as diabetes and hypertension (15).
A major factor secreted by adipocytes is adiponectin, a 30-kDa protein belonging to the collagen superfamily. Serum concentrations of adiponectin are lower in hypertension, obesity, and diabetes, and high blood pressure develops in high-fat diet-fed mice with adiponectin gene deletion (9, 21, 23, 41, 43).
The mechanisms by which PVAT produces its anticontractile effect have yet to be defined but could include activation of the vascular smooth muscle large-conductance Ca2+-activated K+ (BKCa) channel. This channel is known to be involved in the development of hypertension (44), downregulated in diabetes (35), and impaired in metabolic syndrome (2) and insulin resistance (6) and therefore may be involved in mediating PVAT anticontractility. It is a transmembrane channel composed of four α- and four β-subunits. The α-subunits form the pore, and the β-subunits have a regulatory role. The channel is activated by Ca2+ sparks and is located close to the sarcoplasmic reticulum. Therefore it is exposed to relatively high concentrations of Ca2+ ions, which bind to the α-subunit. This process, facilitated by the β-subunit, activates the channel (3, 45). In turn, this leads to K+ efflux and membrane hyperpolarization. Consequently, voltage-gated Ca2+ channels are closed, intracellular Ca2+ concentration is reduced, and vasodilation ensues.
The aim of the present investigation was to identify adipokines responsible for PVAT-induced anticontractility and to determine possible mechanisms of anticontractile transduction in a murine model. Using wire myography and electrophysiology to study mouse knockout models for both the α- and β-subunits of the BKCa channel, we provide evidence that suggests that PVAT-derived adiponectin is responsible for reducing vascular tone and acts by opening myocyte BKCa channels, thereby identifying a critical role for this channel in this important physiological process. We also demonstrate that adipocyte BKCa channels are involved in the response.
METHODS
Animal Models
Procedures were performed in accord with Institutional Guidelines and the United Kingdom Animals (Scientific) Procedures Act of 1986, and the work was conducted under licenses 40/03409 and S1 40/03409, issued by the British Home Office. Kcnma1−/− mice lacking the pore-forming slo1 or α-subunit of the BKCa channel (and wild-type Kcnma1+/+ counterparts expressing the α-subunit) and BKβ1−/− mice lacking the regulatory β1-subunit of the BKCa channel were bred in house at the University of Manchester. Targeted disruption of the Slo gene and the BKβ1 gene has been described previously (3, 36). Genotyping confirmed successful generation of Kcnma1 mice. B6.129-Adipoqtm1Chan/J adiponectin-deficient (adipo−/−) mice were purchased from The Jackson Laboratory and used at 8–10 wk of age. C57BL/6J mice were purchased from Harlan UK and used as wild-type/control animals for BKβ1−/− mice and adipo−/− mice. Male and female Kcnma1+/+, Kcnma1−/−, BKβ1−/−, and C57BL/6J mice (12–18 wk old, ∼25 g body wt) and adipo−/− mice were killed by stunning and cervical dislocation.
Wire Myography
The mesenteric beds from Kcnma1−/−, Kcnma1+/+ counterpart, BKβ1−/−, C57BL/6J, and adipo−/− mice were dissected, and small arteries were isolated. One vascular segment was cleaned of PVAT, while an adjacent section was left with PVAT intact. Both arteries were mounted on 40-μm tungsten wires in a myograph (Danish MyoTech, Aarhus, Denmark) containing physiological saline solution (PSS, in mmol/l: 119 NaCl, 4.7 KCl, 25 NaHCO3, 1.17 KH2PO4, 1.17 MgSO4, 0.026 EDTA, 1.6 CaCl2, and 5.5 glucose). Solutions were equilibrated with 95% air-5% CO2 gas. After an initial 30-min equilibration, vessel wall tension and diameter were normalized in a standardized procedure and stabilized for 1 h. Wire myography was used to assess the norepinephrine (1 × 10−9–3 × 10−5 mol/l)-induced contractile responses of arteries with and without PVAT under a number of conditions: control, in the presence and absence of endothelium, NG-nitro-l-arginine (l-NNA, 10−5 mol/l; Sigma Aldrich), paxilline (10−6 mol/l; Sigma Aldrich), indomethacin (10−6 mol/l; Sigma Aldrich), and adiponectin blocking peptide for PAb to adiponectin receptor 1 (AdipoR1) (5 × 10−3 g/ml; Enzo Life Sciences) (15). Modulators were preincubated for 30 min. Responses were normalized to an initial constriction to 60 mmol/l high-potassium solution (KPSS). The response to exogenously applied globular adiponectin (3 × 10−3 g/ml; Enzo Life Sciences) was also assessed in arteries preconstricted with 3 × 10−5 mol/l norepinephrine.
Solution Transfer Experiments
Arteries with and without PVAT were preconstricted with norepinephrine (3 × 10−5 mol/l). This maximal concentration was used to produce a uniform constriction for each artery and to avoid alteration of norepinephrine concentration in each bath upon transfer. Once a stable constriction had developed, the organ bath solution was taken from a donor wild-type (Kcnma1+/+ or C57BL/6J) small artery with PVAT and used to replace the solution from a recipient artery without PVAT.
PVAT Transfer Experiments
Mesenteric arteries were dissected as described above. PVAT was dissected from the mesenteric bed and stored in ice-cold PSS. Care was taken to ensure that no residual blood vessels remained in the PVAT sample. After the initial constriction to KPSS and cumulative concentration-response curves (CCRC) to norepinephrine, PVAT from a wild-type (C57BL/6J) mouse was positioned in close proximity to a wild-type or BKCa knockout (BKβ1−/−) artery lacking PVAT with a tungsten wire. A CCRC to norepinephrine was performed. After washout, PVAT from a knockout (BKβ1−/−) artery was then placed close to a wild-type (C57BL/6J) or knockout (BKβ1−/−) artery lacking PVAT and the CCRC was repeated. In a separate series of experiments, PVAT from an adipo−/− artery was placed beside a wild-type (C57BL/6J) artery lacking PVAT and a CCRC to norepinephrine was performed.
Electrophysiology
Small segments of mesenteric artery (1st or 2nd order) without PVAT but with intact endothelium, from Kcnma1+/+ and Kcnma1−/− mice, were pinned to the Sylgard base of a thermostatically controlled bath and superfused (10 ml/min) with PSS (and bubbled with 95% air-5% CO2). The myocytes were impaled via the adventitial surface with microelectrodes filled with 3 mol/l KCl (resistance 40–80 MΩ) as described previously (7) and in Table 1. Vessels were exposed to acetylcholine (10−5 mol/l), NS-1619 (3 × 10−5 mol/l), iberiotoxin (10−5 mol/l) and levcromakalim (10−5 mol/l) (see Fig. 5). Because of the high cost of globular adiponectin, flow was stopped and the peptide was added directly to the recording chamber to give a final concentration of 3 μg/ml.
Table 1.
EC50 and log EC50 data calculated from concentration-response curves to norepinephrine where values were normalized to maximum response from within each curve
| No PVAT |
PVAT |
|||||
|---|---|---|---|---|---|---|
| Group | EC50 | Log EC50 | EC50 | Log EC50 | No PVAT n | PVAT n |
| Kcnma1+/+ control | 3.3 × 10−6 | −6.0 ± 0.1 | 2.3 × 10−5 | −5.5 ± 0.1* | 11 | 11 |
| C57BL/6J | 1.1 × 10−6 | −6.1 ± 0.1 | 3.2 × 10−5 | −6.0 ± 0.1 | 10 | 10 |
| C57BL/6J + blocking peptide | 1.1 × 10−6 | −6.1 ± 0.1 | 9.4 × 10−7 | −6.1 ± 0.1 | 5 | 8 |
| adipo−/− | 7.1 × 10−6 | −6.3 ± 0.2 | 7.2 × 10−6 | −5.6 ± 0.8 | 12 | 12 |
| Kcnma1+/+ no endothelium | 5.3 × 10−7 | −6.5 ± 0.2 | 4.1 × 10−5 | −5.7 ± 0.5 | 7 | 6 |
| C57BL/6J no endothelium | 1.1 × 10−6 | −6.1 ± 0.1 | 1.7 × 10−6 | −5.8 ± 0.1 | 6 | 5 |
| Kcnma1+/+ l-NNA | 4.2 × 10−7 | −6.5 ± 0.1 | 8.0 × 10−7 | −5.7 ± 0.5 | 6 | 6 |
| C57BL/6J l-NNA | 1.2 × 10−6 | −6.0 ± 0.1 | 1.5 × 10−6 | −5.9 ± 0.1 | 5 | 6 |
| Kcnma1+/+ indomethacin | 2.4 × 10−6 | −5.0 ± 0.7 | 4.8 × 10−6 | −5.6 ± 0.5 | 6 | 6 |
| Kcnma1+/+ l-NNA + indomethacin | 4.4 × 10−6 | −5.3 ± 0.8 | 1.0 × 10−6 | −6.1 ± 0.2 | 6 | 6 |
| Kcnma1−/− control | 9.7 × 10−6 | −6.1 ± 0.1 | 4.8 × 10−6 | −5.8 ± 0.2 | 10 | 10 |
| BKβ1−/− control | 1.7 × 10−4 | −5.2 ± 0.1 | 4.5 × 10−5 | −5.5 ± 0.3 | 16 | 13 |
| Kcnma1+/+ paxilline | 1.0 × 10−6 | −6.2 ± 0.2 | 1.4 × 10−6 | −6.0 ± 0.2 | 5 | 5 |
| C57BL/6J paxilline | 6.5 × 10−7 | −6.3 ± 0.1 | 3.0 × 10−6 | −5.9 ± 0.1 | 11 | 11 |
| PVAT transfer experiments | ||||||
| C57 control | 1.4 × 10−6 | −6.0 ± 0.2 | 10 | 10 | ||
| +C57 transferred PVAT | 9.5 × 10−7 | −6.2 ± 0.1 | 10 | |||
| +BKβ1−/− PVAT | 5.3 × 10−7† | −7.7 ± 0.7† | 10 | |||
| BKβ1−/− + C57 transferred PVAT | 2.3 × 10−6 | −6.0 ± 0.2 | 10 | |||
| adipo−/− + C57 transferred PVAT | 2.1 × 10−6 | −5.7 ± 0.1 | 6 | |||
PVAT, perivascular adipose tissue; l-NNA, NG-nitro-l-arginine.
P < 0.05, t-test, PVAT vs. no PVAT;
P < 0.05, t-test, vs. PVAT transfer experiments C57 control.
Fig. 5.

Typical traces showing sharp electrode recordings from isolated mesenteric artery without PVAT from Kcnma1+/+ (A) and Kcnma1−/− (B) mice. The presence of BKCa channels was confirmed by hyperpolarization in response to NS-1619 (BKCa channel opener). Acetylcholine (ACh) confirms endothelium presence. Levcromakalim (LK) hyperpolarizes via ATP-sensitive K+ (KATP) channels. Iberiotoxin (IbTx) reverses adiponectin-induced hyperpolarization. m.p., Membrane potential.
Statistical Analyses
Data are expressed as means ± SE. For cumulative response curves, differences between dose-tension relations were analyzed by repeated-measures ANOVA and Bonferroni post hoc tests were used to calculate significance at different concentrations. EC50 and log EC50 analysis was calculated as follows. The maximum response to KPSS was calculated as the difference between baseline and the maximum tension elicited by exposure to KPSS. Constrictions to norepinephrine were normalized to the maximum response within each curve, and the EC50 and log EC50 were calculated from this curve. These data were analyzed with Student's t-test and are presented in Table 1. Responses to exogenous adiponectin and electrophysiology data were analyzed with Student's t-test. A probability value of P < 0.05 was considered statistically significant. Analyses were performed with GraphPad Prism version 3.00 for Windows (GraphPad Software).
RESULTS
Wire Myography: PVAT Has an Anticontractile Effect in Murine Small Arteries
The response to a KPSS constriction performed at the beginning of the protocol was used to normalize the data presented. The maximum response to 60 mmol/l KPSS (ΔmN/mm ± SE) produced by arteries from Kcnma1+/+ mice (n = 57) with PVAT (0.31 ± 0.03) was not significantly different from that produced by arteries from Kcnma1+/+ mice (n = 57) without PVAT (0.38 ± 0.05). Responses to KPSS produced by arteries from Kcnma1−/− mice (n = 36) with PVAT (0.37 ± 0.1) were not significantly different from those from arteries (n = 37) lacking PVAT (0.42 ± 0.04). Comparison of responses to KPSS from arteries from BKβ1−/− mice (n = 34) surrounded by PVAT (0.4 ± 0.07) revealed no significant differences from their PVAT-lacking (n = 34) counterparts (0.34 ± 0.05). Similarly, no differences were detected between arteries from C57BL/6J mice (n = 56) with PVAT (0.33 ± 0.04) and those without PVAT (0.4 ± 0.04) in response to KPSS.
Cumulative concentrations of norepinephrine produced constriction in all groups. Wild-type [Kcnma1+/+ (n = 11) and C57BL/6J (n = 10)] vessels with PVAT constricted significantly less than those without PVAT (Fig. 1, A and B, respectively). A significant shift (P < 0.05, test) was detected in log EC50 values calculated in Kcnma1+/+ arteries (−6.0 ± 0.1 no PVAT vs. −5.5 ± 0.4 PVAT). There was no difference in EC50 values from C57BL/6J arteries with or without PVAT (Table 1).
Fig. 1.
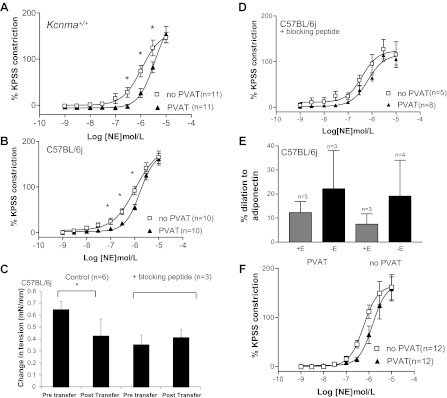
A and B: contractile responses of mesenteric Kcnma1+/+ (A) and C57BL/6J (B) arteries to cumulative concentrations of norepinephrine (NE). PVAT, perivascular adipose tissue; KPSS, high-potassium physiological saline solution. C: solution transfer experiments. D: C57BL/6J arteries in the presence of adiponectin blocking peptide. E: response to exogenous adiponectin in the presence (+E) and absence (−E) of endothelium. F: adipo−/− arteries. *P < 0.05 ANOVA.
Organ Bath Solution Transfer Studies Demonstrate That Adipocytes Release a Vasorelaxing Factor
To ensure that a similar degree of constriction was achieved in each artery (maximum) and that the concentration of norepinephrine remained constant, arteries with and without PVAT were preconstricted with norepinephrine (3 × 10−5 mol/l). Once a stable constriction had developed, the organ bath solution was taken from a donor C57BL/6J small artery with PVAT and used to replace the solution from a recipient artery without PVAT. Control experiments transferring solution to and from wild-type arteries without PVAT (n = 4) were performed to determine whether the transfer maneuver would alter tension. Such a transfer did not significantly alter tension (from 5.5 ± 0.1 to 5.5 ± 0.1 mN/mm). Solutions from wild-type vessels (C57BL/6J) with PVAT (n = 6) caused a significant reduction in tension (P < 0.05, t-test) when transferred onto wild-type arteries (from C57BL/6J mice) without PVAT (Fig. 1C). The presence of adiponectin blocking peptide for AdipoR1 (n = 3) inhibited this effect (Fig. 1C). AdipoR1 peptide blockade (n = 8) also inhibited the PVAT-induced anticontractile response during norepinephrine concentration-response curves in C57BL/6J arteries (Fig. 1D).
Application of Exogenous Adiponectin Induces Vascular Relaxation
Applying globular adiponectin to C57BL/6J arteries preconstricted with norepinephrine (3 × 10−5 mol/l) caused dilations in both the presence and the absence of PVAT and in the presence and absence of endothelium that were significantly different from zero (P < 0.05, paired t-test) (Fig. 1E). Interestingly, arteries from Kcnma1+/+ mice without PVAT (n = 4) also dilated significantly (33.4 ± 10.6%) to adiponectin, but there was no response from arteries from Kcnma1+/+ mice with PVAT.
Anticontractile Effect Is Absent in Adiponectin Knockout Mice
The contractile responses to norepinephrine were assessed in arteries from mice lacking adiponectin. There were no significant differences in responses in the presence or absence of PVAT in arteries from adipo−/− animals (n = 6) (Fig. 1F).
Endothelial Contribution to PVAT Activity
Endothelium removal abolished the anticontractile effects of PVAT response in wild-type (Kcnma1+/+ and C57BL/6J) arteries (Fig. 2, A and B). The anticontractile effect was absent in the presence of the nitric oxide synthase (NOS) inhibitor l-NNA in intact wild-type (Kcnma1+/+ and C57BL/6J) arteries (Fig. 2, C and D). Inhibition of prostacyclins with indomethacin also inhibited the anticontractile response in Kcnma1+/+ arteries (Fig. 2E). Combination of l-NNA and indomethacin confirmed these data (Fig. 2F). The presence of indomethacin and l-NNA abolished the anticontractile effect but also significantly increased tension (P < 0.05, ANOVA) compared with arteries lacking PVAT. There were no significant differences in EC50 values between any of the groups (Table 1).
Fig. 2.
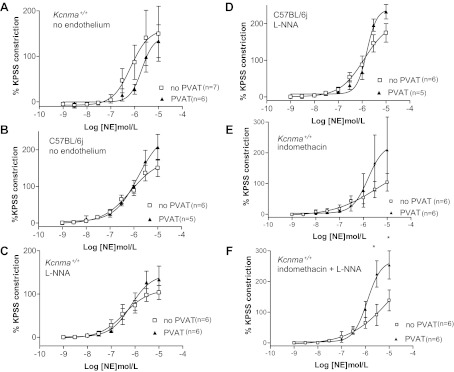
A and B: contractile responses of mesenteric Kcnma1+/+ (A) and C57BL/6J (B) arteries to cumulative concentrations of norepinephrine in the absence of endothelium. C and D: contractile responses of Kcnma1+/+ (C) and C57BL/6J (D) arteries + NG-nitro-l-arginine (l-NNA). E and F: responses of Kcnma1+/+ arteries + indomethacin (E) and + indomethacin and l-NNA (F). *P < 0.05 ANOVA.
Vascular BKCa Activity Is Crucial to PVAT Relaxation
Deletion of the α (Kcnma1−/− mice)- or β (BKβ1−/− mice)-subunit of the BKCa channel resulted in a reduction of the anticontractile response (Fig. 3, A and B). The result remained the same irrespective of whether the endothelium was present or not (data not shown).
Fig. 3.
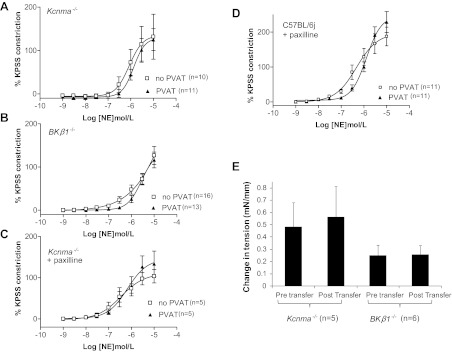
Role of the large-conductance Ca2+-activated K+ (BKCa) channel in mediating the PVAT response. A–D: constrictions produced by Kcnma1−/− (A) and BKβ1−/− (B) arteries to norepinephrine and Kcnma1+/+ (C) and C57BL/6J (D) arteries in the presence of paxilline. E: solution transfer experiments showing the tension produced in Kcnma1−/− and BKβ1−/− arteries constricted with norepinephrine before and after transfer.
Pharmacological inhibition of the BKCa channel with paxilline also abolished the response in both Kcnma1+/+ and C57BL/6J mice (Fig. 3, C and D). There was no significant difference in tension developed regardless of the presence of PVAT. There were no significant differences in EC50 values between any of the groups (Table 1).
Anticontractile Factor Released from PVAT Requires BKCa Channels to Elicit Effects
Transfer of solution from Kcnma1+/+ (n = 5) and C57BL/6J (n = 6) arteries with PVAT to arteries lacking PVAT from Kcnma1−/− and BKβ1−/− mice failed to elicit a change in tension (Fig. 3E).
BKβ1−/− PVAT Does Not Elicit an Anticontractile Effect
In this series of experiments PVAT was taken from a mouse and transferred to arteries lacking PVAT to investigate whether the type of PVAT (wild type or a knockout variety) itself can influence the vascular response. The data are labeled as follows: an artery lacking PVAT in the presence of transferred wild-type or knockout PVAT is labeled “no PVAT (WT)” or “no PVAT (KO),” respectively.
PVAT from a C57BL/6J mouse (n = 10) was placed next to a C57BL/6J artery lacking PVAT [Fig. 4A, no PVAT (WT)]. This significantly reduced tension (P < 0.05, ANOVA) compared with response elicited before PVAT transfer (Fig. 4A, no PVAT). The response in the presence of transferred PVAT was not significantly different from that of a C57BL/6J artery surrounded by its own PVAT (Fig. 4A, PVAT).
Fig. 4.
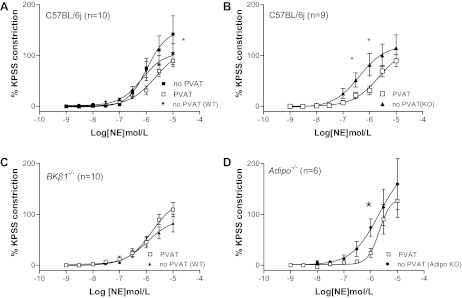
A: effects of transferred C57BL/6J PVAT to arteries lacking PVAT [no PVAT (WT)] (*P < 0.05 ANOVA vs. no PVAT). B: tension elicited by C57BL/6J arteries lacking PVAT in the presence of transferred BKβ1−/− PVAT [no PVAT (KO)] (*P < 0.05 ANOVA vs. PVAT). C: effects of transferred PVAT from C57BL/6J mice to BKβ1−/− arteries lacking PVAT [no PVAT (WT)]. D: constrictions elicited by C57BL/6J arteries lacking PVAT in the presence of transferred PVAT from adipo−/− mice [no PVAT (Adipo KO)] (*P < 0.05 ANOVA vs. PVAT).
When PVAT from a BKβ1−/− mouse (n = 10) was incubated with a C57BL/6J artery lacking PVAT [Fig. 4B, no PVAT (KO)], the tension elicited was significantly greater (P < 0.05, ANOVA) compared with the response of a C57BL/6J artery with PVAT (Fig. 4B, PVAT). EC50 and log EC50 were significantly shifted under these conditions (Table 1).
The presence of C57BL/6J PVAT (n = 10) next to a BKβ1−/− artery lacking PVAT [Fig. 4C, no PVAT (WT)] did not produce a significantly different response compared with a BKβ1−/− artery surrounded by PVAT (Fig. 4C, PVAT).
Finally, incubation of PVAT (n = 6) taken from an adipo−/− mouse failed to induce anticontractility in a C57BL/6J artery lacking PVAT [Fig. 4D, no PVAT (Adipo KO)]. The constrictions under these conditions were significantly greater (P < 0.05, ANOVA) compared with a C57BL/6J artery surrounded by native PVAT (Fig. 4D, PVAT).
Adiponectin Has No Effects in the Absence of the BKCa Channel
Myography.
Addition of exogenous globular adiponectin to preconstricted wild-type arteries (Kcnma1+/+ and C57BL/6J) resulted in a vasodilation (Fig. 1D). Application of adiponectin to arteries with or without PVAT from knockout mice (both Kcnma1−/− and BKβ1−/−) did not elicit a dilation [Kcnma1−/− − PVAT = 8.4 ± 7.4% (n = 4); Kcnma1−/− + PVAT = −7.5 ± 5.4% (n = 4); BKβ1−/− − PVAT = −0.8 ± 1.6% (n = 5); BKβ1−/− + PVAT = −7.1 ± 2.9%; (n = 3)].
Electrophysiology.
The resting membrane potential of smooth muscle cells in Kcnma1+/+ arteries was −53.6 ± 0.4 mV (n = 6). This was not significantly different from that of Kcnma1−/− arteries (−51.4 ± 0.8 mV) (n = 7). The presence of a functional endothelium was confirmed by a hyperpolarization in response to acetylcholine. There was no significant difference in change in membrane potential between Kcnma1+/+ (13.9 ± 1.0 mV) and Kcnma1−/− knockout (16.4 ± 1.4 mV) arteries. Levcromakalim confirmed the presence of ATP-sensitive K+ (KATP) channels in the arteries. There were no differences in the magnitude of the levcromakalim-induced hyperpolarization between Kcnma1+/+ (−23.6 ± 1.7 mV) and Kcnma1−/− (−26.2 ± 1.8 mV) arteries. Furthermore, a hyperpolarization to levcromakalim occurred in Kcnma1−/− arteries, which were unresponsive to adiponectin (Fig. 5B). There were significant differences (P < 0.01, t-test) in the responses of Kcnma1+/+ arteries (−15.2 ± 0.7 mV) and Kcnma1−/− arteries (0.0 ± 0.13 mV) to the BKCa channel opener NS-1619 (3.3 × 10−5 mol/l), confirming the absence of the channel in the Kcnma1−/− mouse. Adiponectin produced a change in membrane potential of −10.5 ± 0.7 mV in Kcnma1+/+ arteries, which was attenuated (−0.1 ± 0.2 mV) by the BKCa channel inhibitor iberiotoxin. The failure of adiponectin to generate myocyte hyperpolarization (−0.4 ± 0.3 mV) in Kcnma1−/− vessels indicated that this channel is essential in mediating the electrical effects of this adipokine (Fig. 5B). The lack of hyperpolarization in responses to adiponectin in the absence of the BKCa channel indicates that this channel is essential in mediating the effect.
DISCUSSION
These studies have been carried out in small arteries from both wild-type mice (Kcnma1+/+ and C57BL/6J) and strains without functional BKCa channels (Kcnma1−/− and BKβ1−/−). The results demonstrate that the white PVAT that surrounds such vessels releases a substance that can reduce tension in preconstricted arteries and that this appears to be mediated by the activation of BKCa channels. In mice without BKCa channels, the relaxant response is significantly reduced when either the functional pore or the regulatory domain is absent. Our hypothesis was further tested pharmacologically in arteries from wild-type mice, in which the effect was abolished with the BKCa channel blocker paxilline. Solution transfer studies confirm that the relaxing factor is released from PVAT. Contractility studies using arteries from adiponectin knockout animals and adiponectin blocking peptide protocols here and in our previously published work (15) indicate that these data are consistent with the involvement of adiponectin. Therefore exogenous globular adiponectin was applied to arteries from wild-type mice and shown to cause relaxation. Average circulating levels of total adiponectin in the human circulation of up to 30 μg/ml have been reported (1, 50), while murine levels are lower, >10 μg/ml (38). Similarly, the application of globular adiponectin caused hyperpolarization that could be abolished with the BKCa channel blocker iberiotoxin, again indicating activation of BKCa, and this effect was not observed in vessels without BKCa. PVAT-induced relaxation also involves endothelial release of nitric oxide (NO) consistent with reported adiponectin effects (4, 5, 47); this appears to be dependent on BKCa activation because no such effect was seen in arterial tissue from BKCa knockout mice. PVAT from BKCa knockout mice was unable to produce an anticontractile effect in wild-type arteries, suggesting that adipocyte BKCa channels are involved in adiponectin signaling.
Our results support the ubiquitous nature of PVAT-induced anticontractility. The demonstration of PVAT-induced anticontractility in murine small arteries adds to the abundance of evidence in other vascular beds including human subcutaneous vessels, internal mammary artery, rat mesenteric, and rat aorta (14, 15). The possible clinical relevance of the effect is highlighted by its absence or reduced efficacy in cardiovascular diseases (15): hypertension is associated with a reduced PVAT response in rats (10), and recently Lu and coworkers (32) reported a functional impairment of the anticontractile response in aortic rings from spontaneously hypertensive rats, which they attributed to angiotensin release. Furthermore, anticontractility is impaired in obesity in both animal and human models (15, 25), and data from diabetic models suggest that anticontractility is also decreased (28).
The most important finding in our study is the pivotal role the BKCa channel appears to play in mediating PVAT-induced anticontractility. Kcnma1−/− mice lacking a functional pore or BKβ1−/− mice lacking the regulatory β-subunit demonstrate a significant reduction in anticontractility, either during concentration-response curves to norepinephrine or via solution transfer studies. It has been reported that the BKCa channel retains some functionality in the absence of the regulatory β1-subunit (3), and because the PVAT effect is reduced in β1-subunit knockout arteries, this would imply that a Ca2+ signal is important in the signaling pathway from adipocyte to myocyte BKCa channel.
The use of an organ bath solution transfer protocol demonstrates that a factor is released from PVAT that influences both endothelial and smooth muscle function. The solution from a preconstricted wild-type artery surrounded with PVAT causes significant relaxation of preconstricted vessels devoid of PVAT. Several studies have used this technique to identify the presence of a transferable adipose-derived relaxing factor (ADRF) (15, 28). A recent review has compared the blossoming field of adipocyte research to that of the endothelial field in the 1980s (46). This may well be the case, the adipocyte being a complex signaling cell with interconnected molecular pathways, each being activated by different stimuli. Such a situation could account for the diverse candidates being proposed as PVAT-derived relaxing factor.
Current literature points to a number of possible candidates for the PVAT-derived relaxing factor, including Ang-(1–7) (27), H2S2 (42), and palmitic acid methyl ester (29). The target channels of PVAT modulators are also the subject of much discussion and debate. Potassium channels such as delayed-rectifier (KV) channels (49), KATP channels (31), as well as small- and intermediate-conductance Ca2+-sensitive potassium channels (12) appear to play a central role in various PVAT pathways.
Previously, we successfully inhibited the PVAT anticontractile effect in human gluteal arteries by using a peptide blocking the effects of adiponectin, thereby suggesting that adiponectin is responsible (15), and achieved similar results in our preparation. Furthermore, contractile experiments performed on adiponectin-deficient mice demonstrated a lack of PVAT anticontractile effect. These findings contrast with those of an earlier study (9) in which an anticontractile effect of PVAT was still observed in mesenteric arteries from adiponectin-deficient mice. However, the superior mesenteric artery and not smaller mesenteric arteries were used in the aforementioned study and arteries were stimulated with serotonin, which suggests that both vessel caliber and stimulators are important when considering experimental design. PVAT from adiponectin-deficient mice failed to reduce tone in wild-type arteries lacking PVAT, suggesting that PVAT-derived adiponectin is required to elicit an anticontractile response. We confirmed the ability of our arteries to respond to adiponectin by observing a relaxation in response to exogenous globular adiponectin. We used globular adiponectin, which is known to act via AdipoR1 and has a low affinity for AdipoR2 (23, 55). In our study there appeared to be a greater effect of adiponectin in the absence of the endothelium, which could potentially be attributed to adiponectin having direct access to the smooth muscle layer (8). However, in vivo one would expect adiponectin to encounter the adventitia first and then the smooth muscle layer. Our results would therefore suggest that a vasocrine signaling system where factors are first released into the bloodstream is at work. Vasocrine signaling mechanisms have been previously reported where factors released from PVAT influence vascular behavior (57). Additionally, we see no effect of adiponectin in arteries from Kcnma+/+ arteries with PVAT, which, again, may reflect an access problem or may be attributable to saturation of native adiponectin receptors with endogenous adiponectin. In our electrophysiology experiments direct application of adiponectin failed to generate myocyte hyperpolarization in vessels derived from Kcnma1−/− mice and also failed to elicit relaxation in bath solution transfer experiments in similar vessels. Our electrophysiology experiments also showed that adiponectin hyperpolarized wild-type arteries and that this was reversible by inhibition of BKCa channels with iberiotoxin. Such results, together with our other findings, demonstrate that adiponectin is likely to be the key anticontractile component liberated from PVAT.
Adiponectin acts via at least three receptors, AdipoR1, AdipoR2, and T-cadherin (5, 23, 47, 55). AdipoR1 has been identified on both smooth muscle and endothelial cells. Therefore inhibition of AdipoR1 may affect not only receptors on the smooth muscle but also those present on the endothelium. Our study demonstrates a considerable role for endothelium-derived factors in mediating the anticontractile effect; both inhibition of NOS and prostacyclin abolish the anticontractile effect. There is a residual (although not significant) effect of PVAT in endothelium-denuded arteries from Kcnma1+/+ mice, which leads us to believe that the effect is not fully mediated via the endothelium but also has a smooth muscle component. Adiponectin has well-documented endothelial protective mechanisms and can increase production of NO (4, 5, 47). This may explain why inhibition of NOS inhibits the PVAT effect in our preparation. In rat aorta, Gao and colleagues (12) have demonstrated a role for endothelium-derived NO in the response and Greenstein et al. (15) reported a blunting of the anticontractility in healthy human arteries in the presence of endothelial NOS inhibitors. We also provide data to show that combination of l-NNA and indomethacin significantly increases tension in arteries surrounded by PVAT. This demonstrates the importance of NO and prostaglandins as mediators of the adiponectin- and PVAT-induced anticontractility. It has been previously reported that salt-fed adiponectin-deficient mice that develop hypertension have low levels of NOS and prostaglandins that can be reversed with adiponectin treatment (37). Additionally, there is also evidence of an interlinked NO pathway cyclooxygenase in endothelial cells where one will compensate for a reduction in levels of the other (48). It may be that a similar pathway exists in our PVAT preparation and that inhibiting both leads to an increase in tension.
Transfer of PVAT from arteries lacking the BKCa channel does not induce an anticontractile effect on arteries carrying the channel, which suggests that adipocyte BKCa channels are involved in the response. Potassium currents (39, 40) and BKCa channel currents have been identified on human preadipocytes and play a role in differentiation (20). It is possible that BKCa channels may influence adipocyte membrane potential and in turn influence the release of adiponectin. It has been suggested by others that adipocyte membrane potential may play a role in the basal metabolic activity of adipocytes (39, 40). Recently, release of PVAT relaxing factor from rat aortic rings has been shown to be dependent on the presence of adipocyte calcium (29). As the BKCa channel requires Ca2+ for activation, this is suggestive of a role for the channel in the release of adiponectin. However, it must be pointed out that in this group's experiments iberiotoxin did not alter responses.
Therefore we propose that adiponectin binds to AdipoR1 on smooth muscle cells, which activates BKCa channels, and the resultant hyperpolarization leads to vasodilation. How adiponectin receptor activation in turn activates BKCa channels remains to be elucidated. A strong candidate for communicating the effects of adiponectin to the BKCa channel is the heterotrimeric serine/threonine protein kinase AMP-activated protein kinase (AMPK) (16). AMPK dysregulation has been linked with obesity and the vascular consequences of metabolic syndrome (17). Also, there is evidence to suggest that this pathway will be altered in obesity because AMPK expression is significantly decreased in obese rats (33). Adiponectin is known to activate AMPK in a number of cell types (4, 26, 54, 56). This results in stimulation of NO production in endothelial cells, which may contribute to PVAT anticontractility, and AMPK-induced release of Ca2+ from smooth muscle cell stores (4, 18, 22, 24, 34, 53, 58). The Ca2+ release may trigger BKCa activation. Indeed, this may explain why the presence of the Ca2+-sensing β-subunit is important in the PVAT anticontractile response. Further studies are in progress to determine whether adiponectin activation of AMPK activates in turn a plasmalemmal channel to allow Ca2+ entry, thereby provoking BKCa opening. NO can also stimulate PKG (19, 30), which in turn is known to activate BKCa channels (51). Data recently published from our lab (52) demonstrated that loss of PKG function inhibits the anticontractile effect.
In summary, the present data demonstrate that the BKCa channel is crucial in mediating the anticontractile effect of PVAT, at least in murine mesenteric arteries. Adiponectin released from PVAT activates AdipoR1 on endothelial and myocytes; the subsequent activation of BKCa leading to myocyte hyperpolarization, along with an endothelial contribution, decreases vascular tone.
GRANTS
This work was funded by the Wellcome Trust and The British Heart Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.M.L., S.B.W., and A.M.H. conception and design of research; F.M.L., S.B.W., Z.Y., M.E.W., G.E., and A.H.W. performed experiments; F.M.L., Z.Y., and A.H.W. analyzed data; F.M.L. and A.M.H. interpreted results of experiments; F.M.L. prepared figures; F.M.L. drafted manuscript; F.M.L., S.B.W., M.E.W., G.E., A.H.W., and A.M.H. edited and revised manuscript; F.M.L., S.B.W., Z.Y., M.E.W., G.E., A.H.W., and A.M.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Meredith, Brenner, and Aldrich for providing the Kcnma1−/−, Kcnma1+/+, and BKβ1−/− mice.
REFERENCES
- 1. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257: 79–83, 1999 [DOI] [PubMed] [Google Scholar]
- 2. Borbouse L, Dick GM, Asano S, Bender SB, Dincer UD, Payne GA, Neeb ZP, Bratz IN, Sturek M, Tune JD. Impaired function of coronary BKCa channels in metabolic syndrome. Am J Physiol Heart Circ Physiol 297: H1629–H1637, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the beta1 subunit of the calcium-activated potassium channel. Nature 407: 870–876, 2000 [DOI] [PubMed] [Google Scholar]
- 4. Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 278: 45021–45026, 2003 [DOI] [PubMed] [Google Scholar]
- 5. Cheng KK, Lam KS, Wang Y, Huang Y, Carling D, Wu D, Wong C, Xu A. Adiponectin-induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 56: 1387–1394, 2007 [DOI] [PubMed] [Google Scholar]
- 6. Dimitropoulou C, Han G, Miller AW, Molero M, Fuchs LC, White RE, Carrier GO. Potassium (BKCa) currents are reduced in microvascular smooth muscle cells from insulin-resistant rats. Am J Physiol Heart Circ Physiol 282: H908–H917, 2002 [DOI] [PubMed] [Google Scholar]
- 7. Edwards G, Gardener MJ, Feletou M, Brady G, Vanhoutte PM, Weston AH. Further investigation of endothelium-derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1-EBIO and ouabain. Br J Pharmacol 128: 1064–1070, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Egner I, Waseton A, Porter E, Edwards G. Hyperpolarising effect of perivascular adipose tissue in rat mesenteric artery myocyte: stimulation by β3 adrenoceptor activation (Abstract). Proceedings of the British Pharmacological Society, 2010 (http://wwwpa2onlineorg/abstracts/vol8issue1) [Google Scholar]
- 9. Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC, Gollasch M. Adiponectin is a novel humoral vasodilator. Cardiovasc Res 75: 719–727, 2007 [DOI] [PubMed] [Google Scholar]
- 10. Galvez B, de Castro J, Herold D, Dubrovska G, Arribas S, Gonzalez MC, Aranguez I, Luft FC, Ramos MP, Gollasch M, Fernandez Alfonso MS. Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 26: 1297–1302, 2006 [DOI] [PubMed] [Google Scholar]
- 11. Gao YJ. Dual modulation of vascular function by perivascular adipose tissue and its potential correlation with adiposity/lipoatrophy-related vascular dysfunction. Curr Pharm Des 13: 2185–2192, 2007 [DOI] [PubMed] [Google Scholar]
- 12. Gao YJ, Lu C, Su LY, Sharma AM, Lee RM. Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol 151: 323–331, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM, Lee RM. Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res 71: 363–373, 2006 [DOI] [PubMed] [Google Scholar]
- 14. Gao YJ, Zeng ZH, Teoh K, Sharma AM, Abouzahr L, Cybulsky I, Lamy A, Semelhago L, Lee RM. Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J Thorac Cardiovasc Surg 130: 1130–1136, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M, Laing I, Yates AP, Pemberton PW, Malik RA, Heagerty AM. Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 119: 1661–1670, 2009 [DOI] [PubMed] [Google Scholar]
- 16. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8: 774–785, 2007 [DOI] [PubMed] [Google Scholar]
- 17. Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 32, Suppl 4: S7–S12, 2008 [DOI] [PubMed] [Google Scholar]
- 18. Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2: 9–19, 2005 [DOI] [PubMed] [Google Scholar]
- 19. Hofmann F. The biology of cyclic GMP-dependent protein kinases. J Biol Chem 280: 1–4, 2005 [DOI] [PubMed] [Google Scholar]
- 20. Hu H, He ML, Tao R, Sun HY, Hu R, Zang WJ, Yuan BX, Lau CP, Tse HF, Li GR. Characterization of ion channels in human preadipocytes. J Cell Physiol 218: 427–435, 2009 [DOI] [PubMed] [Google Scholar]
- 21. Hui X, Lam KS, Vanhoutte PM, Xu A. Adiponectin and cardiovascular health: an update. Br J Pharmacol 165: 574–590, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iwabu M, Yamauchi T, Okada-Iwabu M, Sato K, Nakagawa T, Funata M, Yamaguchi M, Namiki S, Nakayama R, Tabata M, Ogata H, Kubota N, Takamoto I, Hayashi YK, Yamauchi N, Waki H, Fukayama M, Nishino I, Tokuyama K, Ueki K, Oike Y, Ishii S, Hirose K, Shimizu T, Touhara K, Kadowaki T. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca2+ and AMPK/SIRT1. Nature 464: 1313–1319, 2010 [DOI] [PubMed] [Google Scholar]
- 23. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116: 1784–1792, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25, 2005 [DOI] [PubMed] [Google Scholar]
- 25. Ketonen J, Shi J, Martonen E, Mervaala E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ J 74: 1479–1487, 2010 [DOI] [PubMed] [Google Scholar]
- 26. Kubota N, Yano W, Kubota T, Yamauchi T, Itoh S, Kumagai H, Kozono H, Takamoto I, Okamoto S, Shiuchi T, Suzuki R, Satoh H, Tsuchida A, Moroi M, Sugi K, Noda T, Ebinuma H, Ueta Y, Kondo T, Araki E, Ezaki O, Nagai R, Tobe K, Terauchi Y, Ueki K, Minokoshi Y, Kadowaki T. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab 6: 55–68, 2007 [DOI] [PubMed] [Google Scholar]
- 27. Lee RM, Lu C, Su LY, Gao YJ. Endothelium-dependent relaxation factor released by perivascular adipose tissue. J Hypertens 27: 782–790, 2009 [DOI] [PubMed] [Google Scholar]
- 28. Lee RM, Lu C, Su LY, Werstuck G, Gao YJ. Effects of hyperglycemia on the modulation of vascular function by perivascular adipose tissue. J Hypertens 27: 118–131, 2009 [DOI] [PubMed] [Google Scholar]
- 29. Lee YC, Chang HH, Chiang CL, Liu CH, Yeh JI, Chen MF, Chen PY, Kuo JS, Lee TJ. Role of perivascular adipose tissue-derived methyl palmitate in vascular tone regulation and pathogenesis of hypertension. Circulation 124: 1160–1171, 2011 [DOI] [PubMed] [Google Scholar]
- 30. Lohmann SM, Walter U. Tracking functions of cGMP-dependent protein kinases (cGK). Front Biosci 10: 1313–1328, 2005 [DOI] [PubMed] [Google Scholar]
- 31. Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB J 16: 1057–1063, 2002 [DOI] [PubMed] [Google Scholar]
- 32. Lu C, Su LY, Lee RM, Gao YJ. Alterations in perivascular adipose tissue structure and function in hypertension. Eur J Pharmacol 656: 68–73, 2011 [DOI] [PubMed] [Google Scholar]
- 33. Ma L, Ma S, He H, Yang D, Chen X, Luo Z, Liu D, Zhu Z. Perivascular fat-mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high-fat diet-induced obese rats. Hypertens Res 33: 446–453, 2010 [DOI] [PubMed] [Google Scholar]
- 34. Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, Liu F, Dong LQ. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 8: 516–523, 2006 [DOI] [PubMed] [Google Scholar]
- 35. McGahon MK, Dash DP, Arora A, Wall N, Dawicki J, Simpson DA, Scholfield CN, McGeown JG, Curtis TM. Diabetes downregulates large-conductance Ca2+-activated potassium beta 1 channel subunit in retinal arteriolar smooth muscle. Circ Res 100: 703–711, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. J Biol Chem 279: 36746–36752, 2004 [DOI] [PubMed] [Google Scholar]
- 37. Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, Hibuse T, Ryo M, Nishizawa H, Maeda N, Maeda K, Shibata R, Walsh K, Funahashi T, Shimomura I. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension 47: 1108–1116, 2006 [DOI] [PubMed] [Google Scholar]
- 38. Qin Y, Tian YP. Hepatic adiponectin receptor R2 expression is up-regulated in normal adult male mice by chronic exogenous growth hormone levels. Mol Med Report 3: 525–530, 2010 [DOI] [PubMed] [Google Scholar]
- 39. Ramirez-Ponce MP, Mateos JC, Bellido JA. Human adipose cells have voltage-dependent potassium currents. J Membr Biol 196: 129–134, 2003 [DOI] [PubMed] [Google Scholar]
- 40. Ramirez-Ponce MP, Mateos JC, Carrion N, Bellido JA. Voltage-dependent potassium channels in white adipocytes. Biochem Biophys Res Commun 223: 250–256, 1996 [DOI] [PubMed] [Google Scholar]
- 41. Rasmussen MS, Lihn AS, Pedersen SB, Bruun JM, Rasmussen M, Richelsen B. Adiponectin receptors in human adipose tissue: effects of obesity, weight loss, and fat depots. Obesity (Silver Spring) 14: 28–35, 2006 [DOI] [PubMed] [Google Scholar]
- 42. Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T, Crean CS, Luft FC, Huang Y, Schubert R, Gollasch M. Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28: 1875–1882, 2010 [DOI] [PubMed] [Google Scholar]
- 43. Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med 150: 776–783, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stadnicka A, Contney SJ, Moreno C, Weihrauch D, Bosnjak ZJ, Roman RJ, Stekiel TA. Mechanism of differential cardiovascular response to propofol in Dahl salt-sensitive, Brown Norway, and chromosome 13-substituted consomic rat strains: role of large conductance Ca2+ and voltage-activated potassium channels. J Pharmacol Exp Ther 330: 727–735, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sweet TB, Cox DH. Measuring the influence of the BKCa beta1 subunit on Ca2+ binding to the BKCa channel. J Gen Physiol 133: 139–150, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szasz T, Webb RC. Perivascular adipose tissue: more than just structural support. Clin Sci (Lond) 122: 1–12, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tan KC, Xu A, Chow WS, Lam MC, Ai VH, Tam SC, Lam KS. Hypoadiponectinemia is associated with impaired endothelium-dependent vasodilation. J Clin Endocrinol Metab 89: 765–769, 2004 [DOI] [PubMed] [Google Scholar]
- 48. Vassalle C, Domenici C, Lubrano V, L'Abbate A. Interaction between nitric oxide and cyclooxygenase pathways in endothelial cells. J Vasc Res 40: 491–499, 2003 [DOI] [PubMed] [Google Scholar]
- 49. Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y, Gollasch M. Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension 44: 271–276, 2004 [DOI] [PubMed] [Google Scholar]
- 50. Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R, Kadowaki T. Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem 278: 40352–40363, 2003 [DOI] [PubMed] [Google Scholar]
- 51. White RE, Kryman JP, El-Mowafy AM, Han G, Carrier GO. cAMP-dependent vasodilators cross-activate the cGMP-dependent protein kinase to stimulate BKCa channel activity in coronary artery smooth muscle cells. Circ Res 86: 897–905, 2000 [DOI] [PubMed] [Google Scholar]
- 52. Withers SB, Simpson L, Werner ME, Heagerty AM. The role of cGMP dependent protein kinase (PKG) in mediating the anticontractile function of perivascular fat (Abstract). Proc Physiol Soc 25: C11, 2011 [Google Scholar]
- 53. Woods A, Dickerson K, Heath R, Hong SP, Momcilovic M, Johnstone SR, Carlson M, Carling D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2: 21–33, 2005 [DOI] [PubMed] [Google Scholar]
- 54. Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond) 32, Suppl 7: S13–S18, 2008 [DOI] [PubMed] [Google Scholar]
- 55. Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423: 762–769, 2003 [DOI] [PubMed] [Google Scholar]
- 56. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8: 1288–1295, 2002 [DOI] [PubMed] [Google Scholar]
- 57. Yudkin JS, Eringa E, Stehouwer CD. “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet 365: 1817–1820, 2005 [DOI] [PubMed] [Google Scholar]
- 58. Zhou L, Deepa SS, Etzler JC, Ryu J, Mao X, Fang Q, Liu DD, Torres JM, Jia W, Lechleiter JD, Liu F, Dong LQ. Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J Biol Chem 284: 22426–22435, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]